Recent Advances in the Catalyst Design and Mass Transport Control for the Electrochemical Reduction of Carbon Dioxide to Formate


Abstract
1. Introduction
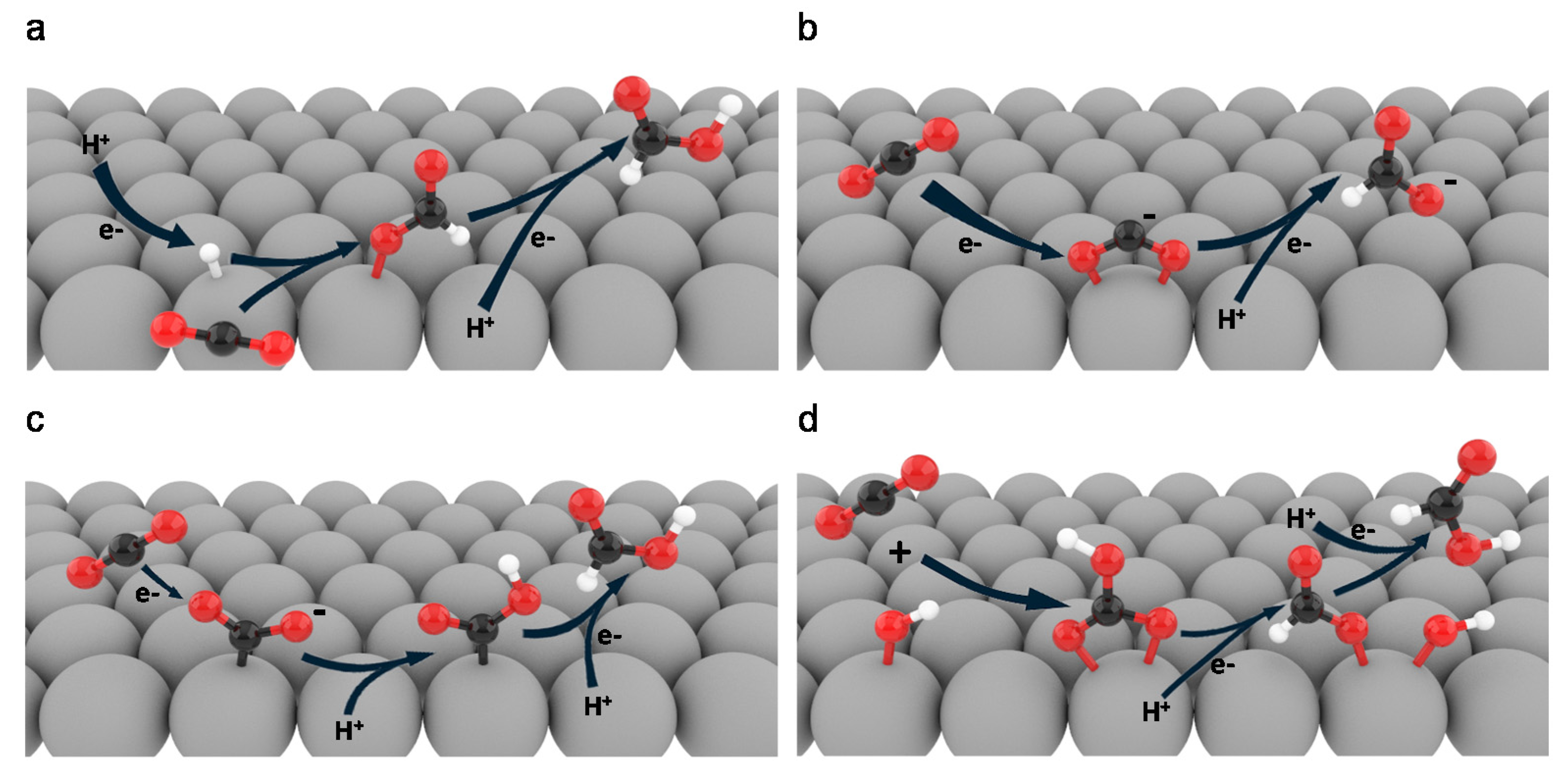
2. Reaction Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
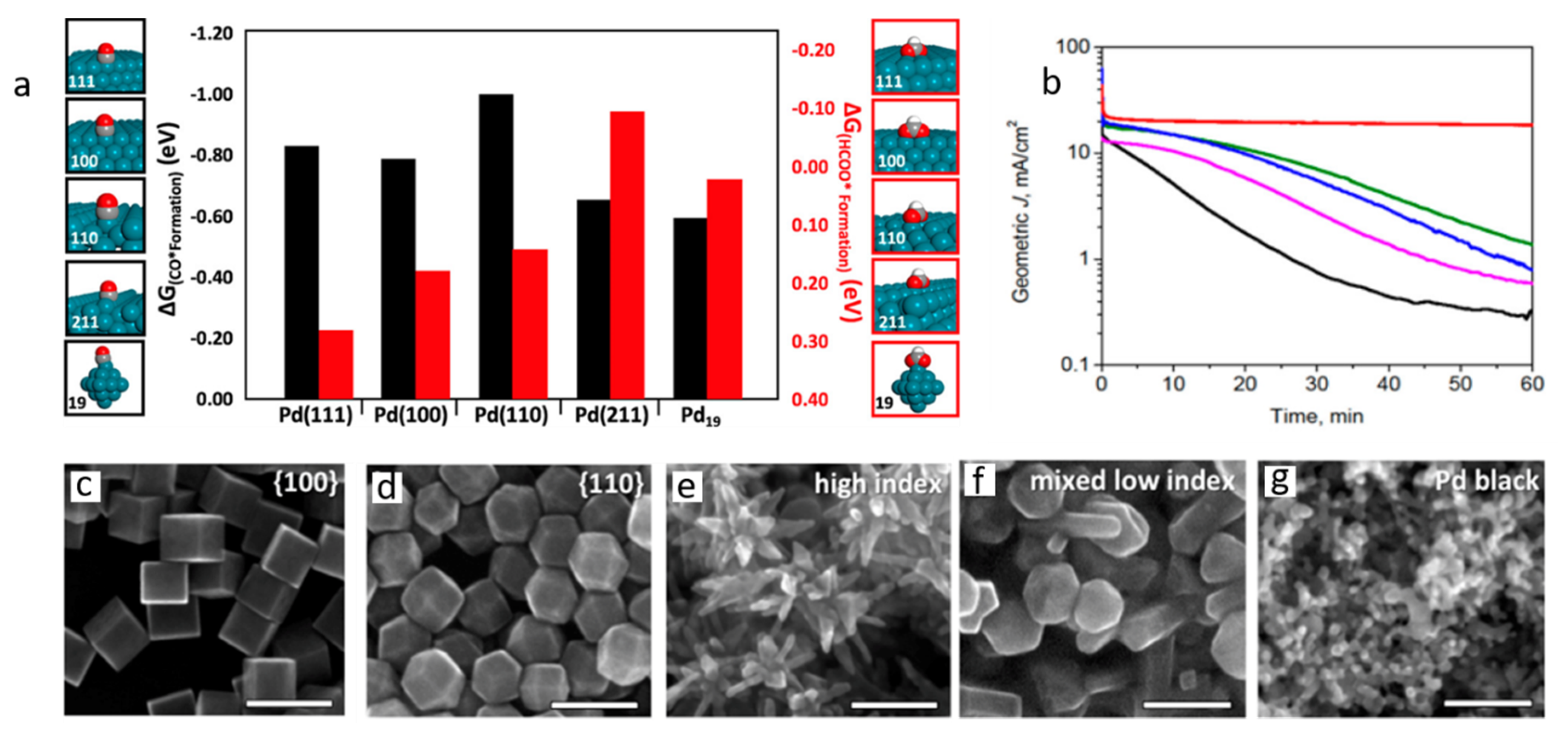
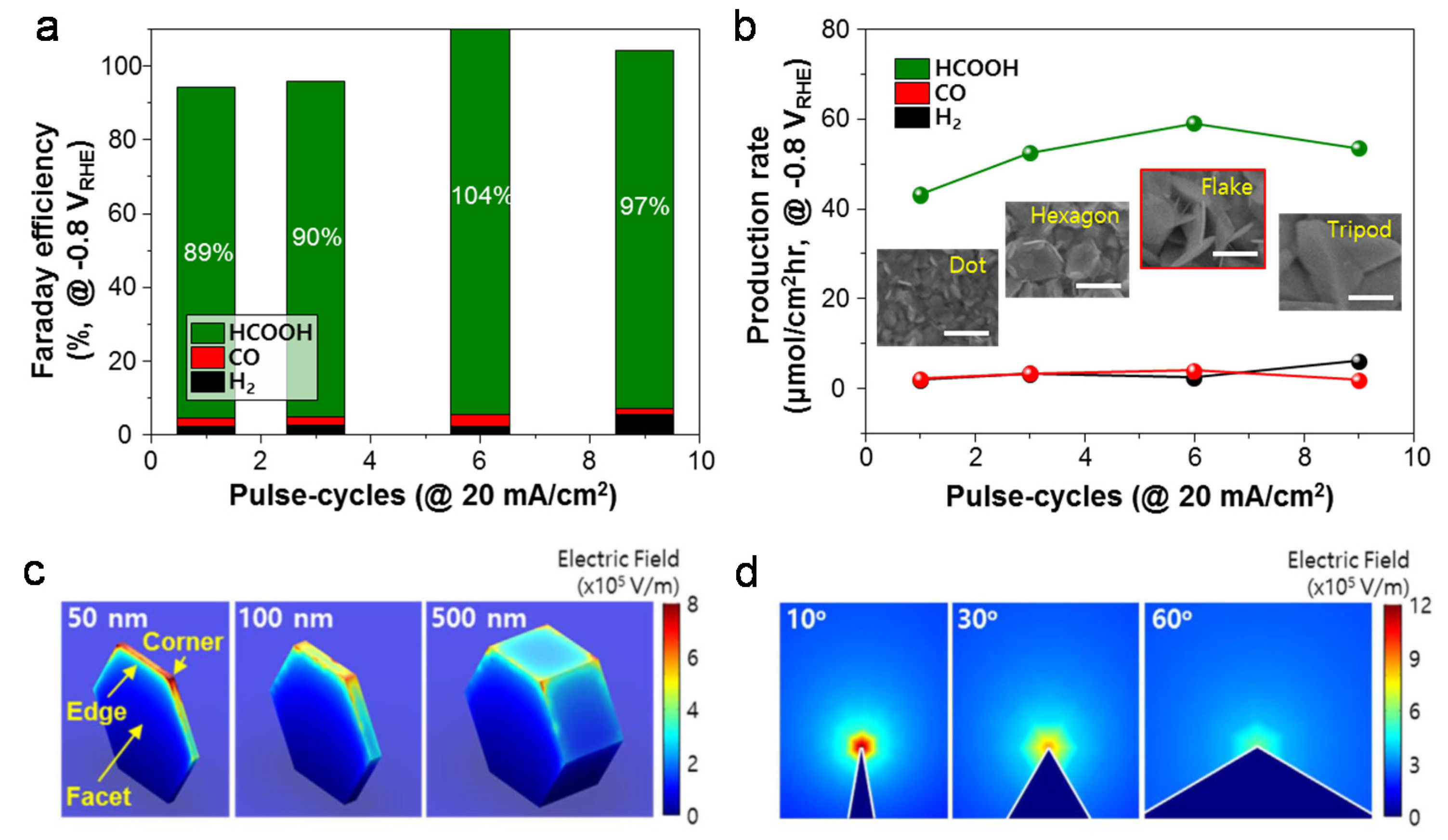
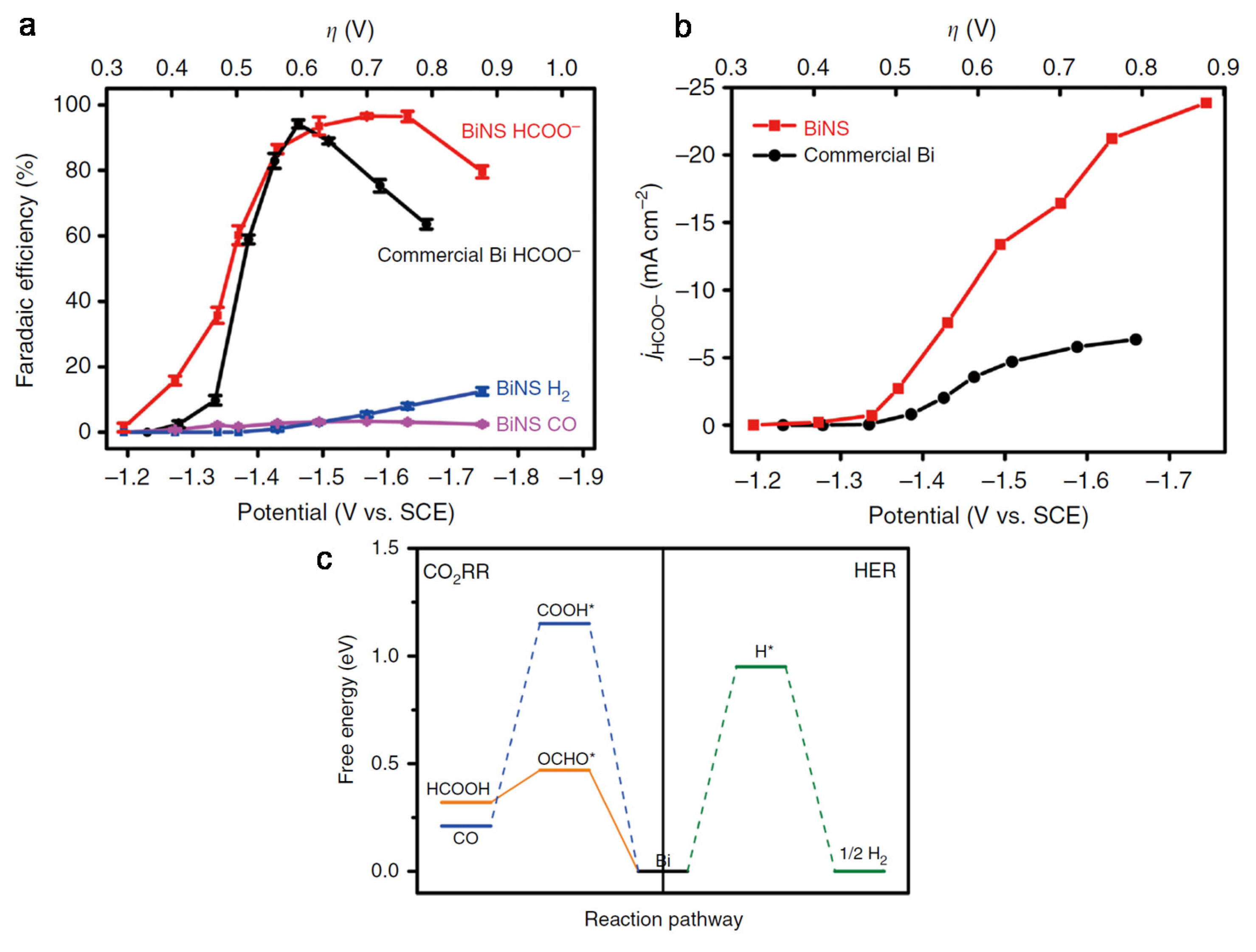
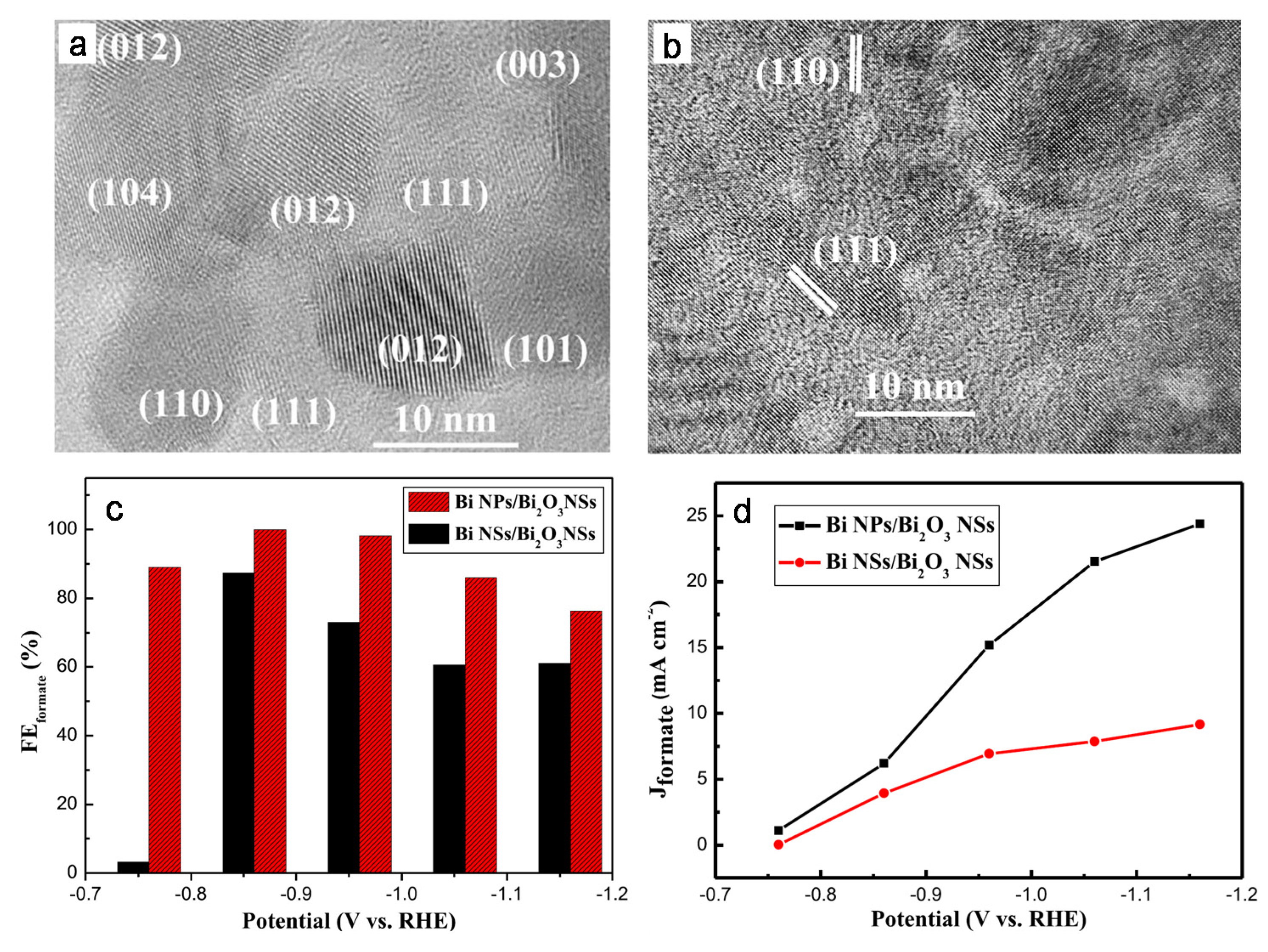
3. Nanostructural Engineering
4. Composition
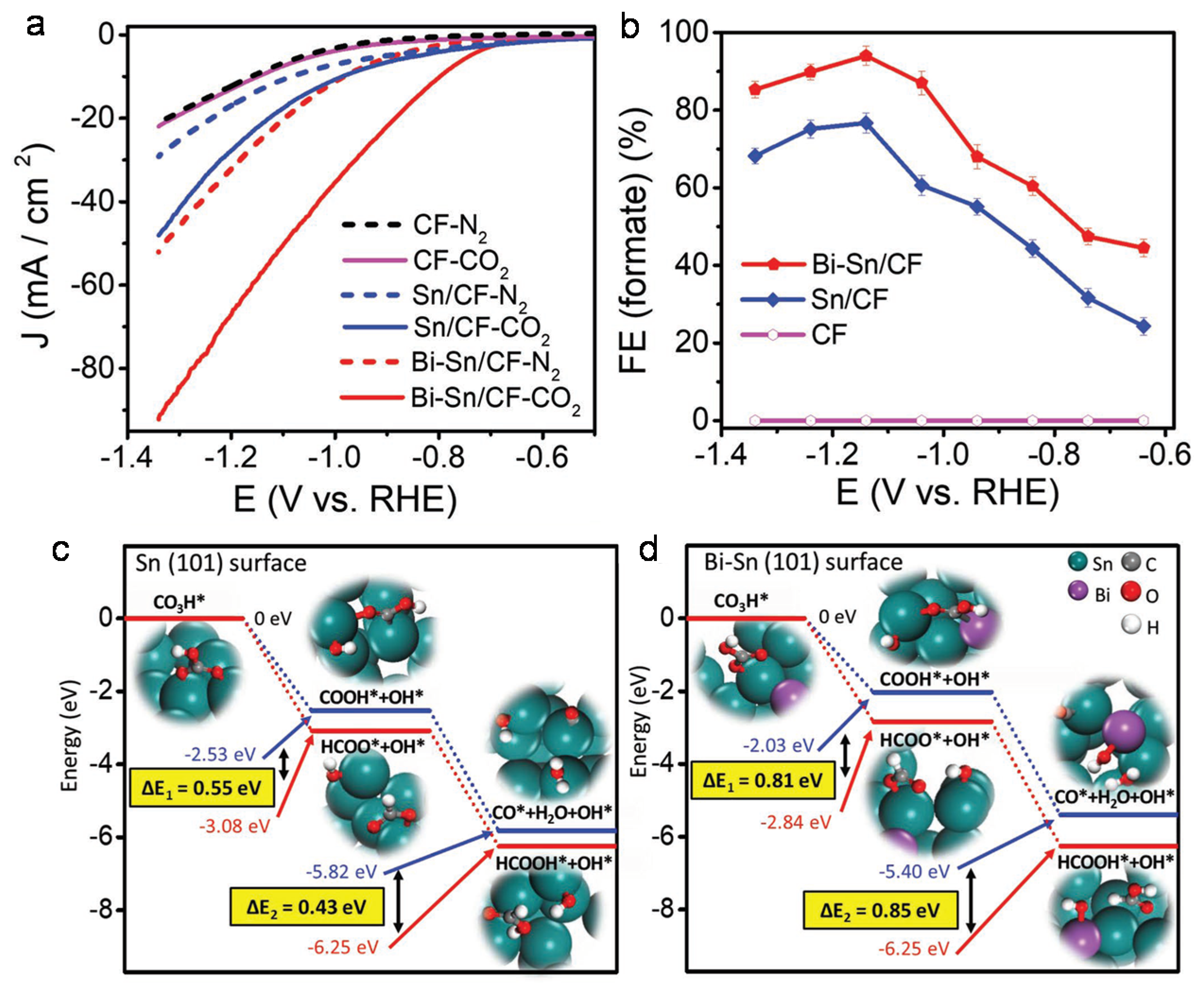
4.1. Bimetallic Compounds
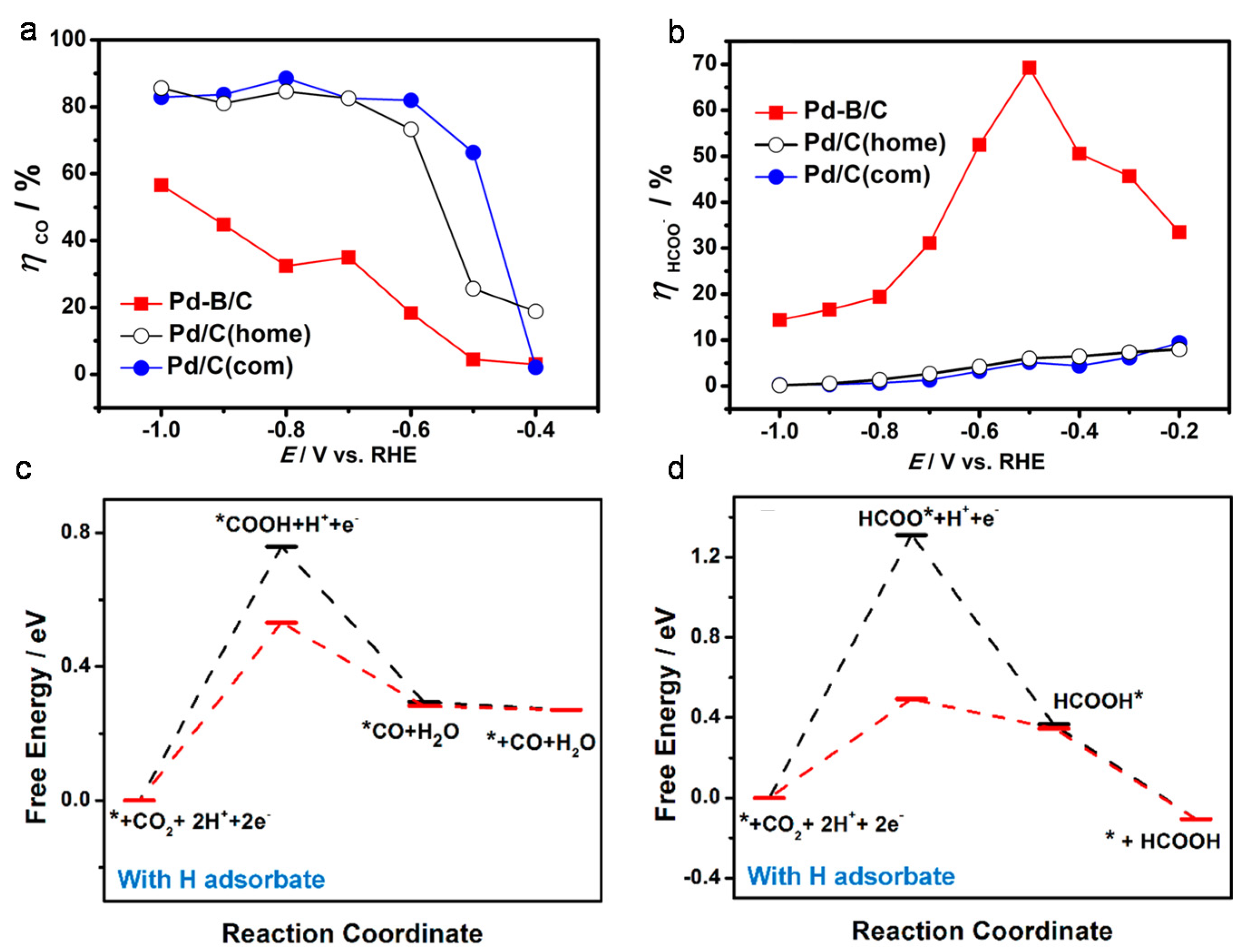
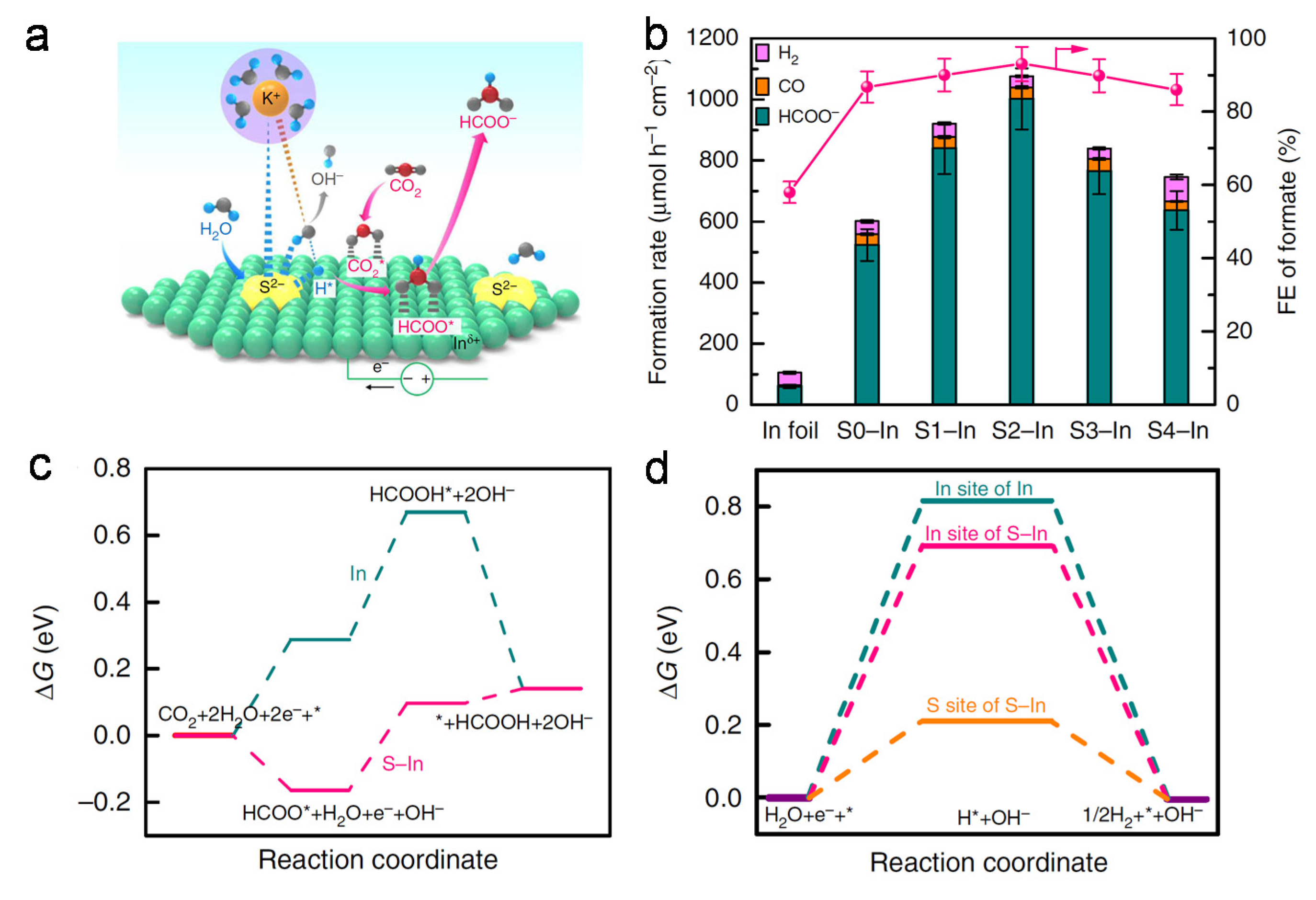
4.2. Doping Materials
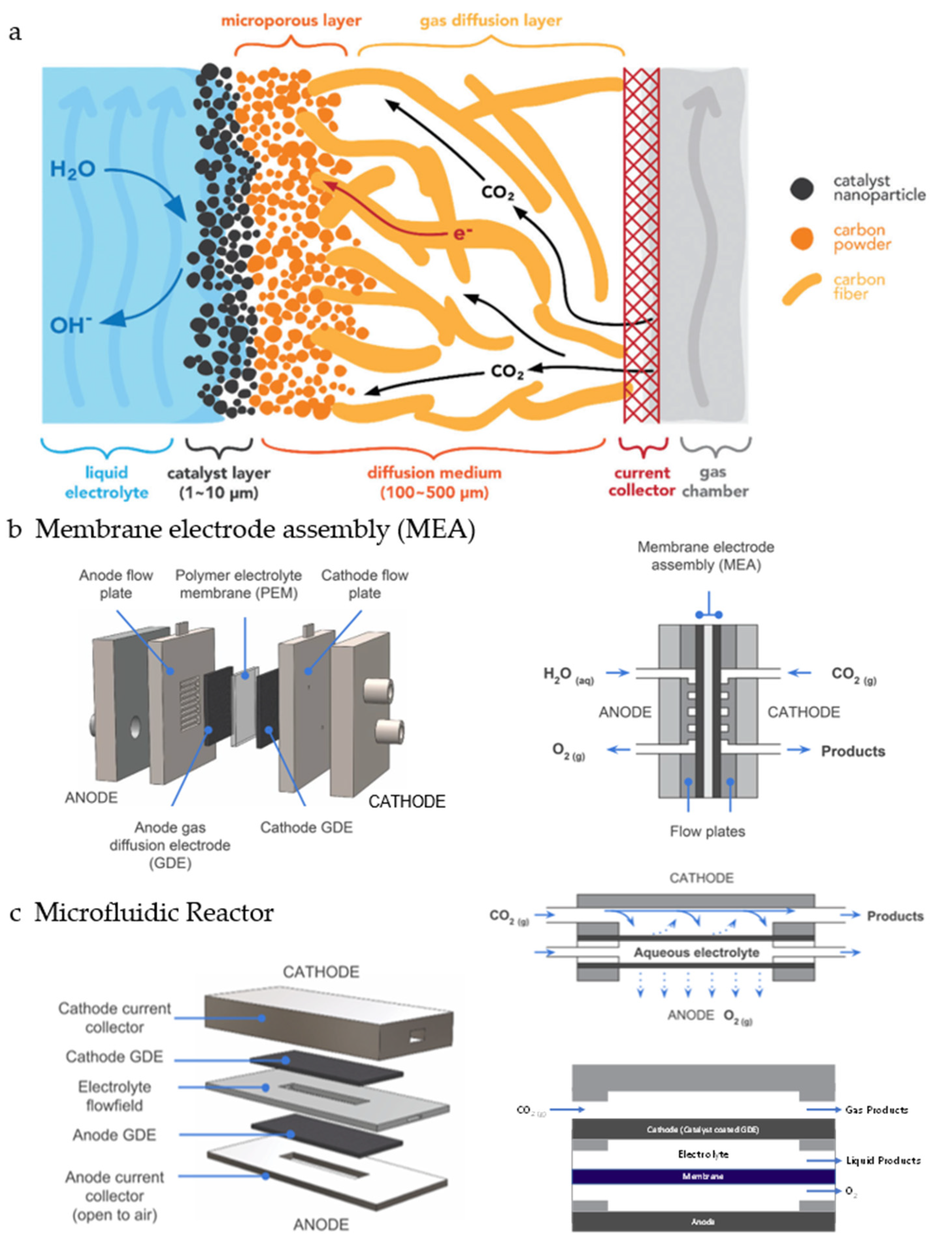
5. Mass Transport
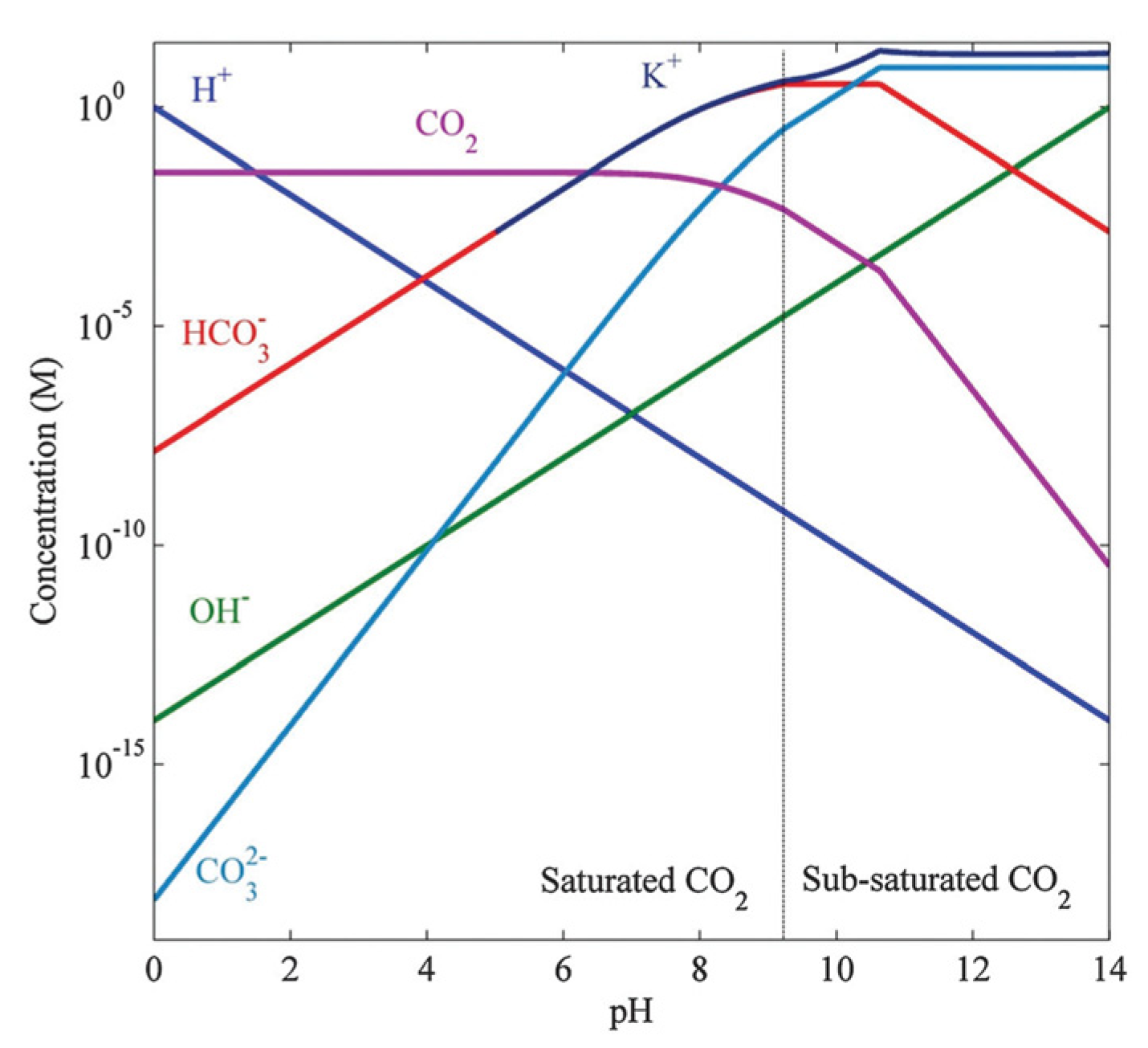
5.1. Local pH Control
5.2. High-Pressure Reactor
5.3. Three-Phase Boundary
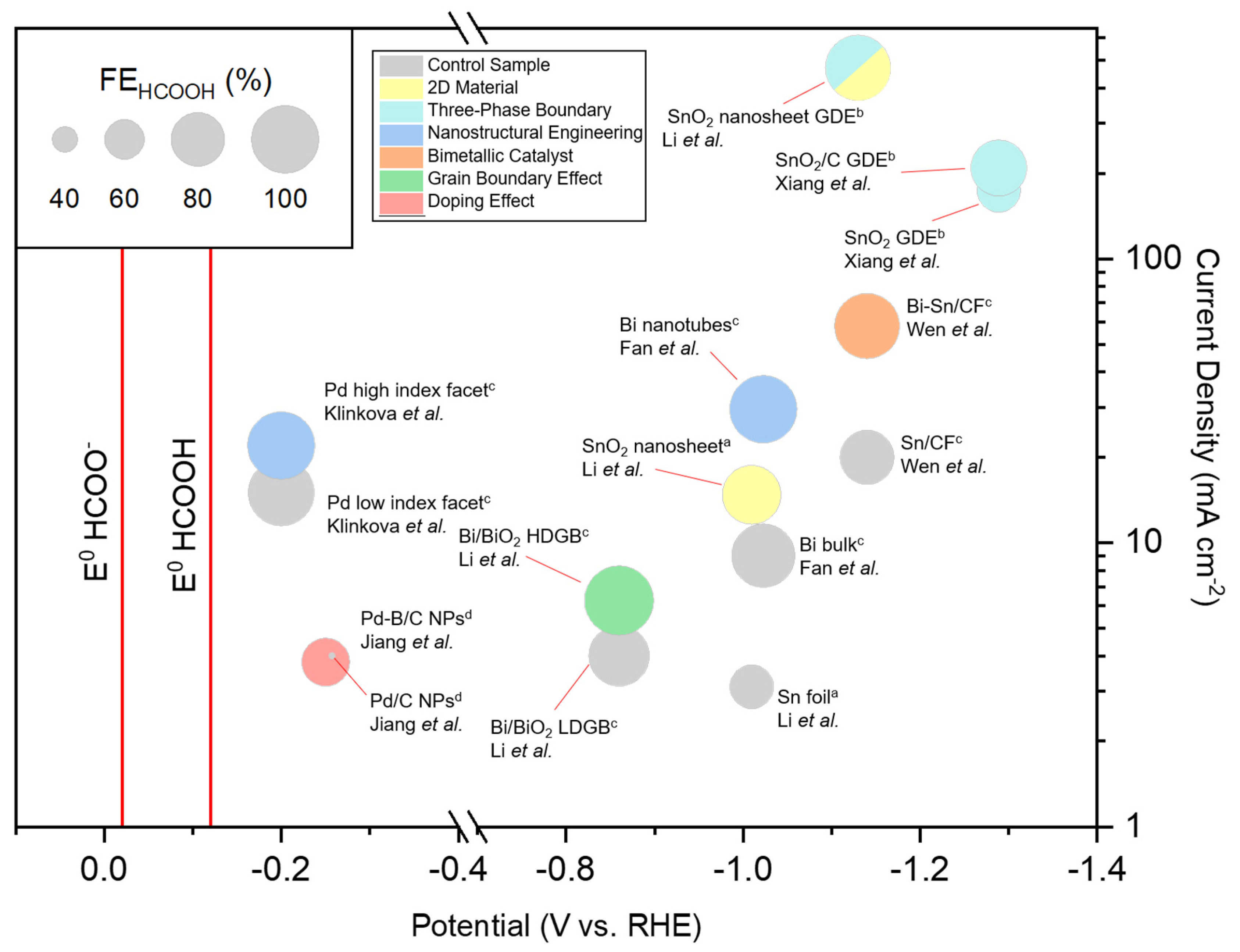
6. Reported Performances
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karl, T.R.; Trenberth, K.E. Modern Global Climate Change. Science 2003, 302, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Pachauri, R.K.; Meyer, L.A. IPCC, 2014: Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2015. [Google Scholar]
- King, A.D.; Karoly, D.J.; Henley, B.J. Australian climate extremes at 1.5 °C and 2 °C of global warming. Nat. Clim. Chang. 2017, 7, 412–416. [Google Scholar] [CrossRef]
- Lee, C.W.; Cho, N.H.; Nam, K.T.; Hwang, Y.J.; Min, B.K. Cyclic two-step electrolysis for stable electrochemical conversion of carbon dioxide to formate. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.K.S.; Viva, F.A.; Olah, G.A. Electrochemical reduction of CO2 over Sn-Nafion® coated electrode for a fuel-cell-like device. J. Power Sources 2013, 223, 68–73. [Google Scholar] [CrossRef]
- Bagotzky, V.S.; Osetrova, N.V. Electrochemical reduction of carbon dioxide. Russ. J. Electrochem. 1995, 31, 409–425. [Google Scholar]
- Agarwal, A.S.; Zhai, Y.; Hill, D.; Sridhar, N. The Electrochemical Reduction of Carbon Dioxide to Formate/Formic Acid: Engineering and Economic Feasibility. ChemSusChem 2011, 4, 1301–1310. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Gärtner, F.; Junge, H.; Beller, M. Catalytic Generation of Hydrogen from Formic acid and its Derivatives: Useful Hydrogen Storage Materials. Top. Catal. 2010, 53, 902–914. [Google Scholar] [CrossRef]
- Fellay, C.; Yan, N.; Dyson, P.J.; Laurenczy, G. Selective Formic Acid Decomposition for High-Pressure Hydrogen Generation: A Mechanistic Study. Chem. A Eur. J. 2009, 15, 3752–3760. [Google Scholar] [CrossRef]
- Xiang, H.; Miller, H.A.; Bellini, M.; Christensen, H.; Scott, K.; Rasul, S.; Yu, E.H. Production of formate by CO2 electrochemical reduction and its application in energy storage. Sustain. Energy Fuels 2019, 4, 277–284. [Google Scholar] [CrossRef]
- Lee, C.W.; Yang, K.D.; Nam, D.H.; Jang, J.H.; Cho, N.H.; Im, S.W.; Nam, K.T. Defining a Materials Database for the Design of Copper Binary Alloy Catalysts for Electrochemical CO2 Conversion. Adv. Mater. 2018, 30, 1–18. [Google Scholar] [CrossRef]
- Lee, C.W.; Kim, C.; Min, B.K. Theoretical insights into selective electrochemical conversion of carbon dioxide. Nano Converg. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [PubMed]
- Rumayor, M.; Dominguez-Ramos, A.; Perez, P.; Irabien, A. A techno-economic evaluation approach to the electrochemical reduction of CO2 for formic acid manufacture. J. CO2 Util. 2019, 34, 490–499. [Google Scholar] [CrossRef]
- Rumayor, M.; Dominguez-Ramos, A.; Irabien, A. Formic Acid Manufacture: Carbon Dioxide Utilization Alternatives. Appl. Sci. 2018, 8, 914. [Google Scholar] [CrossRef]
- Na, J.; Seo, B.; Kim, J.; Lee, C.W.; Lee, H.; Hwang, Y.J.; Min, B.K.; Lee, D.K.; Oh, H.S.; Lee, U. General technoeconomic analysis for electrochemical coproduction coupling carbon dioxide reduction with organic oxidation. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Verma, S.; Kim, B.; Molly Jhong, H.-R.; Ma, S.; Kenis, P.J.A. A Gross-Margin Model for Defining Technoeconomic Benchmarks in the Electroreduction of CO2. ChemSusChem 2016, 9, 1972–1979. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef]
- Yoo, J.S.; Christensen, R.; Vegge, T.; Nørskov, J.K.; Studt, F. Theoretical Insight into the Trends that Guide the Electrochemical Reduction of Carbon Dioxide to Formic Acid. ChemSusChem 2016, 9, 358–363. [Google Scholar] [CrossRef]
- Medina-Ramos, J.; DiMeglio, J.L.; Rosenthal, J. Efficient Reduction of CO2 to CO with High Current Density Using in Situ or ex Situ Prepared Bi-Based Materials. J. Am. Chem. Soc. 2014, 136, 8361–8367. [Google Scholar] [CrossRef]
- Nguyen, D.L.T.; Lee, C.W.; Na, J.; Kim, M.C.; Tu, N.D.K.; Lee, S.Y.; Sa, Y.J.; Won, D.H.; Oh, H.S.; Kim, H.; et al. Mass Transport Control by Surface Graphene Oxide for Selective CO Production from Electrochemical CO2 Reduction. Acs Catal. 2020, 10, 3222–3231. [Google Scholar] [CrossRef]
- Lee, C.W.; Cho, N.H.; Im, S.W.; Jee, M.S.; Hwang, Y.J.; Min, B.K.; Nam, K.T. New challenges of electrokinetic studies in investigating the reaction mechanism of electrochemical CO2 reduction. J. Mater. Chem. A 2018, 6, 14043–14057. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. J. Catal. 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Hoffman, Z.B.; Gray, T.S.; Xu, Y.; Lin, Q.; Gunnoe, T.B.; Zangari, G. High Selectivity Towards Formate Production by Electrochemical Reduction of Carbon Dioxide at Copper–Bismuth Dendrites. ChemSusChem 2019, 12, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Huang, Y.; Ren, D.; Handoko, A.D.; Seh, Z.W.; Hirunsit, P.; Yeo, B.S. On the Role of Sulfur for the Selective Electrochemical Reduction of CO2 to Formate on CuSx Catalysts. Acs Appl. Mater. Interfaces 2018, 10, 28572–28581. [Google Scholar] [CrossRef]
- Lee, C.W.; Shin, S.J.; Jung, H.; Nguyen, D.L.T.; Lee, S.Y.; Lee, W.H.; Won, D.H.; Kim, M.G.; Oh, H.S.; Jang, T.; et al. Metal-Oxide Interfaces for Selective Electrochemical C-C Coupling Reactions. Acs Energy Lett. 2019, 4, 2241–2248. [Google Scholar] [CrossRef]
- He, J.; Johnson, N.J.J.; Huang, A.; Berlinguette, C.P. Electrocatalytic Alloys for CO2 Reduction. ChemSusChem 2018, 11, 48–57. [Google Scholar] [CrossRef]
- Saberi Safaei, T.; Mepham, A.; Zheng, X.; Pang, Y.; Dinh, C.-T.; Liu, M.; Sinton, D.; Kelley, S.O.; Sargent, E.H. High-Density Nanosharp Microstructures Enable Efficient CO2 Electroreduction. Nano Lett. 2016, 16, 7224–7228. [Google Scholar] [CrossRef]
- Lee, H.-E.; Yang, K.D.; Yoon, S.M.; Ahn, H.-Y.; Lee, Y.Y.; Chang, H.; Jeong, D.H.; Lee, Y.-S.; Kim, M.Y.; Nam, K.T. Concave Rhombic Dodecahedral Au Nanocatalyst with Multiple High-Index Facets for CO2 Reduction. Acs Nano 2015, 9, 8384–8393. [Google Scholar] [CrossRef]
- Yang, D.-R.; Liu, L.; Zhang, Q.; Shi, Y.; Zhou, Y.; Liu, C.; Wang, F.-B.; Xia, X.-H. Importance of Au nanostructures in CO2 electrochemical reduction reaction. Sci. Bull. 2020, 65, 796–802. [Google Scholar] [CrossRef]
- Kim, S.; Dong, W.J.; Gim, S.; Sohn, W.; Park, J.Y.; Yoo, C.J.; Jang, H.W.; Lee, J.L. Shape-controlled bismuth nanoflakes as highly selective catalysts for electrochemical carbon dioxide reduction to formate. Nano Energy 2017, 39, 44–52. [Google Scholar] [CrossRef]
- Lamaison, S.; Wakerley, D.; Blanchard, J.; Montero, D.; Rousse, G.; Mercier, D.; Marcus, P.; Taverna, D.; Giaume, D.; Mougel, V.; et al. High-Current-Density CO2-to-CO Electroreduction on Ag-Alloyed Zn Dendrites at Elevated Pressure. Joule 2020, 4, 395–406. [Google Scholar] [CrossRef]
- Kopljar, D.; Inan, A.; Vindayer, P.; Wagner, N.; Klemm, E. Electrochemical reduction of CO2 to formate at high current density using gas diffusion electrodes. J. Appl. Electrochem. 2014, 44, 1107–1116. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Y.; Ma, J.; Zhang, Z.; Hu, W. Recent Advances in Atomic-Level Engineering of Nanostructured Catalysts for Electrochemical CO2 Reduction. Adv. Funct. Mater. 2020, 30, 1910534. [Google Scholar] [CrossRef]
- Lee, C.W.; Cho, N.H.; Yang, K.D.; Nam, K.T. Reaction Mechanisms of the Electrochemical Conversion of Carbon Dioxide to Formic Acid on Tin Oxide Electrodes. ChemElectroChem 2017, 4, 2130–2136. [Google Scholar] [CrossRef]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Kanan, M.W. Pd-Catalyzed Electrohydrogenation of Carbon Dioxide to Formate: High Mass Activity at Low Overpotential and Identification of the Deactivation Pathway. J. Am. Chem. Soc. 2015, 137, 4701–4708. [Google Scholar] [CrossRef] [PubMed]
- Podlovchenko, B.I.; Kolyadko, E.A.; Lu, S. Electroreduction of carbon dioxide on palladium electrodes at potentials higher than the reversible hydrogen potential. J. Electroanal. Chem. 1994, 373, 185–187. [Google Scholar] [CrossRef]
- Stalder, C.J.; Chao, S.; Wrighton, M.S. Electrochemical reduction of aqueous bicarbonate to formate with high current efficiency near the thermodynamic potential at chemically derivatized electrodes. J. Am. Chem. Soc. 1984, 106, 3673–3675. [Google Scholar] [CrossRef]
- Chaplin, R.P.S.; Wragg, A.A. Effects of process conditions and electrode material on reaction pathways for carbon dioxide electroreduction with particular reference to formate formation. J. Appl. Electrochem. 2003, 33, 1107–1123. [Google Scholar] [CrossRef]
- Lai, Q.; Yang, N.; Yuan, G. Highly efficient In–Sn alloy catalysts for electrochemical reduction of CO2 to formate. Electrochem. Commun. 2017, 83, 24–27. [Google Scholar] [CrossRef]
- Lee, C.W.; Hong, J.S.; Yang, K.D.; Jin, K.; Lee, J.H.; Ahn, H.Y.; Seo, H.; Sung, N.E.; Nam, K.T. Selective Electrochemical Production of Formate from Carbon Dioxide with Bismuth-Based Catalysts in an Aqueous Electrolyte. Acs Catal. 2018, 8, 931–937. [Google Scholar] [CrossRef]
- Sullivan, B.P.; Krist, K.; Guard, H.E. Electrochemical and Electrocatalytic Reactions of Carbon Dioxide; Elsevier: Amsterdam, The Netherlands, 1993. [Google Scholar] [CrossRef]
- Jitaru, M.; Lowy, D.A.; Toma, M.; Toma, B.C.; Oniciu, L. Electrochemical reduction of carbon dioxide on flat metallic cathodes. J. Appl. Electrochem. 1997, 27, 875–889. [Google Scholar] [CrossRef]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Komatsu, S.; Tanaka, M.; Okumura, A.; Kungi, A. Preparation of cu-solid polymer electrolyte composite electrodes and application to gas-phase electrochemical reduction of CO2. Electrochim. Acta 1995, 40, 745–753. [Google Scholar] [CrossRef]
- Wen, G.; Lee, D.U.; Ren, B.; Hassan, F.M.; Jiang, G.; Cano, Z.P.; Gostick, J.; Croiset, E.; Bai, Z.; Yang, L.; et al. Orbital Interactions in Bi-Sn Bimetallic Electrocatalysts for Highly Selective Electrochemical CO2 Reduction toward Formate Production. Adv. Energy Mater. 2018, 8, 1802427. [Google Scholar] [CrossRef]
- Sreekanth, N.; Phani, K.L. Selective reduction of CO2 to formate through bicarbonate reduction on metal electrodes: New insights gained from SG/TC mode of SECM. Chem. Commun. 2014, 50, 11143–11146. [Google Scholar] [CrossRef]
- Baruch, M.F.; Pander, J.E.; White, J.L.; Bocarsly, A.B. Mechanistic Insights into the Reduction of CO2 on Tin Electrodes using in Situ ATR-IR Spectroscopy. Acs Catal. 2015, 5, 3148–3156. [Google Scholar] [CrossRef]
- Bai, X.; Chen, W.; Zhao, C.; Li, S.; Song, Y.; Ge, R.; Wei, W.; Sun, Y. Exclusive Formation of Formic Acid from CO2 Electroreduction by a Tunable Pd-Sn Alloy. Angew. Chem. Int. Ed. 2017, 56, 12219–12223. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Meyer, T.J. Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. J. Am. Chem. Soc. 2014, 136, 1734–1737. [Google Scholar] [CrossRef]
- Durand, W.J.; Peterson, A.A.; Studt, F.; Abild-Pedersen, F.; Nørskov, J.K. Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces. Surf. Sci. 2011, 605, 1354–1359. [Google Scholar] [CrossRef]
- Klinkova, A.; De Luna, P.; Dinh, C.T.; Voznyy, O.; Larin, E.M.; Kumacheva, E.; Sargent, E.H. Rational Design of Efficient Palladium Catalysts for Electroreduction of Carbon Dioxide to Formate. Acs Catal. 2016, 6, 8115–8120. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, G.; Zhang, Z.; Jin, M.; Yin, Y. Selectivity on Etching: Creation of High-Energy Facets on Copper Nanocrystals for CO2 Electrochemical Reduction. Acs Nano 2016, 10, 4559–4564. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Han, N.; Deng, J.; Wu, J.; Wang, Y.; Hu, Y.; Ding, P.; Li, Y.; Li, Y.; Lu, J. Selective CO2 Reduction on 2D Mesoporous Bi Nanosheets. Adv. Energy Mater. 2018, 8, 1801536. [Google Scholar] [CrossRef]
- Han, N.; Wang, Y.; Yang, H.; Deng, J.; Wu, J.; Li, Y.; Li, Y. Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Gao, S.; Lin, Y.; Jiao, X.; Sun, Y.; Luo, Q.; Zhang, W.; Li, D.; Yang, J.; Xie, Y. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 2016, 529, 68–71. [Google Scholar] [CrossRef]
- Liu, M.; Pang, Y.; Zhang, B.; de Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C.T.; Fan, F.; Changhong, C.; et al. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 2016, 537, 382–386. [Google Scholar] [CrossRef]
- Li, C.W.; Ciston, J.; Kanan, M.W. Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper. Nature 2014, 508, 504–507. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. Grain-Boundary-Dependent CO2 Electroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. [Google Scholar] [CrossRef]
- Li, L.; Ma, D.K.; Qi, F.; Chen, W.; Huang, S. Bi nanoparticles/Bi2O3 nanosheets with abundant grain boundaries for efficient electrocatalytic CO2 reduction. Electrochim. Acta 2019, 298, 580–586. [Google Scholar] [CrossRef]
- Park, H.; Choi, J.; Kim, H.; Hwang, E.; Ha, D.H.; Ahn, S.H.; Kim, S.K. AgIn dendrite catalysts for electrochemical reduction of CO2 to CO. Appl. Catal. B Environ. 2017, 219, 123–131. [Google Scholar] [CrossRef]
- Luc, W.; Collins, C.; Wang, S.; Xin, H.; He, K.; Kang, Y.; Jiao, F. Ag–Sn Bimetallic Catalyst with a Core–Shell Structure for CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Yang, H.; Deng, J.; Chen, J.; Zhou, Y.; Ding, P.; Li, L.; Han, N.; Li, Y. Copper-Bismuth Bimetallic Microspheres for Selective Electrocatalytic Reduction of CO2 to Formate. Chin. J. Chem. 2019, 37, 497–500. [Google Scholar] [CrossRef]
- Ye, K.; Cao, A.; Shao, J.; Wang, G.; Si, R.; Ta, N.; Xiao, J.; Wang, G. Synergy effects on Sn-Cu alloy catalyst for efficient CO2 electroreduction to formate with high mass activity. Sci. Bull. 2020, 1–9. [Google Scholar] [CrossRef]
- Peng, L.; Wang, Y.; Masood, I.; Zhou, B.; Wang, Y.; Lin, J.; Qiao, J.; Zhang, F.Y. Self-growing Cu/Sn bimetallic electrocatalysts on nitrogen-doped porous carbon cloth with 3D-hierarchical honeycomb structure for highly active carbon dioxide reduction. Appl. Catal. B Environ. 2020, 264, 118447. [Google Scholar] [CrossRef]
- Chatterjee, S.; Griego, C.; Hart, J.L.; Li, Y.; Taheri, M.L.; Keith, J.; Snyder, J.D. Free Standing Nanoporous Palladium Alloys as CO Poisoning Tolerant Electrocatalysts for the Electrochemical Reduction of CO2 to Formate. Acs Catal. 2019, 9, 5290–5301. [Google Scholar] [CrossRef]
- Wang, H.; Maiyalagan, T.; Wang, X. Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications. Acs Catal. 2012, 2, 781–794. [Google Scholar] [CrossRef]
- Chen, Z.; Mou, K.; Wang, X.; Liu, L. Nitrogen-Doped Graphene Quantum Dots Enhance the Activity of Bi2O3 Nanosheets for Electrochemical Reduction of CO2 in a Wide Negative Potential Region. Angew. Chem. Int. Ed. 2018, 57, 12790–12794. [Google Scholar] [CrossRef]
- Zhao, Y.; Liang, J.; Wang, C.; Ma, J.; Wallace, G.G. Tunable and Efficient Tin Modified Nitrogen-Doped Carbon Nanofibers for Electrochemical Reduction of Aqueous Carbon Dioxide. Adv. Energy Mater. 2018, 8. [Google Scholar] [CrossRef]
- Gao, D.; Zhou, H.; Cai, F.; Wang, D.; Hu, Y.; Jiang, B.; Cai, W.-B.; Chen, X.; Si, R.; Yang, F.; et al. Switchable CO2 electroreduction via engineering active phases of Pd nanoparticles. Nano Res. 2017, 10, 2181–2191. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, X.G.; Jiang, K.; Wu, D.Y.; Cai, W. Bin. Boosting Formate Production in Electrocatalytic CO2 Reduction over Wide Potential Window on Pd Surfaces. J. Am. Chem. Soc. 2018, 140, 2880–2889. [Google Scholar] [CrossRef]
- Ma, W.; Xie, S.; Zhang, X.G.; Sun, F.; Kang, J.; Jiang, Z.; Zhang, Q.; Wu, D.Y.; Wang, Y. Promoting electrocatalytic CO2 reduction to formate via sulfur-boosting water activation on indium surfaces. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Raciti, D.; Mao, M.; Wang, C. Mass transport modelling for the electroreduction of CO2 on Cu nanowires. Nanotechnology 2018, 29, 044001. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Huo, G.; Chen, W.Y.; Fu, X.Z.; Luo, J.L. Boosting formate production at high current density from CO2 electroreduction on defect-rich hierarchical mesoporous Bi/Bi2O3 junction nanosheets. Appl. Catal. B Environ. 2020, 271, 118957. [Google Scholar] [CrossRef]
- Schouten, K.J.P.; Pérez Gallent, E.; Koper, M.T.M. The influence of pH on the reduction of CO and CO2 to hydrocarbons on copper electrodes. J. Electroanal. Chem. 2014, 716, 53–57. [Google Scholar] [CrossRef]
- Singh, M.R.; Clark, E.L.; Bell, A.T. Effects of electrolyte, catalyst, and membrane composition and operating conditions on the performance of solar-driven electrochemical reduction of carbon dioxide. Phys. Chem. Chem. Phys. 2015, 17, 18924–18936. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Zhang, J.; Li, M.; Züttel, A. Boosting CO Production in Electrocatalytic CO2 Reduction on Highly Porous Zn Catalysts. Acs Catal. 2019, 9, 3783–3791. [Google Scholar] [CrossRef]
- Ma, M.; Trześniewski, B.J.; Xie, J.; Smith, W.A. Selective and Efficient Reduction of Carbon Dioxide to Carbon Monoxide on Oxide-Derived Nanostructured Silver Electrocatalysts. Angew. Chem. Int. Ed. 2016, 55, 9748–9752. [Google Scholar] [CrossRef]
- Todoroki, M.; Hara, K.; Kudo, A.; Sakata, T. Electrochemical reduction of high pressure CO2 at Pb, Hg and In electrodes in an aqueous KHCO3 solution. J. Electroanal. Chem. 1995, 394, 199–203. [Google Scholar] [CrossRef]
- Ramdin, M.; Morrison, A.R.T.; de Groen, M.; van Haperen, R.; de Kler, R.; van den Broeke, L.J.P.; Trusler, J.P.M.; de Jong, W.; Vlugt, T.J.H. High Pressure Electrochemical Reduction of CO2 to Formic Acid/Formate: A Comparison between Bipolar Membranes and Cation Exchange Membranes. Ind. Eng. Chem. Res. 2019, 58, 1834–1847. [Google Scholar] [CrossRef]
- Merino-Garcia, I.; Alvarez-Guerra, E.; Albo, J.; Irabien, A. Electrochemical membrane reactors for the utilisation of carbon dioxide. Chem. Eng. J. 2016, 305, 104–120. [Google Scholar] [CrossRef]
- Pletcher, D. The cathodic reduction of carbon dioxide—What can it realistically achieve? A mini review. Electrochem. Commun. 2015, 61, 97–101. [Google Scholar] [CrossRef]
- Motoo, S.; Watanabe, M.; Furuya, N. Gas diffusion electrode of high performance. J. Electroanal. Chem. Interfacial Electrochem. 1984, 160, 351–357. [Google Scholar] [CrossRef]
- Weekes, D.M.; Salvatore, D.A.; Reyes, A.; Huang, A.; Berlinguette, C.P. Electrolytic CO2 Reduction in a Flow Cell. Acc. Chem. Res. 2018, 51, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Delafontaine, L.; Asset, T.; Atanassov, P. Metal–Nitrogen–Carbon Electrocatalysts for CO2 Reduction towards Syngas Generation. ChemSusChem 2020, 13, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.C.; Bell, A.T.; Weber, A.Z. Modeling gas-diffusion electrodes for CO2 reduction. Phys. Chem. Chem. Phys. 2018, 20, 16973–16984. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Song, H.; Kim, B.; Oh, J. Towards Higher Rate Electrochemical CO2 Conversion: From Liquid-Phase to Gas-Phase Systems. Catalysts 2019, 9, 224. [Google Scholar] [CrossRef]
- Del Castillo, A.; Alvarez-Guerra, M.; Solla-Gullón, J.; Sáez, A.; Montiel, V.; Irabien, A. Sn nanoparticles on gas diffusion electrodes: Synthesis, characterization and use for continuous CO2 electroreduction to formate. J. Co2 Util. 2017, 18, 222–228. [Google Scholar] [CrossRef]
- Jayashree, R.S.; Yoon, S.K.; Brushett, F.R.; Lopez-Montesinos, P.O.; Natarajan, D.; Markoski, L.J.; Kenis, P.J.A. On the performance of membraneless laminar flow-based fuel cells. J. Power Sources 2010, 195, 3569–3578. [Google Scholar] [CrossRef]
- Whipple, D.T.; Finke, E.C.; Kenis, P.J.A. Microfluidic Reactor for the Electrochemical Reduction of Carbon Dioxide: The Effect of pH. Electrochem. Solid-State Lett. 2010, 13, B109. [Google Scholar] [CrossRef]
- Li, J.; Jiao, J.; Zhang, H.; Zhu, P.; Ma, H.; Chen, C.; Xiao, H.; Lu, Q. Two-Dimensional SnO2 Nanosheets for Efficient Carbon Dioxide Electroreduction to Formate. Acs Sustain. Chem. Eng. 2020, 8, 4975–4982. [Google Scholar] [CrossRef]
- Fan, K.; Jia, Y.; Ji, Y.; Kuang, P.; Zhu, B.; Liu, X.; Yu, J. Curved Surface Boosts Electrochemical CO2 Reduction to Formate via Bismuth Nanotubes in a Wide Potential Window. Acs Catal. 2020, 10, 358–364. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfath, M.; Lee, C.W. Recent Advances in the Catalyst Design and Mass Transport Control for the Electrochemical Reduction of Carbon Dioxide to Formate. Catalysts 2020, 10, 859. https://doi.org/10.3390/catal10080859
Alfath M, Lee CW. Recent Advances in the Catalyst Design and Mass Transport Control for the Electrochemical Reduction of Carbon Dioxide to Formate. Catalysts. 2020; 10(8):859. https://doi.org/10.3390/catal10080859
Chicago/Turabian StyleAlfath, Muhammad, and Chan Woo Lee. 2020. "Recent Advances in the Catalyst Design and Mass Transport Control for the Electrochemical Reduction of Carbon Dioxide to Formate" Catalysts 10, no. 8: 859. https://doi.org/10.3390/catal10080859
APA StyleAlfath, M., & Lee, C. W. (2020). Recent Advances in the Catalyst Design and Mass Transport Control for the Electrochemical Reduction of Carbon Dioxide to Formate. Catalysts, 10(8), 859. https://doi.org/10.3390/catal10080859