The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2

 ,
, 
 and
and 
Abstract
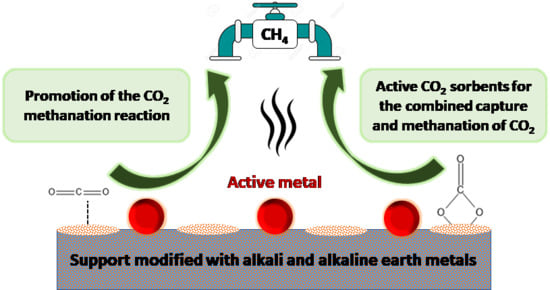
:
1. Introduction
2. Alkali and Alkaline Earth Metals as Promoters of the CO2 Methanation Reaction
2.1. Alkalimetals (Li, Na, K and Cs)
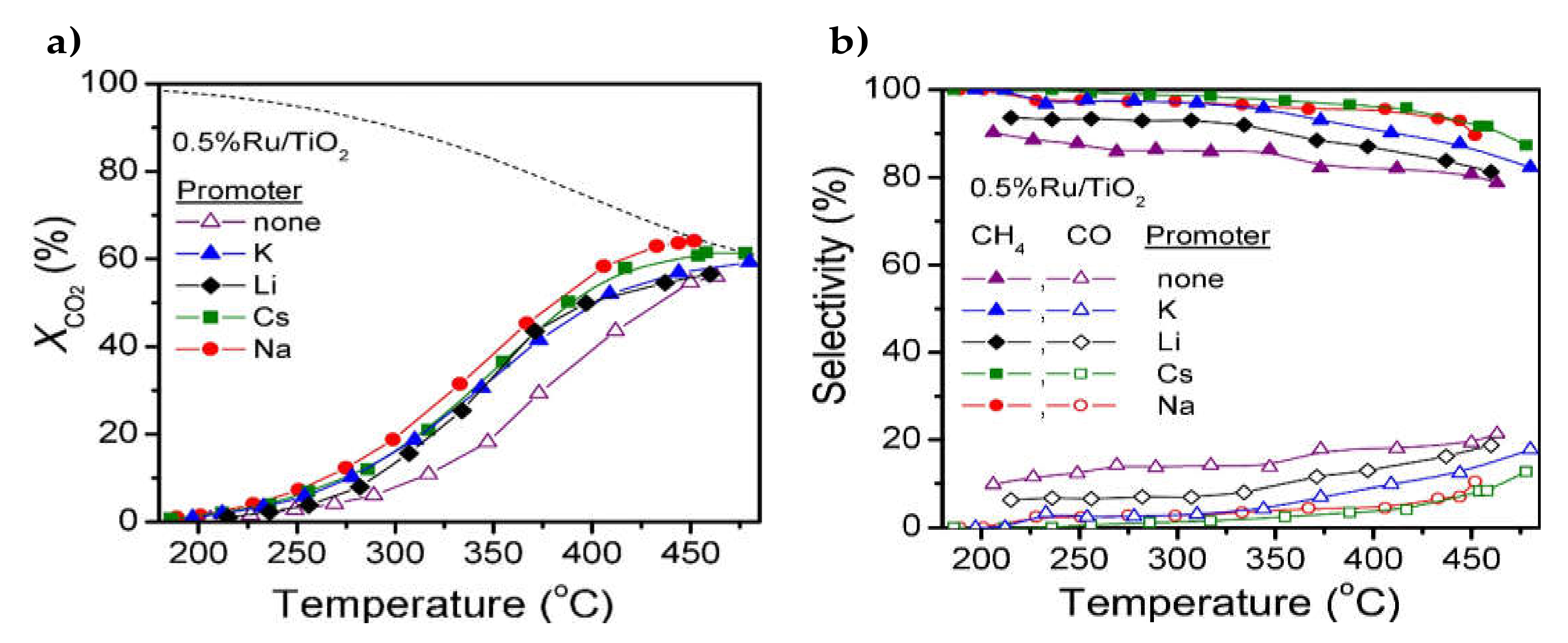
2.1.1. Screenings of Various Alkalimetals
2.1.2. Sodium (Na)
2.1.3. Pottasium (K)
2.2. Alkaline Earth Metals (Mg, Ca, Sr and Ba)
2.2.1. Screenings of Various Alkaline Earth Metals
2.2.2. Magnesium (Mg)
2.2.3. Calcium (Ca)
2.2.4. Strontium (Sr) and Barium (Ba)
2.3. Hydrotalcite-Derived and Zeolite Supported Catalysts
2.3.1. Hydrotalcite-Derived Catalysts
2.3.2. Zeolite Supported Catalysts
2.4. Other Promoters that Favour CO2 Chemisorption
3. Alkali and Alkaline Earth Metals as Active Sorbents Phases in the Combined Capture and Methanation of CO2
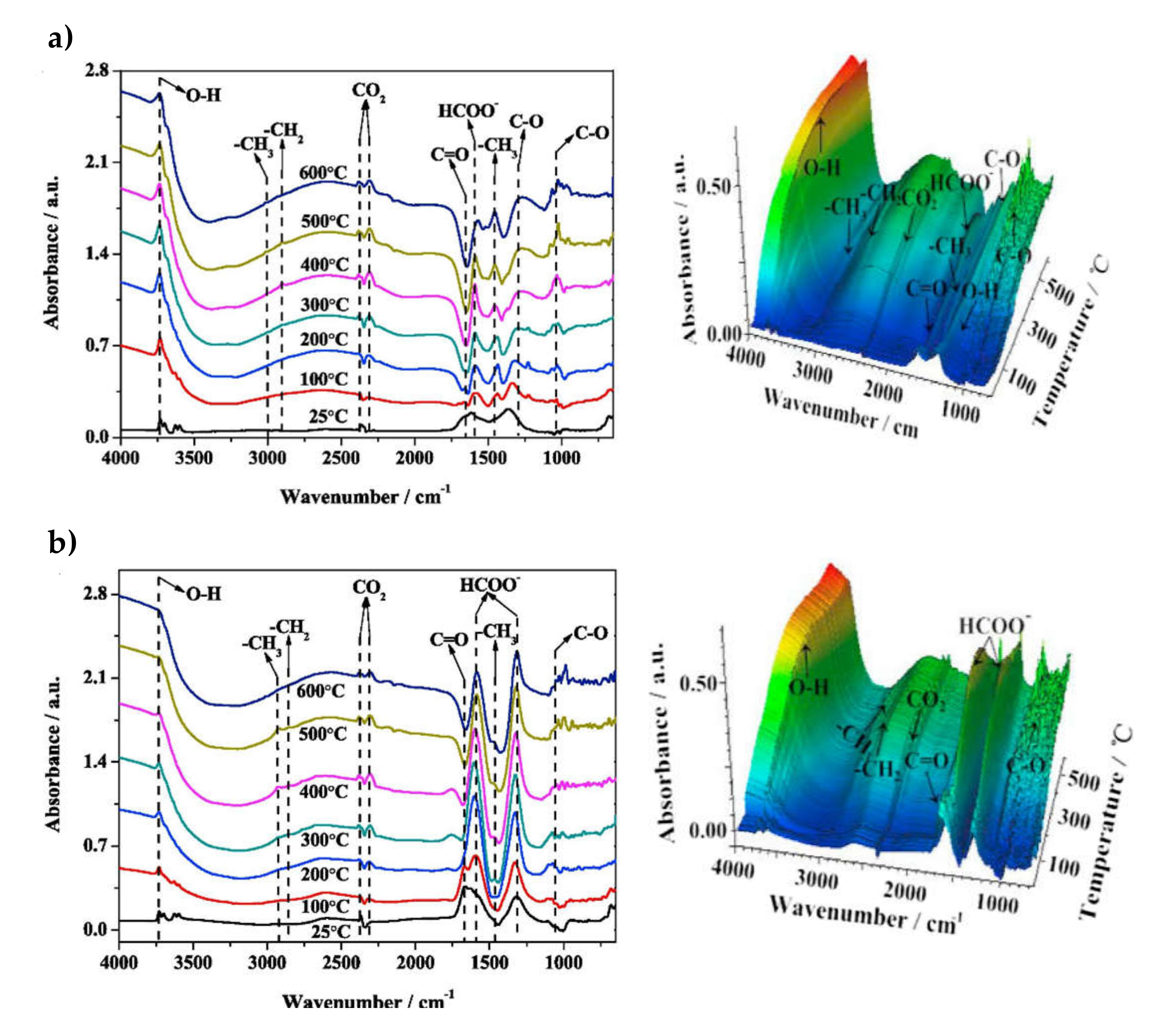
3.1. Ru-Based Dual-Function Materials
3.2. Ni-Based Dual-Function Materials
3.3. Dual-Function Materials for Products Other than Methane
3.4. Configuartions Using a Seperate Sorbent and Methanation Catalyst
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mardani, A.; Streimikiene, D.; Cavallaro, F.; Loganathan, N.; Khoshnoudi, M. Carbon dioxide (CO2) emissions and economic growth: A systematic review of two decades of research from 1995 to 2017. Sci. Total Environ. 2019, 649, 31–49. [Google Scholar] [CrossRef]
- Szulejko, J.E.; Kumar, P.; Deep, A.; Kim, K.H. Global warming projections to 2100 using simple CO2 greenhouse gas modeling and comments on CO2 climate sensitivity factor. Atmos. Pollut. Res. 2017, 8, 136–140. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Dong, F. Grand challenges for Catalytic Remediation in environmental and energy applications towards a cleaner and sustainable future. Front. Environ. Chem. 2020, 1, 5. [Google Scholar] [CrossRef]
- L’Orange Seigo, S.; Dohle, S.; Siegrist, M. Public perception of carbon capture and storage (CCS): A review. Renew. Sustain. Energy Rev. 2014, 38, 848–863. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Krishnamurthy, A.; Rownaghi, A.A.; Rezaei, F. Carbon capture and utilization update. Energy Technol. 2017, 5, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef] [Green Version]
- Thema, M.; Bauer, F.; Sterner, M. Power-to-Gas: Electrolysis and methanation status review. Renew. Sustain. Energy Rev. 2019, 112, 775–787. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P.L. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Vogt, C.; Monai, M.; Kramer, G.J.; Weckhuysen, B.M. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Ye, R.P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, C.; Monai, M.; Sterk, E.B.; Palle, J.; Melcherts, A.E.M.; Zijlstra, B.; Groeneveld, E.; Berben, P.H.; Boereboom, J.M.; Hensen, E.J.M.; et al. Understanding carbon dioxide activation and carbon–carbon coupling over nickel. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048–4058. [Google Scholar] [CrossRef]
- Cárdenas-Arenas, A.; Quindimil, A.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Isotopic and in situ DRIFTS study of the CO2 methanation mechanism using Ni/CeO2 and Ni/Al2O3 catalysts. Appl. Catal. B Environ. 2020, 265, 118538. [Google Scholar] [CrossRef]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrog. Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Boaro, M.; Colussi, S.; Trovarelli, A. Ceria-based materials in hydrogenation and reforming reactions for CO2 valorization. Front. Chem. 2019, 7, 28. [Google Scholar] [CrossRef]
- Choi, S.; Drese, J.H.; Jones, C.W. Adsorbent materials for carbon dioxide capture from large anthropogenic point sources. ChemSusChem 2009, 2, 796–854. [Google Scholar] [CrossRef]
- Kierzkowska, A.M.; Pacciani, R.; Müller, C.R. CaO-based CO2 sorbents: From fundamentals to the development of new, highly effective materials. ChemSusChem 2013, 6, 1130–1148. [Google Scholar] [CrossRef]
- Duyar, M.S.; Arellano-Treviño, M.A.; Farrauto, R.J. Dual function materials for CO2 capture and conversion using renewable H2. Appl. Catal. B Environ. 2015, 168–169, 370–376. [Google Scholar] [CrossRef]
- Melo Bravo, P.; Debecker, D.P. Combining CO2 capture and catalytic conversion to methane. Waste Dispos. Sustain. Energy 2019, 1, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Wang, S.; Schrunk, E.T.; Mahajan, H.; Farrauto, R.J. The role of ruthenium in CO2 capture and catalytic conversion to fuel by dual function materials (DFM). Catalysts 2017, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Arellano-Treviño, M.A.; Kanani, N.; Jeong-Potter, C.W.; Farrauto, R.J. Bimetallic catalysts for CO2 capture and hydrogenation at simulated flue gas conditions. Chem. Eng. J. 2019, 375, 121953. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Mechanism of the CO2 storage and in situ hydrogenation to CH4. Temperature and adsorbent loading effects over Ru-CaO/Al2O3 and Ru-Na2CO3/Al2O3 catalysts. Appl. Catal. B Environ. 2019, 256, 117845. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Guan, S.; Huang, J.; Wu, C. Direct and highly selective conversion of captured CO2 into methane through integrated carbon capture and utilization over dual functional materials. J. CO2 Util. 2020, 38, 262–272. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Lee, W.J.; Li, C.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.; Lim, S. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today 2020, in press. [Google Scholar] [CrossRef]
- Lv, C.; Xu, L.; Chen, M.; Cui, Y.; Wen, X.; Li, Y.; Wu, C.E.; Yang, B.; Miao, Z.; Hu, X.; et al. Recent Progresses in Constructing the Highly Efficient Ni Based Catalysts With Advanced Low-Temperature Activity Toward CO2 Methanation. Front. Chem. 2020, 8, 1–32. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Sebastian, V.; Monzon, A.; Baker, M.A.; Hinder, S.J.; Polychronopoulou, K.; Yentekakis, I.V.; Goula, M.A. An in depth investigation of deactivation through carbon formation during the biogas dry reforming reaction for Ni supported on modified with CeO2 and La2O3 zirconia catalysts. Int. J. Hydrog. Energy 2018, 43, 18955–18976. [Google Scholar] [CrossRef]
- Liu, K.; Xu, X.; Xu, J.; Fang, X.; Liu, L.; Wang, X. The distributions of alkaline earth metal oxides and their promotional effects on Ni/CeO2 for CO2 methanation. J. CO2 Util. 2020, 38, 113–124. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Baklavaridis, A.; Papadakis, V.G.; Goula, M.A. Synthesis Gas Production via the Biogas Reforming Reaction Over Ni/MgO–Al2O3 and Ni/CaO–Al2O3 Catalysts. Waste Biomass Valorization 2016, 7, 725–736. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Vernoux, P.; Goula, G.; Caravaca, A. Electropositive promotion by alkalis or alkaline earths of Pt-group metals in emissions control catalysis: A status report. Catalysts 2019, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Petala, A.; Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: I. Effect of alkali additives on catalytic activity and selectivity. Appl. Catal. B Environ. 2018, 224, 919–927. [Google Scholar] [CrossRef]
- Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: II. Effect of alkali additives on the reaction pathway. Appl. Catal. B Environ. 2018, 236, 162–170. [Google Scholar] [CrossRef]
- Cimino, S.; Boccia, F.; Lisi, L. Effect of alkali promoters (Li, Na, K) on the performance of Ru/Al2O3 catalysts for CO2 capture and hydrogenation to methane. J. CO2 Util. 2020, 37, 195–203. [Google Scholar] [CrossRef]
- Liang, C.; Ye, Z.; Dong, D.; Zhang, S.; Liu, Q.; Chen, G.; Li, C.; Wang, Y.; Hu, X. Methanation of CO2: Impacts of modifying nickel catalysts with variable-valence additives on reaction mechanism. Fuel 2019, 254, 115654. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Zhang, L.; Gao, J.; Shao, Y.; Dong, D.; Zhang, S.; Liu, Q.; Xu, L.; Hu, X. Impacts of alkali or alkaline earth metals addition on reaction intermediates formed in methanation of CO2 over cobalt catalysts. J. Energy Inst. 2020, 93, 1581–1596. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Yentekakis, I.V.; Lintz, H.G. Non-faradaic electrochemical modification of catalytic activity: A status report. Catal. Today 1992, 11, 303–438. [Google Scholar] [CrossRef]
- Klaitzidou, I.; Makri, M.; Theleritis, D.; Katsaounis, A.; Vayenas, C.G. Comparative study of the electrochemical promotion of CO2 hydrogenation on Ru using Na+, K+, H+ and O2- conducting solid electrolytes. Surf. Sci. 2016, 646, 194–203. [Google Scholar] [CrossRef]
- Theleritis, D.; Makri, M.; Souentie, S.; Caravaca, A.; Κatsaounis, A.; Vayenas, C.G. Comparative study of the electrochemical promotion of CO2 hydrogenation over Ru-supported catalysts using electronegative and electropositive promoters. ChemElectroChem 2014, 1, 254–262. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, T.W.; Lee, S.H.; Park, E.D. Effects of Na content in Na/Ni/SiO2 and Na/Ni/CeO2 catalysts for CO and CO2 methanation. Catal. Today 2018, 303, 159–167. [Google Scholar] [CrossRef]
- Beierlein, D.; Häussermann, D.; Pfeifer, M.; Schwarz, T.; Stöwe, K.; Traa, Y.; Klemm, E. Is the CO2 methanation on highly loaded Ni-Al2O3 catalysts really structure-sensitive? Appl. Catal. B Environ. 2019, 247, 200–219. [Google Scholar] [CrossRef]
- Büchel, R.; Baiker, A.; Pratsinis, S.E. Effect of Ba and K addition and controlled spatial deposition of Rh in Rh/Al2O3 catalysts for CO2 hydrogenation. Appl. Catal. A Gen. 2014, 477, 93–101. [Google Scholar] [CrossRef]
- Iloy, R.A.; Jalama, K. Effect of Operating Temperature, Pressure and Pottasium Loading on the Performance of Silica-Supported Cobalt Catalyst in CO2 Hydrogenation to Hydrocarbon Fuel. Catalysts 2019, 9, 807. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Hu, X.; Wang, Y.; Hu, S.; Xiang, J.; Li, C.; Chen, G.; Liu, Q.; Wei, T.; Dong, D. Regulation the reaction intermediates in methanation reactions via modification of nickel catalysts with strong base. Fuel 2019, 237, 566–579. [Google Scholar] [CrossRef]
- He, L.; Lin, Q.; Liu, Y.; Huang, Y. Unique catalysis of Ni-Al hydrotalcite derived catalyst in CO2 methanation: Cooperative effect between Ni nanoparticles and a basic support. J. Energy Chem. 2014, 23, 587–592. [Google Scholar] [CrossRef]
- Konsolakis, M.; Yentekakis, I.V. Strong promotional effects of Li, K, Rb and Cs on the Pt-catalysed reduction of NO by propene. Appl. Catal. B 2001, 29, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Yentekakis, I.V.; Konsolakis, M.; Lambert, R.M.; Palermo, A.; Tikhov, M. Successful application of the electrochemical promotion to the design of effective conventional catalyst formulations. Solid State Ion. 2000, 136, 783–790. [Google Scholar] [CrossRef]
- Vogt, C.; Wijten, J.; Madeira, C.L.; Kerkenaar, O.; Xu, K.; Holzinger, R.; Monai, M.; Weckhuysen, B.M. Alkali Promotion in the Formation of CH4 from CO2 and Renewably Produced H2 over Supported Ni Catalysts. ChemCatChem 2020, 12, 2792–2800. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, G.; Liu, Y. Perovskite-Type Oxides as the Catalyst Precursors for Preparing Supported Metallic Nanocatalysts: A Review. Ind. Eng. Chem. Res. 2018, 57, 1–17. [Google Scholar] [CrossRef]
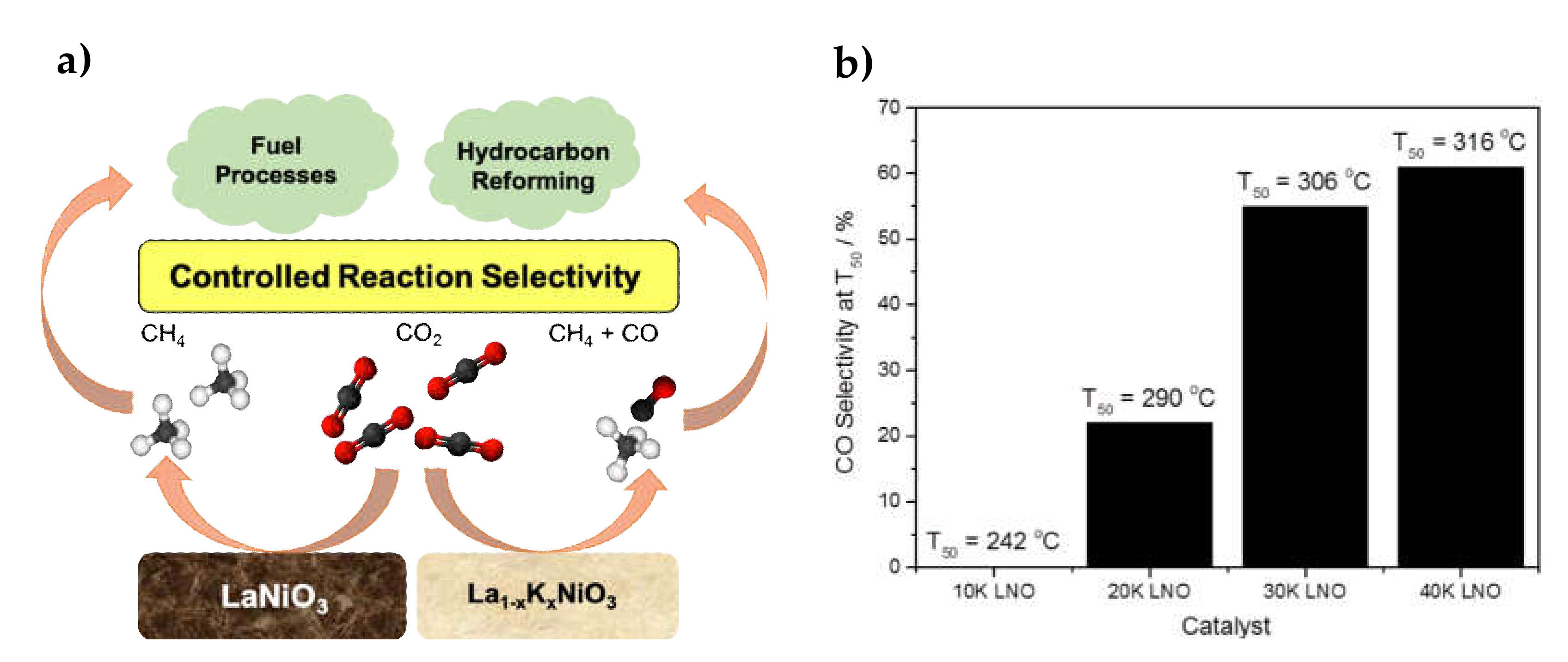
- Tsounis, C.; Wang, Y.; Arandiyan, H.; Wong, R.J.; Toe, C.Y.; Amal, R.; Scott, J. Tuning the Selectivity of LaNiO3 Perovskites for CO2 hydrogenation through potassium substitution. Catalysts 2020, 10, 409. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Lu, G. The difference of roles of alkaline-earth metal oxides on silica-supported nickel catalysts for CO2 methanation. RSC Adv. 2014, 4, 58171–58177. [Google Scholar] [CrossRef]
- Liang, C.; Hu, X.; Wei, T.; Jia, P.; Zhang, Z.; Dong, D.; Zhang, S.; Liu, Q.; Hu, G. Methanation of CO2 over Ni/Al2O3 modified with alkaline earth metals: Impacts of oxygen vacancies on catalytic activity. Int. J. Hydrog. Energy 2019, 44, 8197–8213. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Hatzisymeon, M.; Betsi-Argyropoulou, I.; Botzolaki, G.; Kousi, K.; Kondarides, D.I.; Taylor, M.J.; Parlett, C.M.A.; Osatiashtiani, A.; et al. Effect of support oxygen storage capacity on the catalytic performance of Rh nanoparticles for CO2 reforming of methane. Appl. Catal. B Environ. 2019, 243, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Goula, M.A.; Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Tsiaoussis, I.; Panagiotopoulou, P.; Goula, G.; Yentekakis, I.V. Syngas production via the biogas dry reforming reaction over Ni supported on zirconia modified with CeO2 or La2O3 catalysts. Int. J. Hydrog. Energy 2017, 42, 13724–13740. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.; Wang, X.; Fang, X.; Wang, H.; Xu, X. New insights into CO2 methanation mechanisms on Ni/MgO catalysts by DFT calculations: Elucidating Ni and MgO roles and support effects. J. CO2 Util. 2019, 33, 55–63. [Google Scholar] [CrossRef]
- Li, Y.; Lu, G.; Ma, J. Highly active and stable nano NiO-MgO catalyst encapsulated by silica with a core-shell structure for CO2 methanation. RSC Adv. 2014, 4, 17420–17428. [Google Scholar] [CrossRef]
- Park, J.N.; McFarland, E.W. A highly dispersed Pd-Mg/SiO2 catalyst active for methanation of CO2. J. Catal. 2009, 266, 92–97. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.M.; Park, J.N. Bifunctional mechanism of CO2 methanation on Pd-MgO/SiO2 catalyst: Independent roles of MgO and Pd on CO2 methanation. J. Phys. Chem. C 2010, 114, 7128–7131. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kovarik, L.; Szanyi, J. Heterogeneous catalysis on atomically dispersed supported metals: CO2 reduction on multifunctional Pd catalysts. ACS Catal. 2013, 3, 2094–2100. [Google Scholar] [CrossRef]
- Alcalde-Santiago, V.; Davó-Quiñonero, A.; Lozano-Castelló, D.; Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Ni/LnOx catalysts (Ln = La, Ce or Pr) for CO2 methanation. ChemCatChem 2019, 11, 810–819. [Google Scholar]
- Cárdenas-Arenas, A.; Quindimil, A.; Davó-Quinonero, A.; Bailón-García, E.; Lozano-Castelló, D.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Design of active sites in Ni/CeO2 catalysts for the methanation of CO2: Tailoring the Ni-CeO2 contact. Appl. Mater. Today 2020, 19, 100591. [Google Scholar] [CrossRef]
- Guo, M.; Lu, G. The effect of impregnation strategy on structural characters and CO2 methanation properties over MgO modified Ni/SiO2 catalysts. Catal. Commun. 2014, 54, 55–60. [Google Scholar] [CrossRef]
- Tan, J.; Wang, J.; Zhang, Z.; Ma, Z.; Wang, L.; Liu, Y. Highly dispersed and stable Ni nanoparticles confined by MgO on ZrO2 for CO2 methanation. Appl. Surf. Sci. 2019, 481, 1538–1548. [Google Scholar] [CrossRef]
- Xu, L.; Wang, F.; Chen, M.; Yang, H.; Nie, D.; Qi, L.; Lian, X. Alkaline-promoted Ni based ordered mesoporous catalysts with enhanced low-temperature catalytic activity toward CO2 methanation. RSC Adv. 2017, 7, 18199–18210. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Y.; Zhang, G.; Sun, W.; Bai, Y.; Zheng, L.; Han, X.; Wu, L. Influence of Mg-promoted Ni-based Catalyst Supported on Coconut Shell Carbon for CO2 Methanation. ChemistrySelect 2019, 4, 838–845. [Google Scholar] [CrossRef]
- Xiao-liu, W.; Meng, Y.; Ling-jun, Z.; Xiao-nan, Z.; Shu-rong, W. CO2 methanation over Ni /Mg@MCM-41 prepared by in-situ synthesis method. J. Fuel Chem. Technol. 2020, 48, 456–465. [Google Scholar]
- Bacariza, M.C.; Graça, I.; Bebiano, S.S.; Lopes, J.M.; Henriques, C. Magnesium as Promoter of CO2 Methanation on Ni-Based USY Zeolites. Energy Fuels 2017, 31, 9776–9789. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xu, L.; Chen, M.; Lv, C.; Cui, Y.; Wen, X.; Wu, C.E.; Yang, B.; Miao, Z.; Hu, X.; et al. Facilely fabricating highly dispersed Ni-based catalysts supported on mesoporous MFI nanosponge for CO2 methanation. Microporous Mesoporous Mater. 2020, 302, 110250. [Google Scholar] [CrossRef]
- Zhang, L.; Bian, L.; Zhu, Z.; Li, Z. La-promoted Ni/Mg-Al catalysts with highly enhanced low-temperature CO2 methanation performance. Int. J. Hydrog. Energy 2018, 43, 2197–2206. [Google Scholar] [CrossRef]
- Jingyu, Z.; Hongfang, M.; Fangyu, J.; Haitao, Z.; Weiyong, Y. Mn and Mg dual promoters modified Ni/α-Al2O3 catalysts for high temperature syngas methanation. Fuel Process. Technol. 2018, 172, 225–232. [Google Scholar]
- Baysal, Z.; Kureti, S. CO2 methanation on Mg-promoted Fe catalysts. Appl. Catal. B Environ. 2020, 262, 118300. [Google Scholar] [CrossRef]
- Kirchner, J.; Baysal, Z.; Kureti, S. Activity and Structural Changes of Fe-based Catalysts during CO2 Hydrogenation towards CH4—A Mini Review. ChemCatChem 2020, 12, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yang, H.; Chen, M.; Wang, F.; Nie, D.; Qi, L.; Lian, X.; Chen, H.; Wu, M. CO2 methanation over Ca doped ordered mesoporous Ni-Al composite oxide catalysts: The promoting effect of basic modifier. J. CO2 Util. 2017, 21, 200–210. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zhao, G.; Yang, H.; Yuan, M.; An, X.; Zhou, H.; Qiao, Y.; Tian, Y. CO2 methanation over ordered mesoporous NiRu-doped CaO-Al2O3 nanocomposites with enhanced catalytic performance. Int. J. Hydrog. Energy 2018, 43, 239–250. [Google Scholar] [CrossRef]
- Do, P.N.U.; Luu, C.L.; Nguyen, P.A.; Van Nguyen, T.T.; Hoang, T.C.; Nguyen, T. Promotion effect of CO on methanation of CO2-rich gas over nanostructured modified NiO/γ-Al2O3 catalysts. Adv. Nat. Sci. Nanosci. Nanotechnol. 2019, 10, 035004. [Google Scholar] [CrossRef]
- Yang, W.; Feng, Y.; Chu, W. Promotion effect of CaO modification on mesoporous Al2O3-supported Ni catalysts for CO2 methanation. Int. J. Chem. Eng. 2016, 2016. 2041821. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.F.; Yang, W.; Wang, N.; Luo, S.Z.; Chu, W. Catalytic properties of Ni/CNTs and Ca-Promoted Ni/CNTs for methanation reaction of carbon dioxide. Adv. Mater. Res. 2014, 924, 217–226. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, W.; Chu, W. Effect of Ca modification on the catalytic performance of Ni/AC for CO2 methanation. Integr. Ferroelectr. 2016, 172, 40–48. [Google Scholar] [CrossRef]
- Zhou, M.; Ahmad, A. Synthesis, processing and characterization of calcia-stabilized zirconia solid electrolytes for oxygen sensing applications. Mater. Res. Bull. 2006, 41, 690–696. [Google Scholar] [CrossRef]
- Takano, H.; Shinomiya, H.; Izumiya, K.; Kumagai, N.; Habazaki, H.; Hashimoto, K. CO2 methanation of Ni catalysts supported on tetragonal ZrO2 doped with Ca2+ and Ni2+ ions. Int. J. Hydrog. Energy 2015, 40, 8347–8355. [Google Scholar] [CrossRef]
- Everett, O.E.; Zonetti, P.C.; Alves, O.C.; de Avillez, R.R.; Appel, L.G. The role of oxygen vacancies in the CO2 methanation employing Ni/ZrO2 doped with Ca. Int. J. Hydrog. Energy 2020, 45, 6352–6359. [Google Scholar] [CrossRef]
- Jia, C.; Gao, J.; Li, J.; Gu, F.; Xu, G.; Zhong, Z.; Su, F. Nickel catalysts supported on calcium titanate for enhanced CO methanation. Catal. Sci. Technol. 2013, 3, 490–499. [Google Scholar] [CrossRef]
- Do, J.Y.; Park, N.K.; Seo, M.W.; Lee, D.; Ryu, H.J.; Kang, M. Effective thermocatalytic carbon dioxide methanation on Ca-inserted NiTiO3 perovskite. Fuel 2020, 271, 117624. [Google Scholar] [CrossRef]
- Toemen, S.; Abu Bakar, W.A.W.; Ali, R. Effect of ceria and strontia over Ru/Mn/Al2O3 catalyst: Catalytic methanation, physicochemical and mechanistic studies. J. CO2 Util. 2016, 13, 38–49. [Google Scholar] [CrossRef]
- Steiger, P.; Kröcher, O.; Ferri, D. Increased nickel exsolution from LaFe0.8Ni0.2O3 perovskite-derived CO2 methanation catalysts through strontium doping. Appl. Catal. A Gen. 2020, 590, 117328. [Google Scholar] [CrossRef]
- Huynh, H.L.; Yu, Z. CO2 Methanation on Hydrotalcite-Derived Catalysts and Structured Reactors: A Review. Energy Technol. 2020, 8, 1901475. [Google Scholar] [CrossRef] [Green Version]
- Bacariza, M.C.; Graça, I.; Lopes, J.M.; Henriques, C. Tuning Zeolite Properties towards CO2 Methanation: An Overview. ChemCatChem 2019, 11, 2388–2400. [Google Scholar] [CrossRef]
- Bette, N.; Thielemann, J.; Schreiner, M.; Mertens, F. Methanation of CO2 over a (Mg,Al)Ox Supported Nickel Catalyst Derived from a (Ni,Mg,Al)-Hydrotalcite-like Precursor. ChemCatChem 2016, 8, 2903–2906. [Google Scholar] [CrossRef]
- Liu, J.; Bing, W.; Xue, X.; Wang, F.; Wang, B.; He, S.; Zhang, Y.; Wei, M. Alkaline-assisted Ni nanocatalysts with largely enhanced low-temperature activity toward CO2 methanation. Catal. Sci. Technol. 2016, 6, 3976–3983. [Google Scholar] [CrossRef]
- Ren, J.; Mebrahtu, C.; Palkovits, R. Ni-based catalysts supported on Mg-Al hydrotalcites with different morphologies for CO2 methanation: Exploring the effect of metal-support interaction. Catal. Sci. Technol. 2020, 10, 1902–1913. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Debek, R.; Motak, M.; Grzybek, T.; Gálvez, M.E.; Da Costa, P. Novel Ni-La-hydrotalcite derived catalysts for CO2 methanation. Catal. Commun. 2016, 83, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Wierzbicki, D.; Motak, M.; Grzybek, T.; Gálvez, M.E.; Da Costa, P. The influence of lanthanum incorporation method on the performance of nickel-containing hydrotalcite-derived catalysts in CO2 methanation reaction. Catal. Today 2018, 307, 205–211. [Google Scholar] [CrossRef]
- Martins, J.A.; Faria, A.C.; Soria, M.A.; Miguel, C.V.; Rodrigues, A.E.; Madeira, L.M. CO2 methanation over hydrotalcite-derived nickel/ruthenium and supported ruthenium catalysts. Catalysts 2019, 9, 1008. [Google Scholar] [CrossRef] [Green Version]
- Mebrahtu, C.; Krebs, F.; Perathoner, S.; Abate, S.; Centi, G.; Palkovits, R. Hydrotalcite based Ni-Fe/(Mg, Al)Ox catalysts for CO2 methanation-tailoring Fe content for improved CO dissociation, basicity, and particle size. Catal. Sci. Technol. 2018, 8, 1016–1027. [Google Scholar] [CrossRef]
- Graça, I.; González, L.V.; Bacariza, M.C.; Fernandes, A.; Henriques, C.; Lopes, J.M.; Ribeiro, M.F. CO2 hydrogenation into CH4 on NiHNaUSY zeolites. Appl. Catal. B Environ. 2014, 147, 101–110. [Google Scholar] [CrossRef]
- Westermann, A.; Azambre, B.; Bacariza, M.C.; Graça, I.; Ribeiro, M.F.; Lopes, J.M.; Henriques, C. Insight into CO2 methanation mechanism over NiUSY zeolites: An operando IR study. Appl. Catal. B Environ. 2015, 174, 120–125. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Bértolo, R.; Graça, I.; Lopes, J.M.; Henriques, C. The effect of the compensating cation on the catalytic performances of Ni/USY zeolites towards CO2 methanation. J. CO2 Util. 2017, 21, 280–291. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Graça, I.; Lopes, J.M.; Henriques, C. Enhanced activity of CO2 hydrogenation to CH4 over Ni based zeolites through the optimization of the Si/Al ratio. Microporous Mesoporous Mater. 2018, 267, 9–19. [Google Scholar] [CrossRef]
- Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ni catalysts with La as promoter supported over Y- and BETA- zeolites for CO2 methanation. Appl. Catal. B Environ. 2018, 238, 393–403. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Graça, I.; Lopes, J.M.; Henriques, C. Ni-Ce/Zeolites for CO2 Hydrogenation to CH4: Effect of the Metal Incorporation Order. ChemCatChem 2018, 10, 2773–2781. [Google Scholar] [CrossRef]
- Zhang, F.; Lu, B.; Sun, P. Highly stable Ni-based catalysts derived from LDHs supported on zeolite for CO2 methanation. Int. J. Hydrog. Energy 2020, 45, 16183–16192. [Google Scholar] [CrossRef]
- Xu, L.; Wang, F.; Chen, M.; Nie, D.; Lian, X.; Lu, Z.; Chen, H.; Zhang, K.; Ge, P. CO2 methanation over rare earth doped Ni based mesoporous catalysts with intensified low-temperature activity. Int. J. Hydrog. Energy 2017, 42, 15523–15539. [Google Scholar] [CrossRef]
- Zhao, K.; Li, Z.; Bian, L. CO2 methanation and co-methanation of CO and CO2 over Mn-promoted Ni/Al2O3 catalysts. Front. Chem. Sci. Eng. 2016, 10, 273–280. [Google Scholar] [CrossRef]
- Liu, Q.; Bian, B.; Fan, J.; Yang, J. Cobalt doped Ni based ordered mesoporous catalysts for CO2 methanation with enhanced catalytic performance. Int. J. Hydrog. Energy 2018, 43, 4893–4901. [Google Scholar] [CrossRef]
- Mihet, M.; Lazar, M.D. Methanation of CO2 on Ni/γ-Al2O3: Influence of Pt, Pd or Rh promotion. Catal. Today 2018, 306, 294–299. [Google Scholar] [CrossRef]
- Guilera, J.; Del Valle, J.; Alarcón, A.; Díaz, J.A.; Andreu, T. Metal-oxide promoted Ni/Al2O3 as CO2 methanation micro-size catalysts. J. CO2 Util. 2019, 30, 11–17. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Cavattoni, T.; Finocchio, E.; Riani, P.; Flytzani-Stephanopoulos, M.; Busca, G. A study of Ni/La-Al2O3 catalysts: A competitive system for CO2 methanation. Appl. Catal. B Environ. 2019, 248, 286–297. [Google Scholar] [CrossRef]
- Liang, C.; Wei, T.; Wang, H.; Yu, Z.; Dong, D.; Zhang, S.; Liu, Q.; Hu, G.; Hu, X. Impacts of La addition on formation of the reaction intermediates over alumina and silica supported nickel catalysts in methanation of CO2. J. Energy Inst. 2020, 93, 723–738. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, L.; Zhuo, Y.; Zhu, Y.; Wang, S. Enhancement of CO2 Methanation over La-Modified Ni/SBA-15 Catalysts Prepared by Different Doping Methods. ACS Sustain. Chem. Eng. 2019, 7, 14647–14660. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, Q. Perovskite LaNiO3 Nanocrystals inside Mesostructured Cellular Foam Silica: High Catalytic Activity and Stability for CO2 Methanation. Energy Technol. 2020, 8, 1–12. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ni loading effects on dual function materials for capture and in-situ conversion of CO2 to CH4 using CaO or Na2CO3. J. CO2 Util. 2019, 34, 576–587. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Zheng, Q.; Farrauto, R.; Chau Nguyen, A. Adsorption and Methanation of Flue Gas CO2 with Dual Functional Catalytic Materials: A Parametric Study. Ind. Eng. Chem. Res. 2016, 55, 6768–6776. [Google Scholar] [CrossRef]
- Wang, S.; Farrauto, R.J.; Karp, S.; Jeon, J.H.; Schrunk, E.T. Parametric, cyclic aging and characterization studies for CO2 capture from flue gas and catalytic conversion to synthetic natural gas using a dual functional material (DFM). J. CO2 Util. 2018, 27, 390–397. [Google Scholar] [CrossRef]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Wang, F.; He, S.; Chen, H.; Wang, B.; Zheng, L.; Wei, M.; Evans, D.G.; Duan, X. Active Site Dependent Reaction Mechanism over Ru/CeO2 Catalyst toward CO2 Methanation. J. Am. Chem. Soc. 2016, 138, 6298–6305. [Google Scholar] [CrossRef]
- Zhen, W.; Li, B.; Lu, G.; Ma, J. Enhancing catalytic activity and stability for CO2 methanation on Ni–Ru/γ-Al2O3 via modulating impregnation sequence and controlling surface active species. RSC Adv. 2014, 4, 16472–16479. [Google Scholar] [CrossRef]
- Chai, K.H.; Leong, L.K.; Wong, D.S.H.; Tsai, D.H.; Sethupathi, S. Effect of CO2 adsorbents on the Ni-based dual-function materials for CO2 capturing and in situ methanation. J. Chin. Chem. Soc. 2020, 67, 1–11. [Google Scholar] [CrossRef]
- Hu, L.; Urakawa, A. Continuous CO2 capture and reduction in one process: CO2 methanation over unpromoted and promoted Ni/ZrO2. J. CO2 Util. 2018, 25, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Sun, N.; Wang, B.; Han, Z.; Cao, S.; Hu, D.; Zhu, T.; Shen, Q.; Wei, W. 2D-Layered Ni–MgO–Al2O3 Nanosheets for Integrated Capture and Methanation of CO2. ChemSusChem 2020, 13, 360–368. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Riesco-García, J.M.; Penelás-Pérez, G.; Urakawa, A. Enabling continuous capture and catalytic conversion of flue gas CO2 to syngas in one process. J. CO2 Util. 2016, 14, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Hyakutake, T.; Van Beek, W.; Urakawa, A. Unravelling the nature, evolution and spatial gradients of active species and active sites in the catalyst bed of unpromoted and K/Ba-promoted Cu/Al2O3 during CO2 capture-reduction. J. Mater. Chem. A 2016, 4, 6878–6885. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Rownaghi, A.A.; Rezaei, F. Combined Capture and Utilization of CO2 for Syngas Production over Dual-Function Materials. ACS Sustain. Chem. Eng. 2018, 6, 13551–13561. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Zhao, J.; Shen, B.; Shi, J.; Huang, J.; Wu, C. Dual functional catalytic materials of Ni over Ce-modified CaO sorbents for integrated CO2 capture and conversion. Appl. Catal. B Environ. 2019, 244, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Veselovskaya, J.V.; Parunin, P.D.; Netskina, O.V.; Kibis, L.S.; Lysikov, A.I.; Okunev, A.G. Catalytic methanation of carbon dioxide captured from ambient air. Energy 2018, 159, 766–773. [Google Scholar] [CrossRef]
- Veselovskaya, J.V.; Parunin, P.D.; Netskina, O.V.; Okunev, A.G. A Novel Process for Renewable Methane Production: Combining Direct Air Capture by K2CO3/Alumina Sorbent with CO2 Methanation over Ru/Alumina Catalyst. Top. Catal. 2018, 61, 1528–1536. [Google Scholar] [CrossRef]
- Veselovskaya, J.V.; Lysikov, A.I.; Netskina, O.V.; Kuleshov, D.V.; Okunev, A.G. K2CO3-Containing Composite Sorbents Based on Thermally Modified Alumina: Synthesis, Properties, and Potential Application in a Direct Air Capture/Methanation Process. Ind. Eng. Chem. Res. 2020, 59, 7130–7139. [Google Scholar] [CrossRef]
- Miguel, C.V.; Soria, M.A.; Mendes, A.; Madeira, L.M. A sorptive reactor for CO2 capture and conversion to renewable methane. Chem. Eng. J. 2017, 322, 590–602. [Google Scholar] [CrossRef]
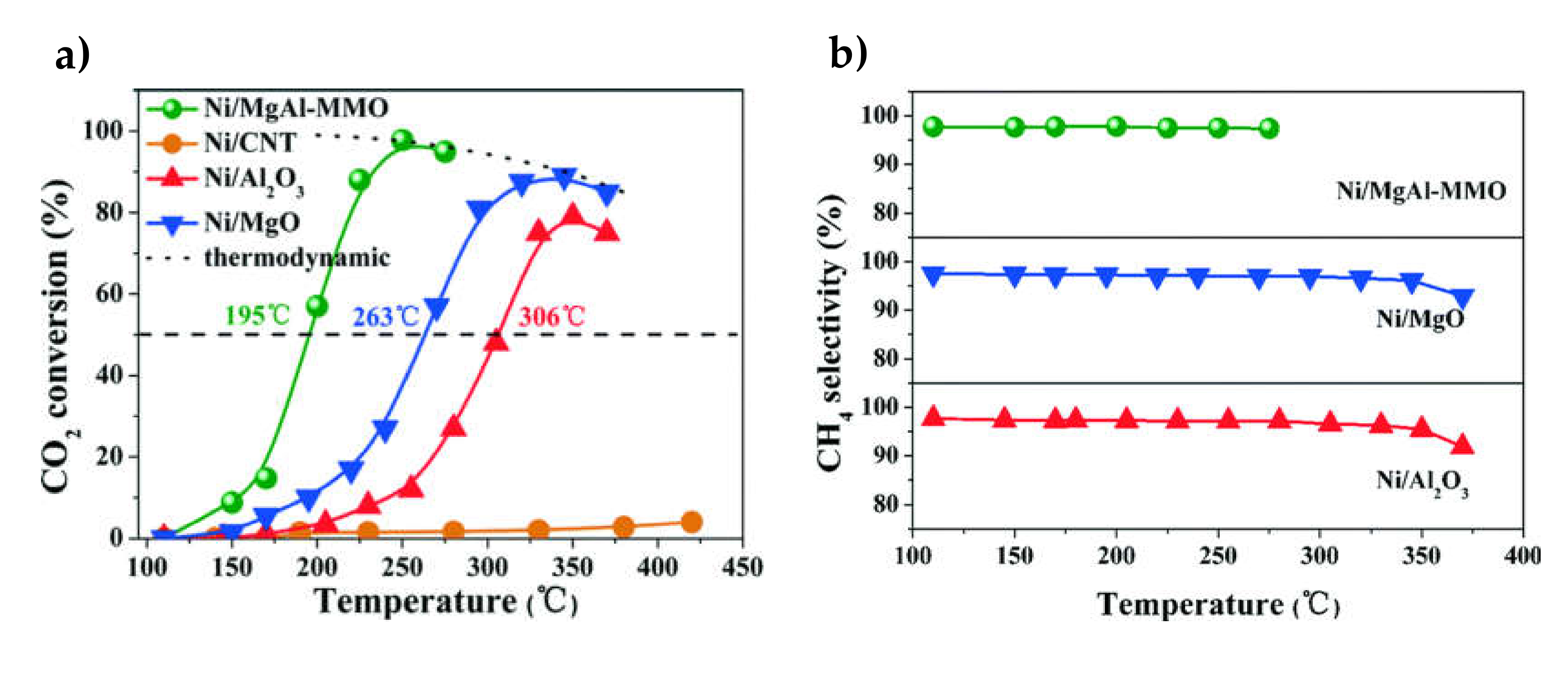
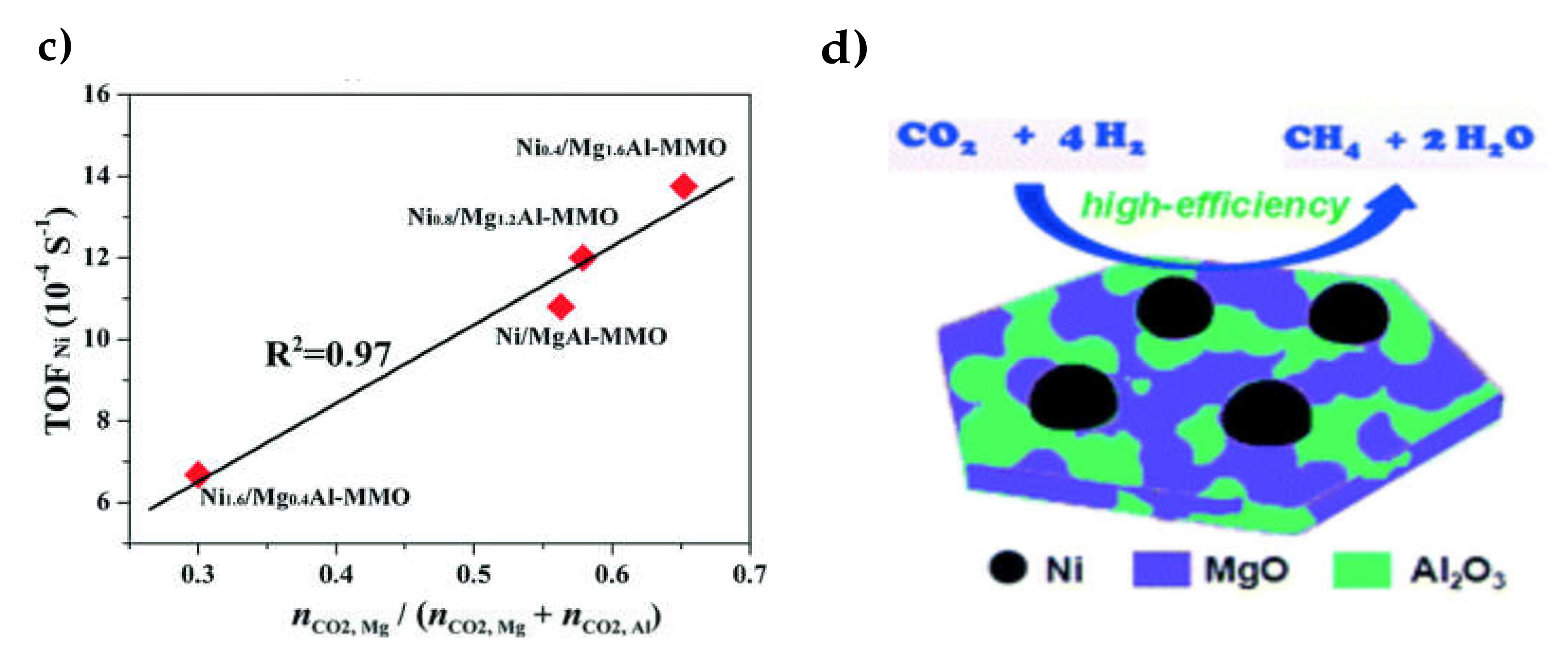

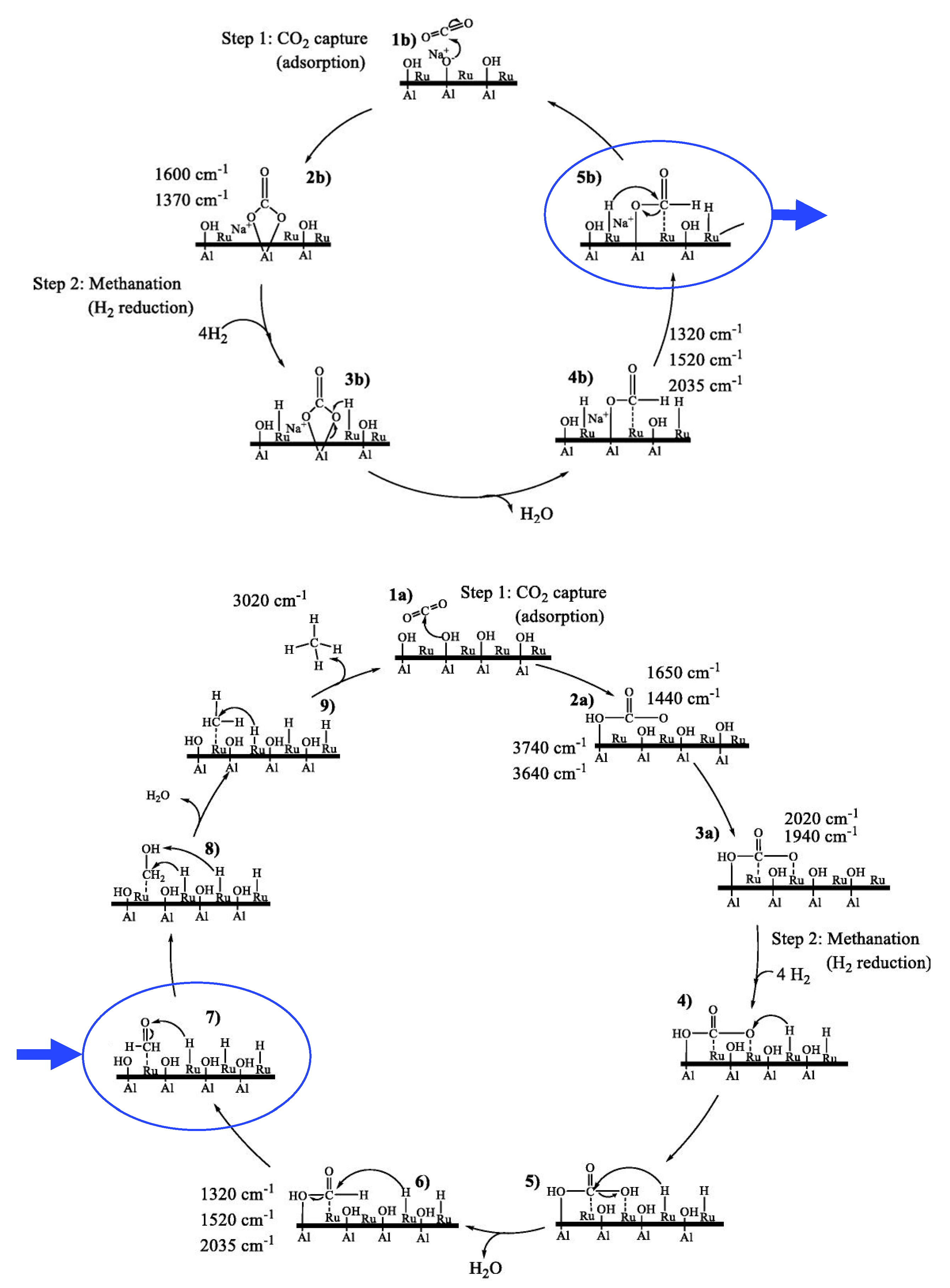

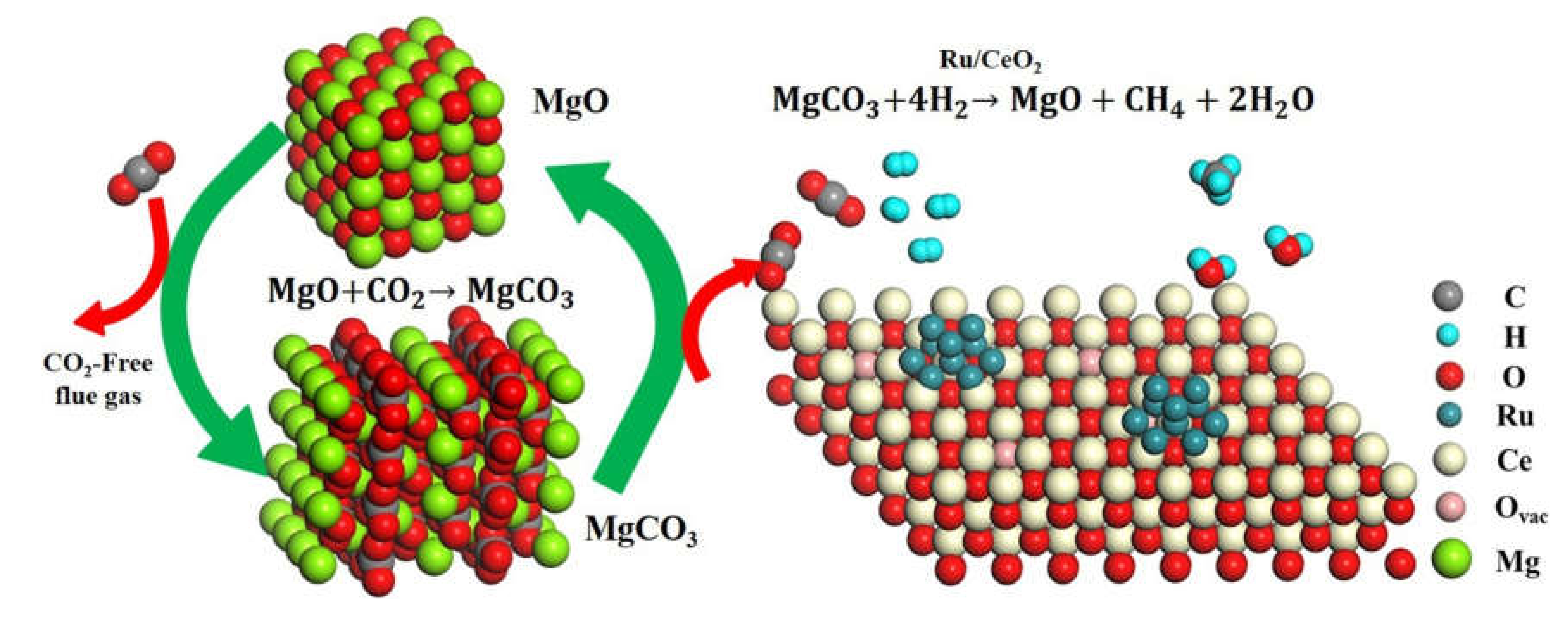


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promoter | Catalyst | Synthesis Method | Conditions | Performance | Comments | Ref. |
|---|---|---|---|---|---|---|
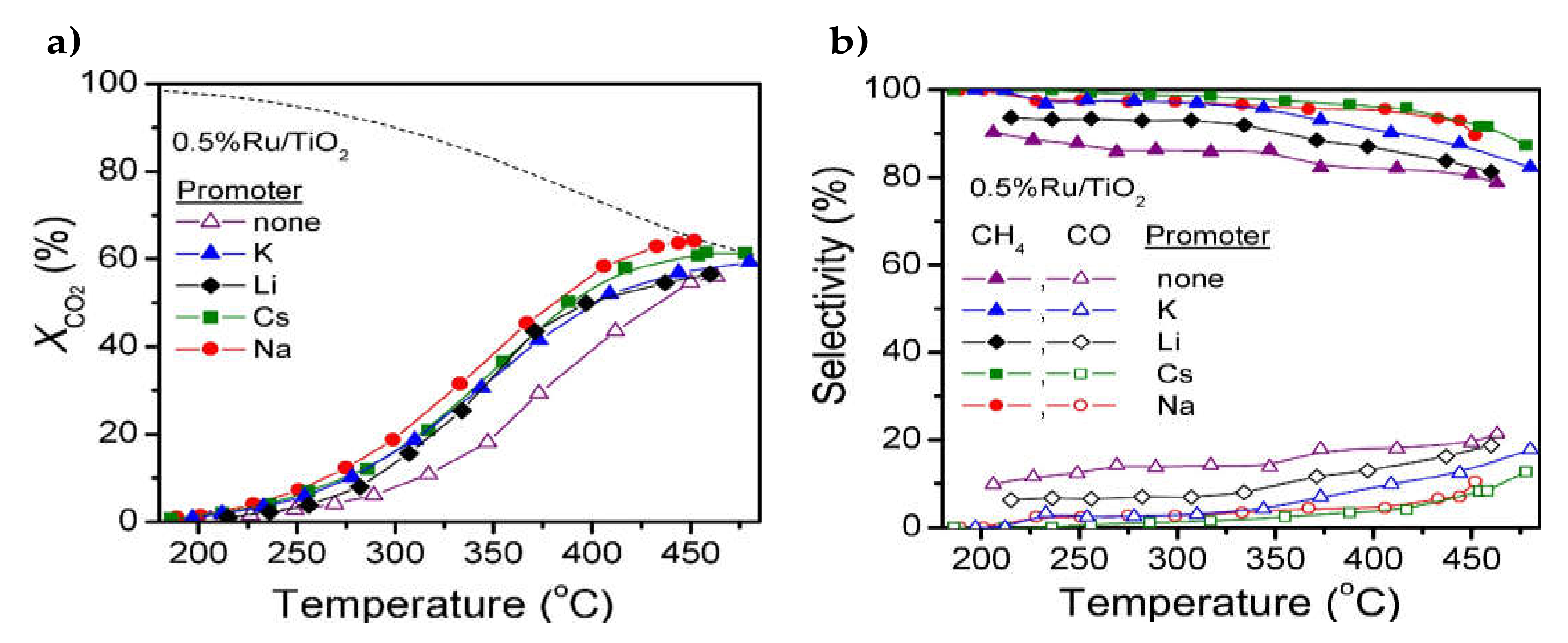
| 0.06–0.4% Li, Na, K and Cs | 0.5% and 5% Ru/TiO2 | Wet impregnation | GHSV = 56,000 h−1 H2/CO2 = 4/1 | XCO2 = 60%, SCH4 = 95% at 400 °C (0.2% Na on 0.5% Ru/TiO2) | Catalytic activity improved after addition of small amounts of alkalimetals on 0.5% Ru/TiO2. Less pronounced effect for 5% Ru/TiO2 | [33] |
| 10% LiNO3, NaNO3, Na2CO3 and K2CO3 | 1% Ru/Al2O3 | Incipient wetness impregnation | GHSV = 25,000 h−1 H2/CO2 = 4/1 | XCO2 = 60%, SCH4 = 99% at 350 °C (10% LiNO3 on 1% Ru/Al2O3) | Only LiNO3 significantly promoted activity. Na2CO3 and K2CO3 masked Ru particles and suppressed methanation activity | [35] |
| 5% Na, K, Mg and Ca | 20% Co/Al2O3 | Incipient wetness impregnation | H2/CO2 = 4/1 | XCO2 = 52%, SCH4 = 67% at 400 °C (5% Na on 10% Co/Al2O3) | Na and K increased the population of basic sites but promoted CO selectivity. Mg and Ca did not favour methanation activity either, but prevented Co nanoparticle sintering | [37] |
| 0.1–1% Na | 33% Ni/SiO2 and 13% Ni/CeO2 | Wet impregnation | H2/CO2 = 50/1 | XCO2 = 85%, SCH4 = 85% at 250 °C (0.1% Na on 33% Ni/SiO2) | Ni active surface area decreased upon Na addition. Negative impact on activity for Ni/CeO2, but slightly positive impact for Ni/SiO2 | [41] |
| 16.5% K and Ba | 1% Rh/Al2O3 | Flame spray pyrolysis | GHSV = 6000 h−1 H2/CO2 = 4/1 | XCO2 = 25%, SCH4 = 0% at 375 °C (16.5 % K on 1% Rh/Al2O3) | K-addition led to pure CO production over the entire temperature range. Ba-modified catalyst slightly inferior to Rh/Al2O3 for CO2 methanation | [43] |
| 0.5–20% KOH | 0.5–20% Ni/Al2O3 | Incipient wetness impregnation | H2/CO2 = 4/1 | XCO2 = 40%, SCH4 = 45% at 400 °C (10% K on 12% Ni/Al2O3) | Strongly adsorbed formate intermediates over KOH-modified catalysts favoured CO selectivity | [45] |
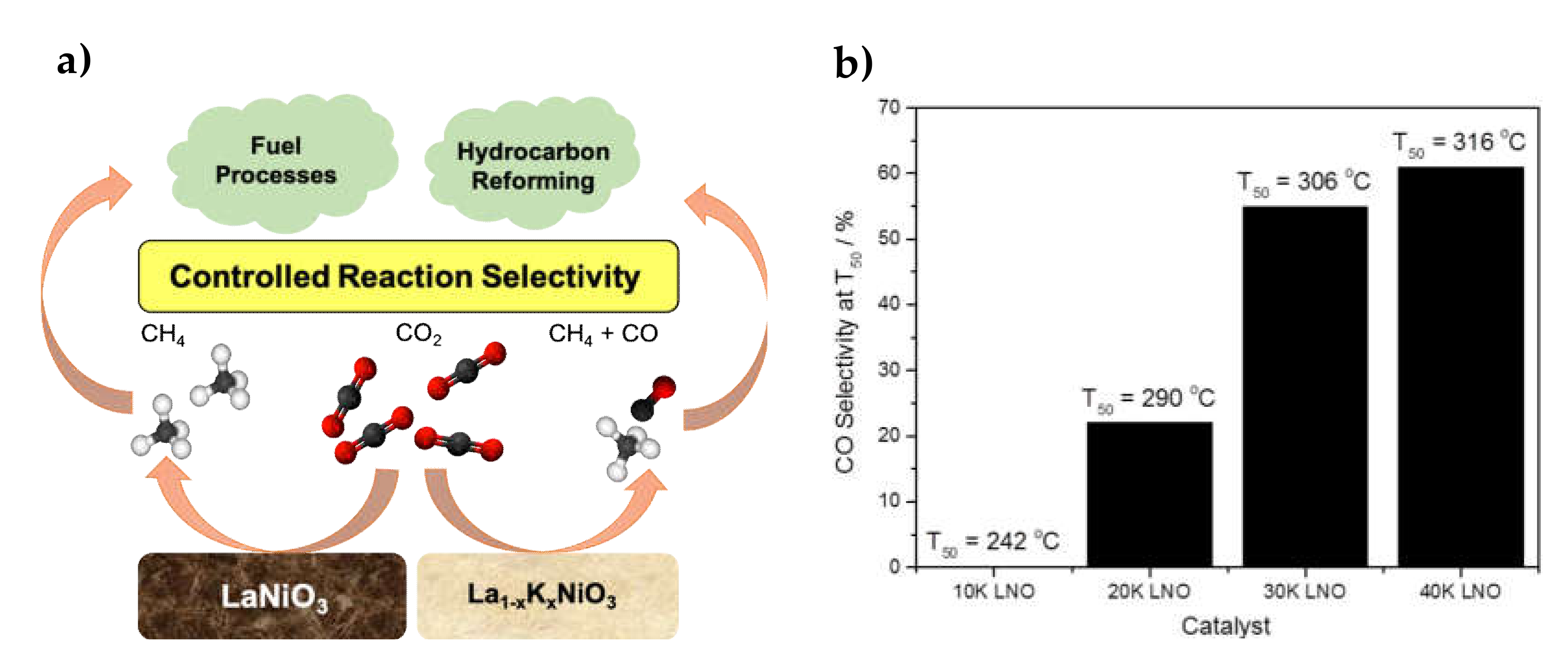
| 1.7–7.6% K (x = 0.1–0.4 for La1-xKxNiO3) | 24–28.5% Ni/La2O3 | Citrate sol-gel La1-xKxNiO3 synthesis and reduction | GHSV = 48,000 h−1 H2/CO2 = 12.5/1 | XCO2 = 50%, SCH4 = 45% at 306 °C (for x= 0.3 => La0.7K0.3NiO3) | Easy-to-desorb CO species formed after K-doping stirred the reaction towards RWGS and CO selectivity | [51] |
| 4% MgO, CaO, SrO and BaO | 10% Ni/SiO2 | Wet impregnation | GHSV = 15,000 h−1 H2/CO2 = 4/1 | XCO2 = 71%, SCH4 = 99% at 300 °C (4% SrO on 10% Ni/SiO2) | MgO inhibited activity, CaO had little effect, SrO and BaO enhanced activity, while SrO also promoted the catalyst stability | [52] |
| 1–7.5% Mg, Ca, Sr and Ba | 20% Ni/Al2O3 | Incipient wetness impregnation | GHSV = 16,000 h−1 H2/CO2 = 4/1 | XCO2 = 80%, SCH4 = 100% at 300 °C (5% Ba on 20% Ni/Al2O3) | Lighter elements Mg and Ca promoted CO selectivity, while heavier elements Sr and Ba favoured methanation | [53] |
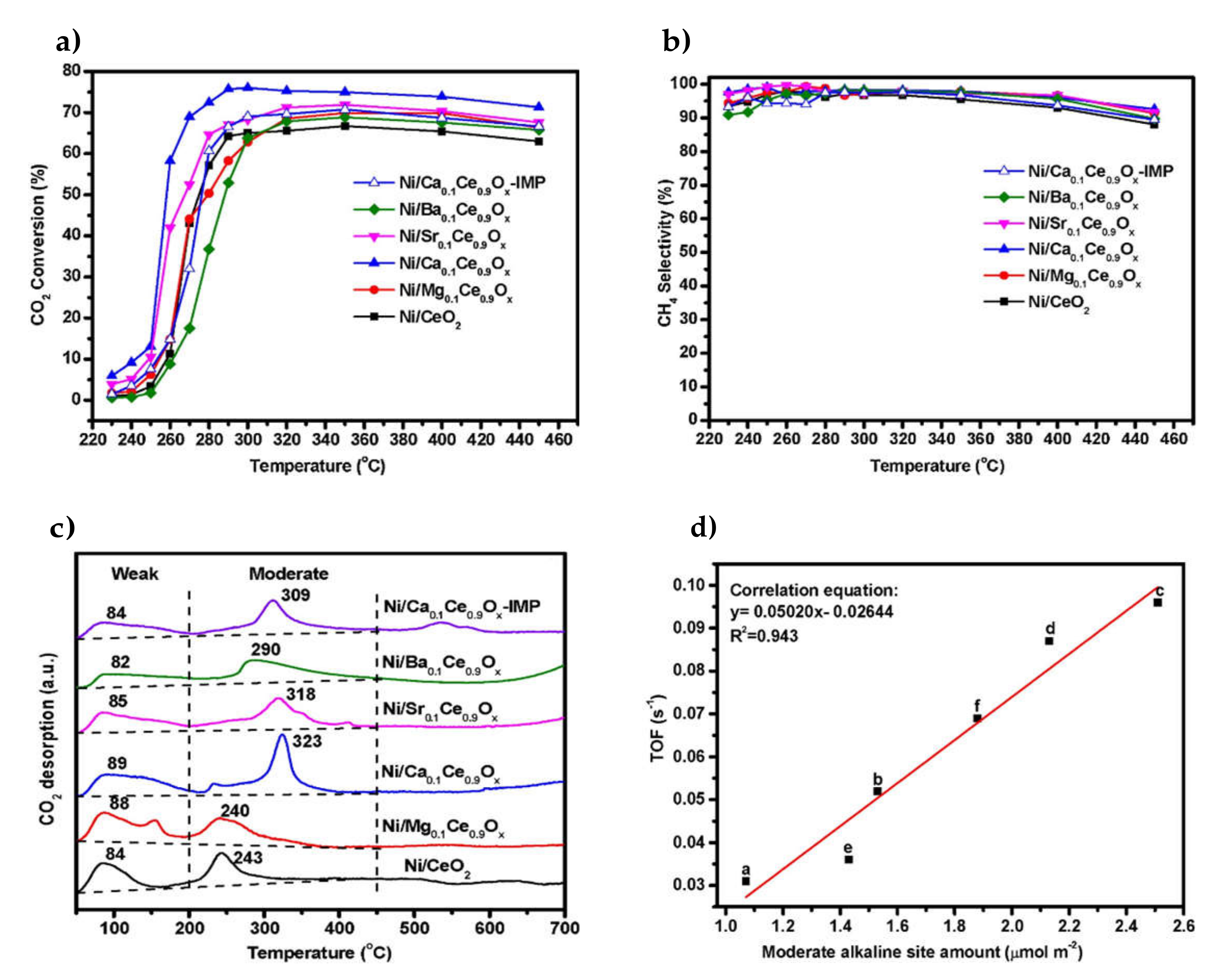
| 1.4% Mg, 2.2% Ca, 4.7% Sr and 7.2% Ba (Ce0.9M0.1Ox support) | 10% Ni/Ce0.9M0.1Ox | Citrate sol-gel modified CeO2 synthesis and Ni incipient wetness impregnation | WHSV = 36,000 mL h−1 g−1 H2/CO2 = 4/1 | XCO2 = 70%, SCH4 = 95% at 270 °C (10% Ni/Ce0.9Ca0.1Ox) | Ca-doping led to the highest methanation activity (with Sr being second-to-best). TOFNi could be linearly correlated to the amount of oxygen vacancies and moderately strong basic sites on the support surface | [30] |
| 1–4% MgO | 10% Ni/SiO2 | Sequential wet impregnations and co-impregnation | WHSV = 36,000 mL h−1 g−1 H2/CO2 = 4/1 | XCO2 = 65%, SCH4 = 98% at 400 °C (1% Mg on 10% Ni/SiO2) | 1% MgO introduced by co-impregantion led to the highest catalytic activity and stability. Higher MgO contents blocked some Ni active sites | [63] |
| 0.4–2.5% Mg (Change in Ni/Mg molar ratio) | 6% Ni/ZrO2 | Citrate complexing method | WHSV = 15,000 mL h−1 g−1, p = 0,1 MPa, H2/CO2 = 4/1 | XCO2 = 90%, SCH4 = 100% at 250 °C (1% Mg on 6% Ni/ZrO2) | Mg-addition led to Ni nanoparticle confinement, as well as resistance to sintering and coke formation | [64] |
| 0.5–5% Mg | 12% Ni/Al2O3 (OMA) | Evaporation-induced self-assembly (EISA) | GHSV = 15,000 h−1 H2/CO2 = 4/1 | XCO2 = 65%, SCH4 = 96% at 300 °C (2.5% Mg on 12% Ni/Al2O3) | Mg-dopant increased the surface basicity of the catalysts and facilitated the chemisorption and activation of CO2 | [65] |
| 1% Mg | α-Fe2O3 | Incipient wetness impregnation | GHSV = 150,000 h−1, p = 8 bar, H2/CO2 = 4/1 | XCO2 = 22%, SCH4 = 9% at 400 °C after 18 h (2.% Mg on α-Fe2O3) | Improved CO2 adsorption over the Mg-modified Fe catalyst led to highly active Fe carbides and better methanation performance | [72] |
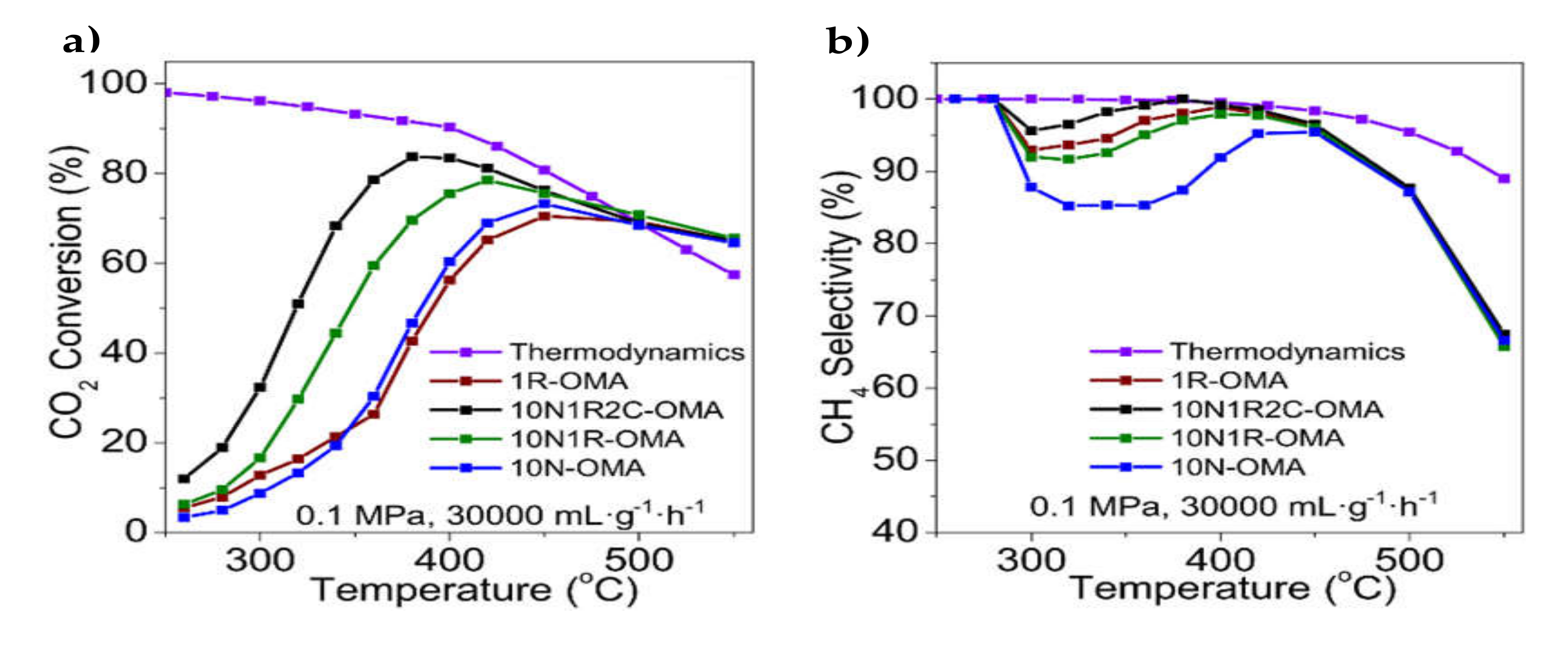
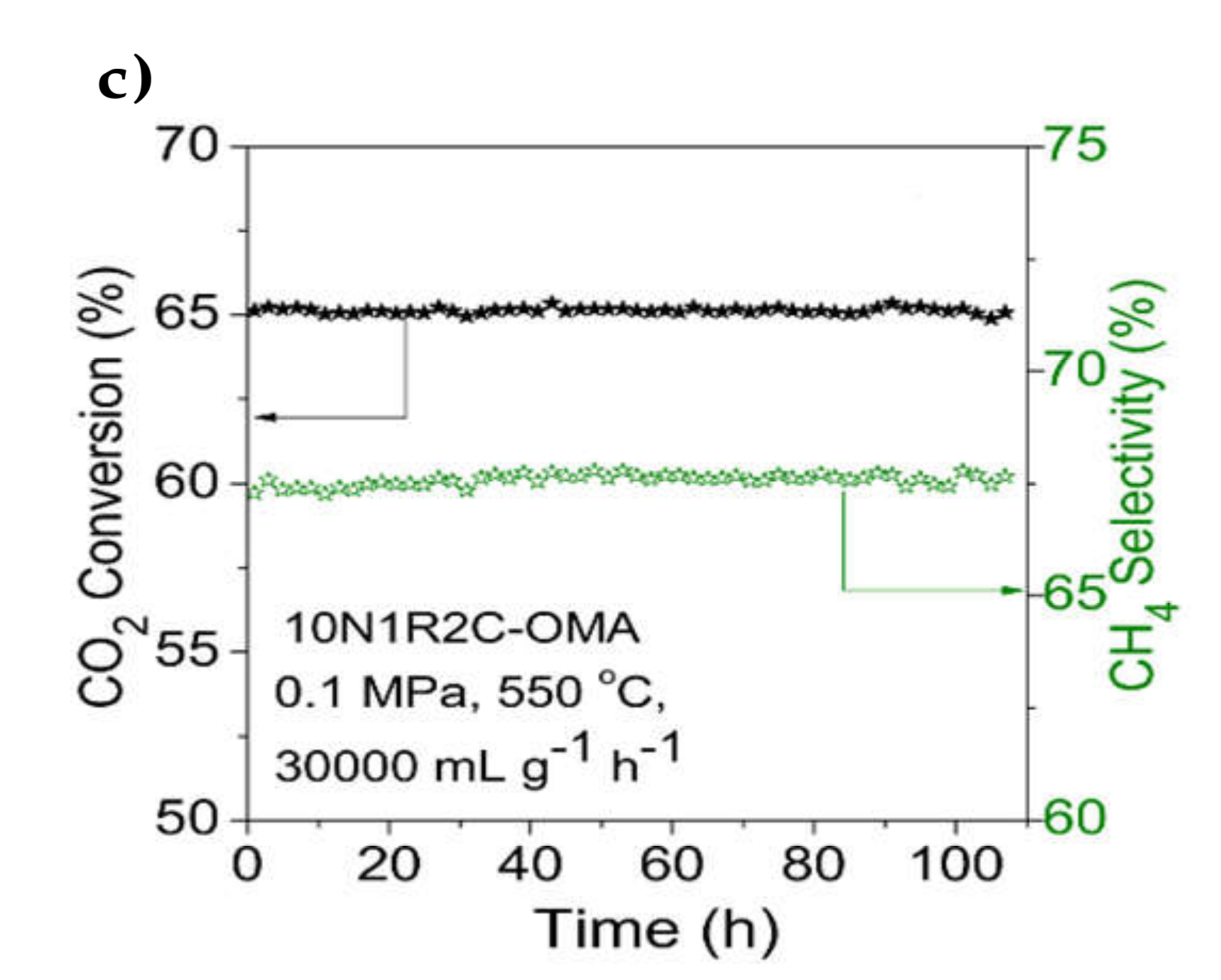
| 0.8–8% Ca | 12% Ni/Al2O3 (OMA) | Evaporation-induced self-assembly (EISA) | GHSV = 15,000 h−1 H2/CO2 = 4/1 | XCO2 = 70%, SCH4 = 98% at 300 °C (6.4% Ca on 12% Ni/Al2O3) | Increased surface alkalinity of the support led to enhanced CO2 chemisorption and lower activation energy for CO2 methanation | [74] |
| 2% CaO | 1% Ru, 10% Ni/Al2O3 (OMA) | Evaporation-induced self-assembly (EISA) | WHSV = 30,000 mL h−1 g−1 H2/CO2 = 4/1 | XCO2 = 80%, SCH4 = 98% at 360 °C (2% CaO on 0.8% Ru, 8% Ni/Al2O3) | Step increase in the CO2 conversion and CH4 selectivity after adding Ru second metal and CaO promoter | [75] |
| 0.7% Ca | 5% Ni/CaxZr1-xO2-δ | Wet impregnation | H2/CO2 = 4/1 | XCO2 = 72%, SCH4 = 100% at 350 °C (0.7% Ca on 5% Ni/CaxZr1-xO2-δ) | Oxygen vacancies-coordinately unsaturated sites (cus) pairs facilitated the C-O bond scission, a crucial step during CO2 methanation | [82] |
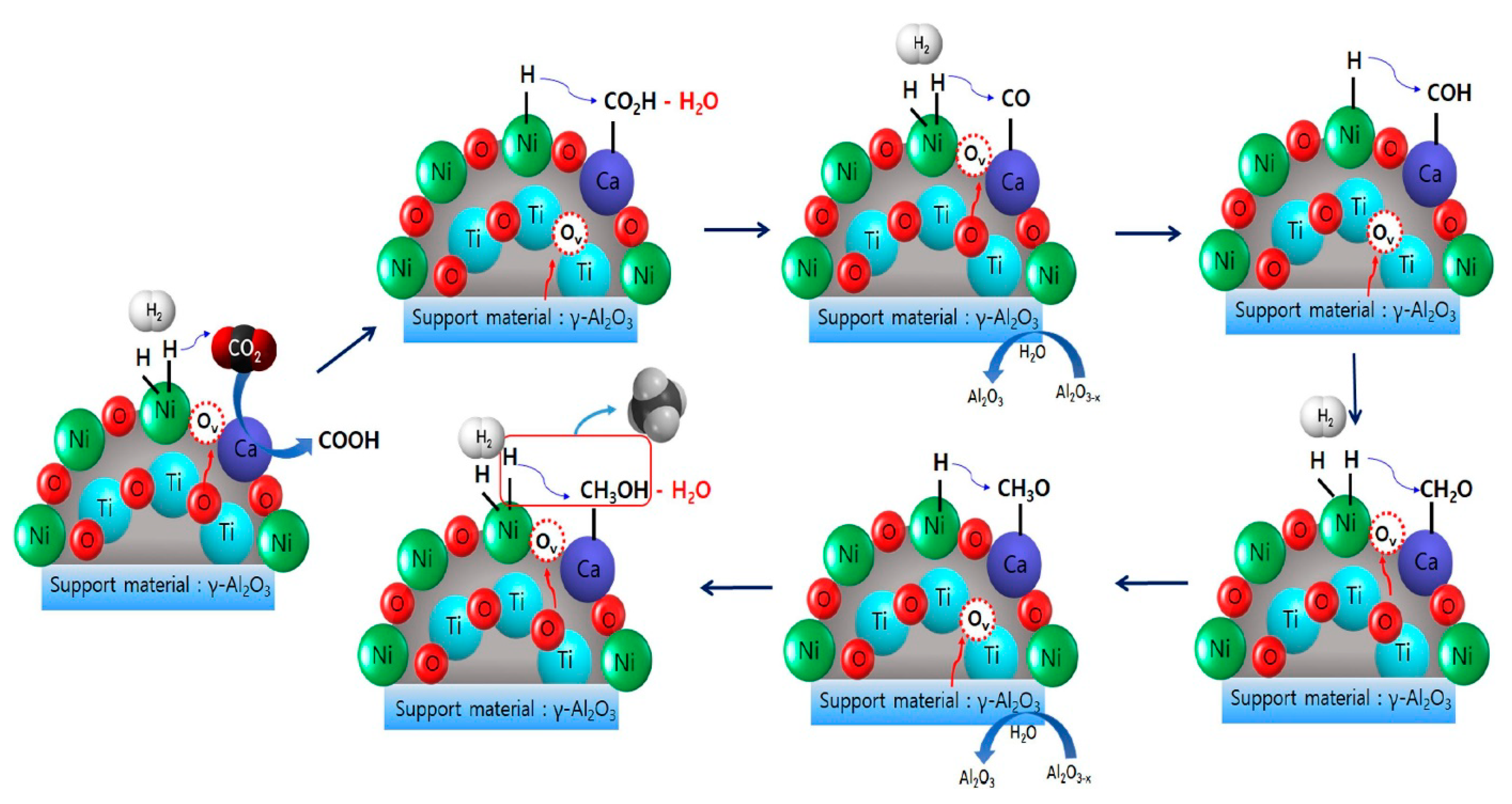
| 0.1–1% Ca (x = 0.01–0.1 for CaxNi1-xTiO3-δ) | 30% Ca-NiTiO3/Al2O3 (11.5% Ni) | Coprecipitation Ca-NiTiO3 synthesis and wet impregnation | GHSV = 5000 h−1 H2/CO2 = 4/1 | XCO2 = 55%, SCH4 = 99.7% at 350 °C (0.5% Ca on 30% Ca-NiTiO3/Al2O3) | Ca-doping on NiTiO3 A-site promoted the formation of oxygen vacancies in close proximity to active Ni and therefore the chemisorption of CO2 | [84] |
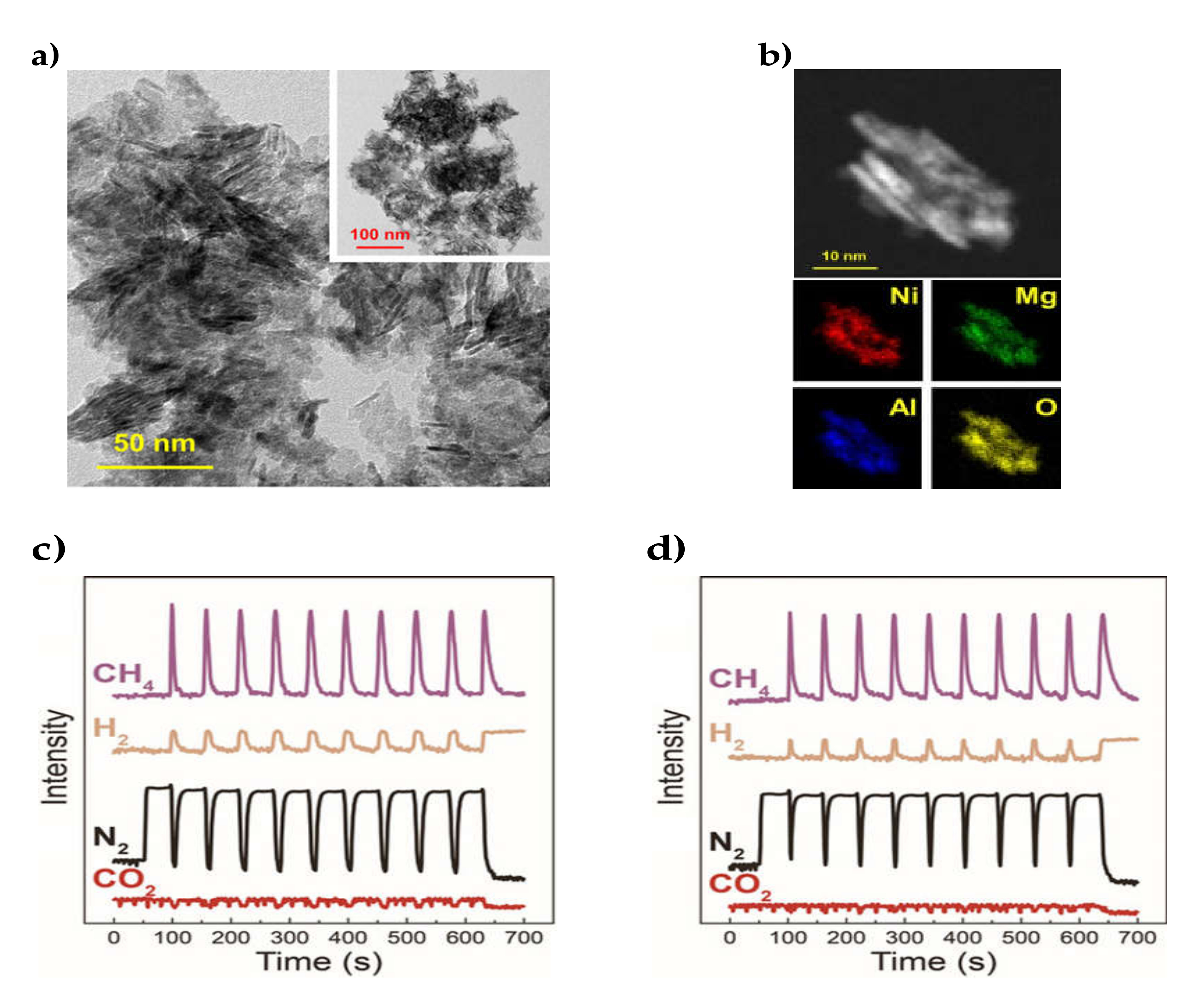
| 14% Mg | 59% Ni/MgO- Al2O3 | Coprecipitation hydrotalcite synthesis and reduction | WHSV = 1100 min-1 g−1 H2/CO2 = 4/1 | XCO2 = 70%, SCH4 = 90% at 315 °C(14% Mg on 59% Ni/MgO- Al2O3) | Stable performance, as well as sintering and coking resistance for the hydrotalcite-derived catalysts | [89] |
| 6.1–28.5% Mg | 17.2–57.8% Ni/MgO- Al2O3 | Coprecipitation hydrotalcite synthesis and reduction | GHSV = 2400 h−1 H2/CO2 = 4/1 | XCO2 = 97.9%, SCH4 = 97.5% at 250 °C (17.3% Mg on 37.5% Ni/MgO-Al2O3 | Far superior low-temperature CO2 conversion for Mg-containing hydrotalcite-derived catalysts, due to the increased surface alkalinity | [90] |
| 0.3–12% Li, Na, K, Cs, Mg, Ca and Ba | 15% Ni/USY zeolite | Ion-exchange for cations and Ni incipient wetness impregnation | GHSV = 43,000 h−1 H2/CO2 = 4/1 | XCO2 = 62%, SCH4 = 92% at 375 °C (12.1% Cs in 15% Ni/Cs-exchanged USY) | Greater size for monovalent cations improved CO2 adsorption (Cs+ > Na+ > Li+ > H+), while Mg2+ benefited Ni dispersion | [98] |
| 0.9–13% Mg | 4.8–15% Ni/USY zeolite | Ion-exchange or impregnation for Mg and Ni incipient wetness impregnation | GHSV = 43,000 h−1 H2/CO2 = 4/1 | XCO2 = 65%, SCH4 = 93% at 400 °C (0.7% Mg in 15% Ni/Mg-exchanged USY) | Finer and better-dispersed Ni nanoparticles after Mg impregnation or Mg ion-exchange | [68] |
| CO2 Sorbent | Catalyst | CO2 Capture Conditions | CO2 Methanation Conditions | CH4 Production [mmol CH4/g DFM] | Comments | Reference |
|---|---|---|---|---|---|---|
| 1–10% CaO | 1–10% Ru/Al2O3 | 10% CO2/N2, 320 °C, 30 min | 4% H2/N2, 320 °C, 2 h | 0.50 (5% Ru, 10% CaO/Al2O3) | High sorbent loading increased CO2 spillover to Ru active sites. 5% Ru, 10%CaO/Al2O3 as best composition. Cyclic operation at 320 °C | [19] |
| 10% CaO, MgO, Na2CO3 and K2CO3 | 0.1–10% Rh and 5% Ru/Al2O3 | 10% CO2/N2, 320 °C, 30 min | 4% H2/N2, 320 °C, 2 h | 0.4 (0.1% Rh, 10% CaO/Al2O3), 1.05 (5% Ru, 10% Na2CO3/Al2O3) | 0.1% Rh was quite active but Rh is pricy. Na2CO3 constituted a better adsorbent than CaO for Ru-based DFMs | [113] |
| 10% CaO, MgO, Na2CO3 (6.1% “Na2O”) and K2CO3 (7.01% “Κ2O”) | 0.5% Rh and 5% Ru/Al2O3 | 10% CO2/N2, 320 °C, 30 min | 10% H2/N2, 320 °C, 1 h | 0.614 (5% Ru, 6.1% “Na2O”/Al2O3) | Ru-based DFMs with Na2CO3 achieved the fastest hydrogenation rates | [21] |
| 5–15% CaO and Na2CO3 | 4% Ru/Al2O3 | 11% CO2/Ar, 280–400 °C, 1 min | 10% H2/Ar, 280–400 °C, 2 min | 0.414 at 400 °C (4% Ru, 15% CaO/Al2O3), 0.383 at 310 °C (4% Ru, 10% Na2CO3/Al2O3) | Higher sorbent loading promoted CH4 yield. More stable carbonates formed on CaO required higher temperature to decompose | [24] |
| 10% LiNO3, NaNO3, Na2CO3 and K2CO3 | 1% Ru/Al2O3 | 15% CO2/Ar, 230 °C, 30 min | 12% H2/Ar, 230 °C, 30 min | Not reported | Li-promoted DFM presented faster methanation kinetics. Operation at just 230 °C, a lower temperature compared with other DFMs | [35] |
| 200% High-capacity MgO sorbent | 2.5–10% Ru/CeO2 | 65% CO2/N2, 300 °C, 1 h | 5% H2/N2, 300 °C, | 6.60, 1. cycle 3.36, 10. cycle (5% Ru/CeO2 + 200% MgO) | High CO2 capture capacity due to a large MgO content. Mechanism: oxygen vacancy catalyzed CO2 hydrogenation through a formate intermediate | [25] |
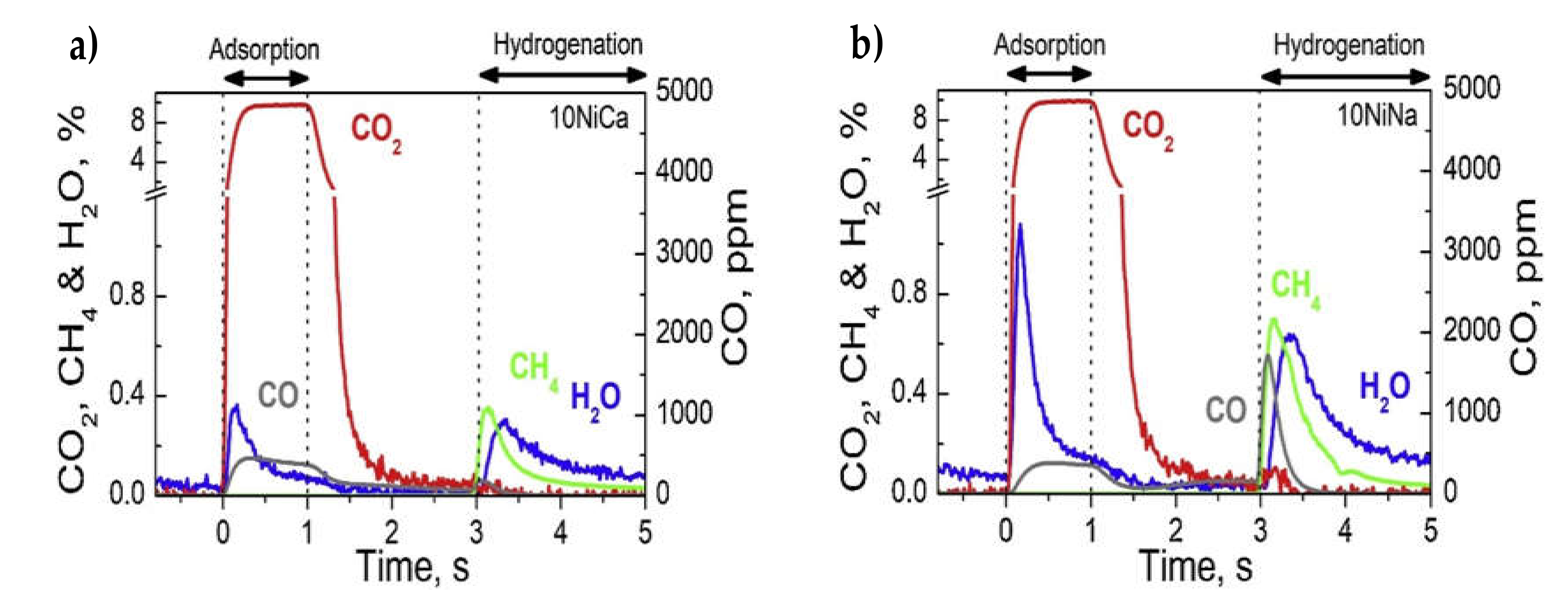
| 10% Na2CO3 (6.1% “Na2O”) | 10% Ni/Al2O3 | 7.5% CO2/N2 and 7.5% CO2, 4.5% O2, 15% H2O/N2, 320 °C, 20 min | 15% H2/N2, 320 °C, 1 h | 0.276, Without O2 during CO2 capture 0, With O2 during CO2 capture (10% Ni, 6.1% “Na2O”/Al2O3) | Ni-based DFMs required a prereduction/activation step at 650 °C and an O2-free gas during CO2 capture in order to produce CH4 | [21] |
| 10% Na2CO3 (6.1% “Na2O”) | 0.1–1% Ru, Pt, Pd with 10% Ni/Al2O3 | 7.5% CO2, 4.5% O2, 15% H2O/N2, 320 °C, 20 min | 15% H2/N2, 320 °C | 0.38 (1% Ru, 10% Ni, 6.1% “Na2O”/Al2O3) | Pt and Ru addition as second metals promoted NiO reducibility. Ni and Ru synergy led to the highest CH4 yield | [23] |
| 5–15% CaO and Na2CO3 | 5–15% Ni/Al2O3 | 10% CO2/Ar, 280–520 °C, 1 min | 10% H2/Ar, 280–520 °C, 2 min | 0.142 at 520 °C (15% Ni, 15% CaO/Al2O3), 0.185 at 400 °C (10% Ni, 10% Na2CO3/Al2O3) | The adsorbent phase enhanced NiO reducibility. Methanation was favoured at higher temperatures when CaO was used as sorbent | [112] |
| 10% CaO, MgO, Na2CO3 and K2CO3 | 10% Ni/Al2O3 | 9.5% CO2/N2, 320 °C, 25 min | 10% H2/N2, 320 °C, 20 min | 0.140 (10% Ni, 10% CaO/Al2O3) 0.178 (10% Ni, 10% Na2CO3/Al2O3) | Easier CO2 release and higher methanation capacity when Na2CO3 was used as sorbent | [119] |
| 5% K and La | 15% Ni/ZrO2 | 4.7% CO2/He, 250–450 °C, 5 min | Pure H2, 250–450 °C, 5 min | Not reported | K-promotion greatly enhanced CO2 adsorption and promoted methanation at higher temperatures, while La-promotion favoured faster methanation kinetics | [120] |
| 15–30% Mg | 17–50% Ni/MgO- Al2O3 | 15% CO2/N2, 200 and 250 °C, various times | Pure H2, 200 and 250 °C, various times | Not reported | Hydrotalcite-derived layered oxides with high Ni and Mg contents provided a high CH4 yield and favourable methanation kinetics | [121] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2. Catalysts 2020, 10, 812. https://doi.org/10.3390/catal10070812
Tsiotsias AI, Charisiou ND, Yentekakis IV, Goula MA. The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2. Catalysts. 2020; 10(7):812. https://doi.org/10.3390/catal10070812
Chicago/Turabian StyleTsiotsias, Anastasios I., Nikolaos D. Charisiou, Ioannis V. Yentekakis, and Maria A. Goula. 2020. "The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2" Catalysts 10, no. 7: 812. https://doi.org/10.3390/catal10070812
APA StyleTsiotsias, A. I., Charisiou, N. D., Yentekakis, I. V., & Goula, M. A. (2020). The Role of Alkali and Alkaline Earth Metals in the CO2 Methanation Reaction and the Combined Capture and Methanation of CO2. Catalysts, 10(7), 812. https://doi.org/10.3390/catal10070812