Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol


Abstract
1. Introduction
2. Results and Discussion
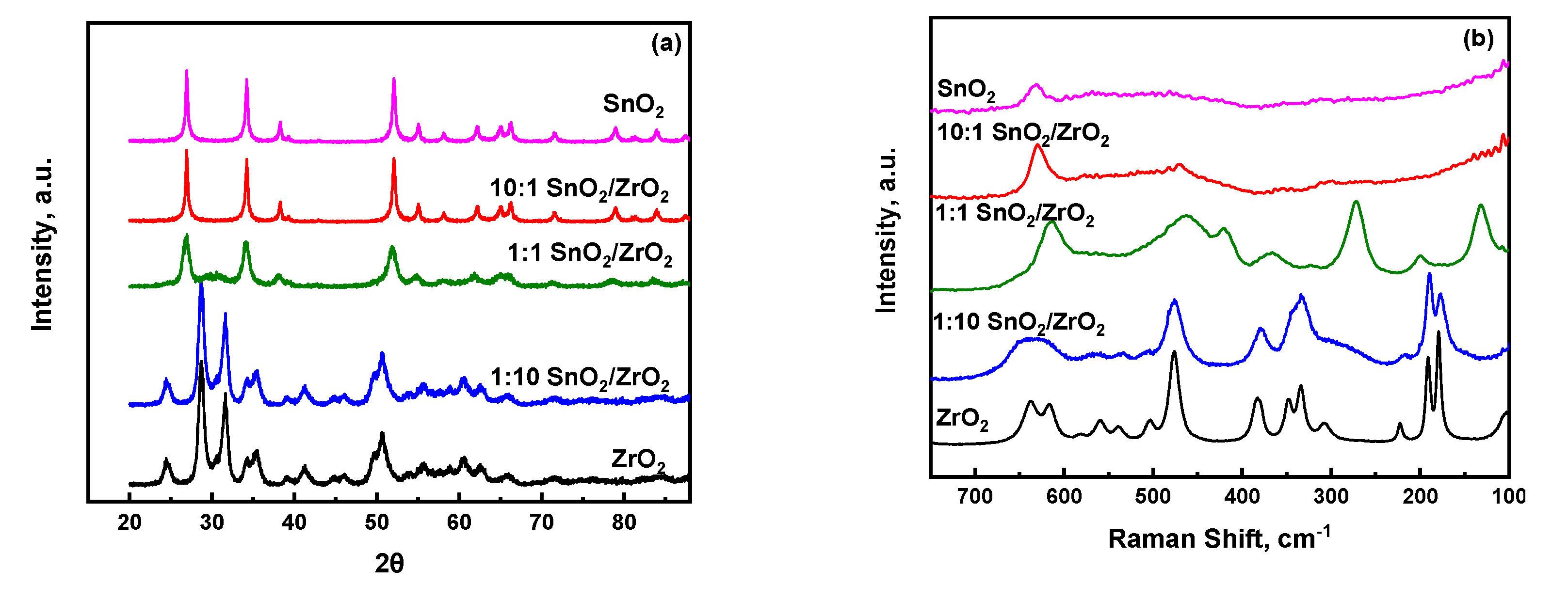
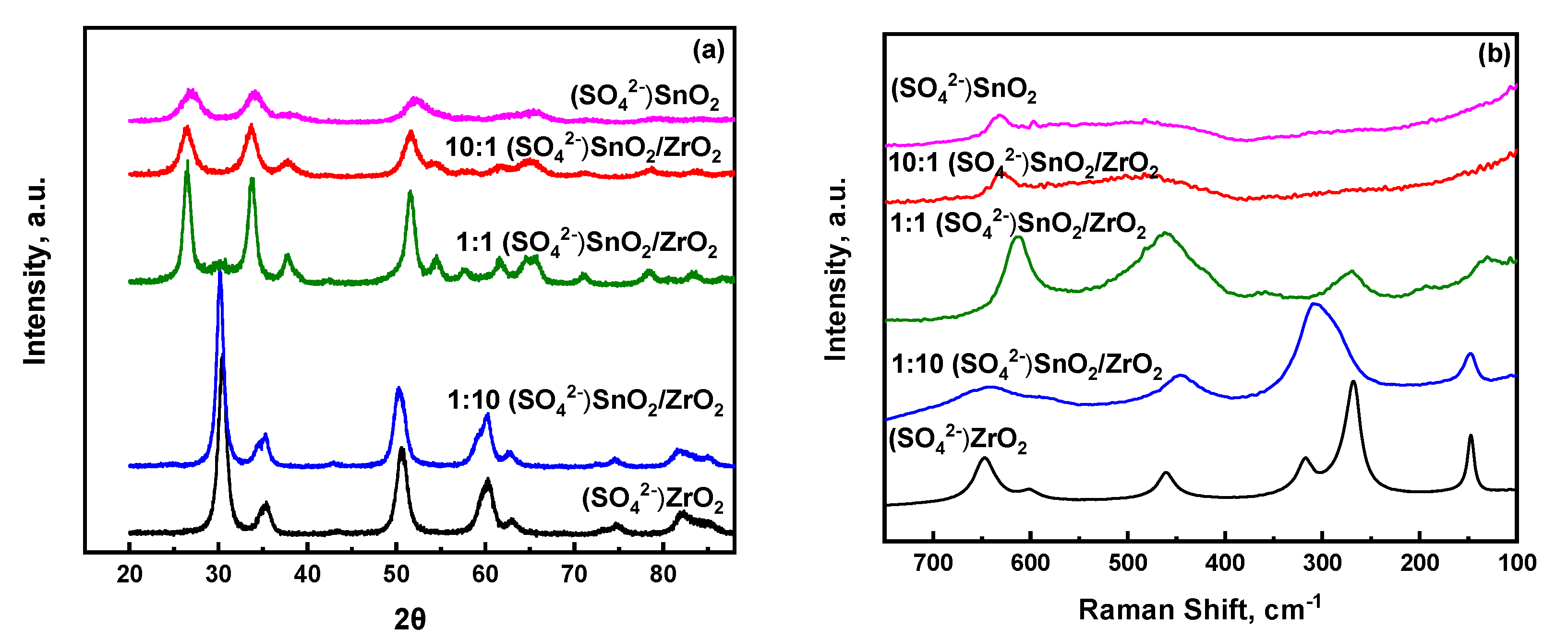
2.1. Physicochemical Properties of SnO2/ZrO2 and S-SnO2/ZrO2 Mixed Metal Oxide Catalysts
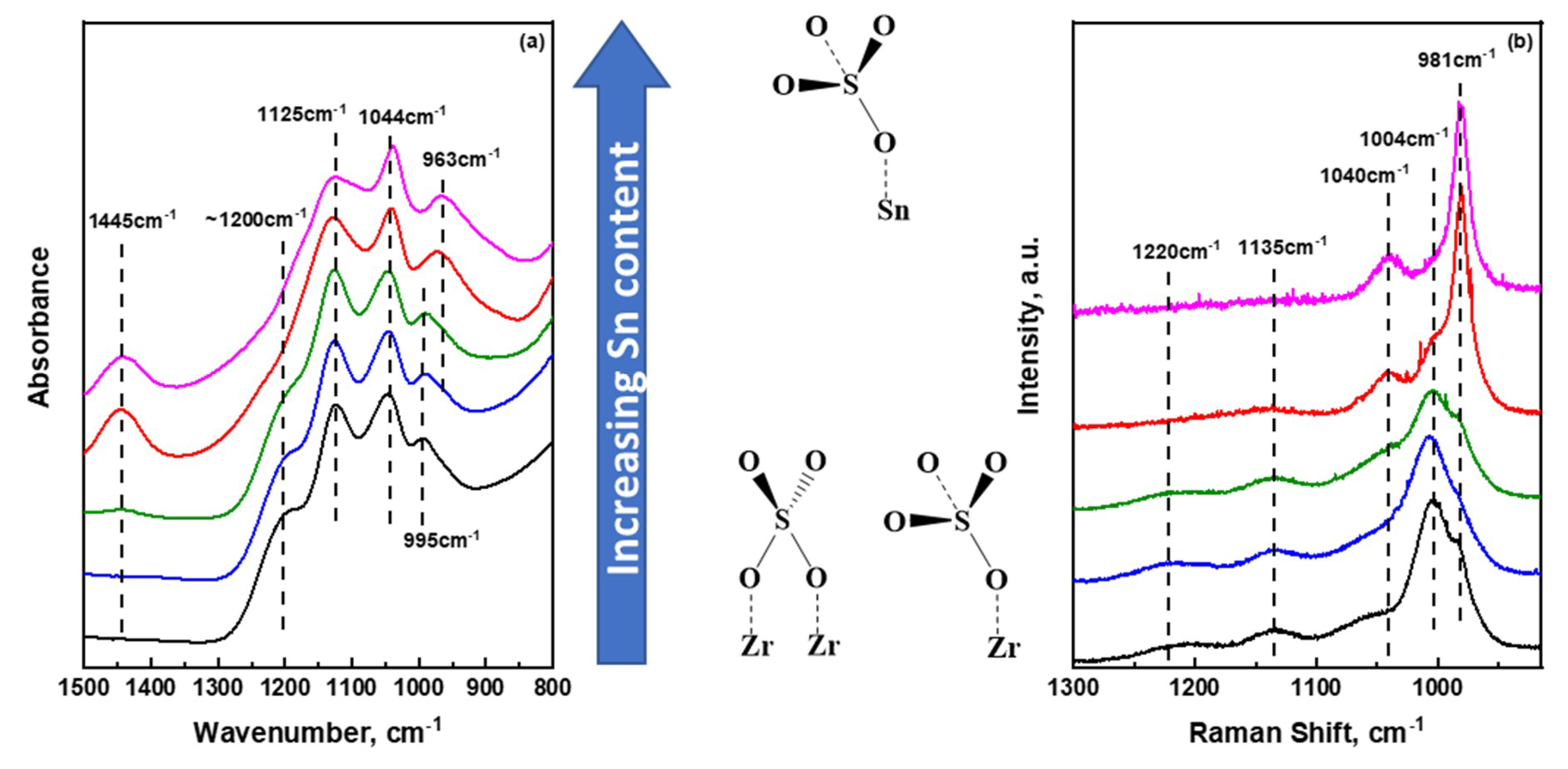
2.2. Insights into the Molecular Structure of Surface Sulfate Species on Mixed SnO2/ZrO2 Hydroxides
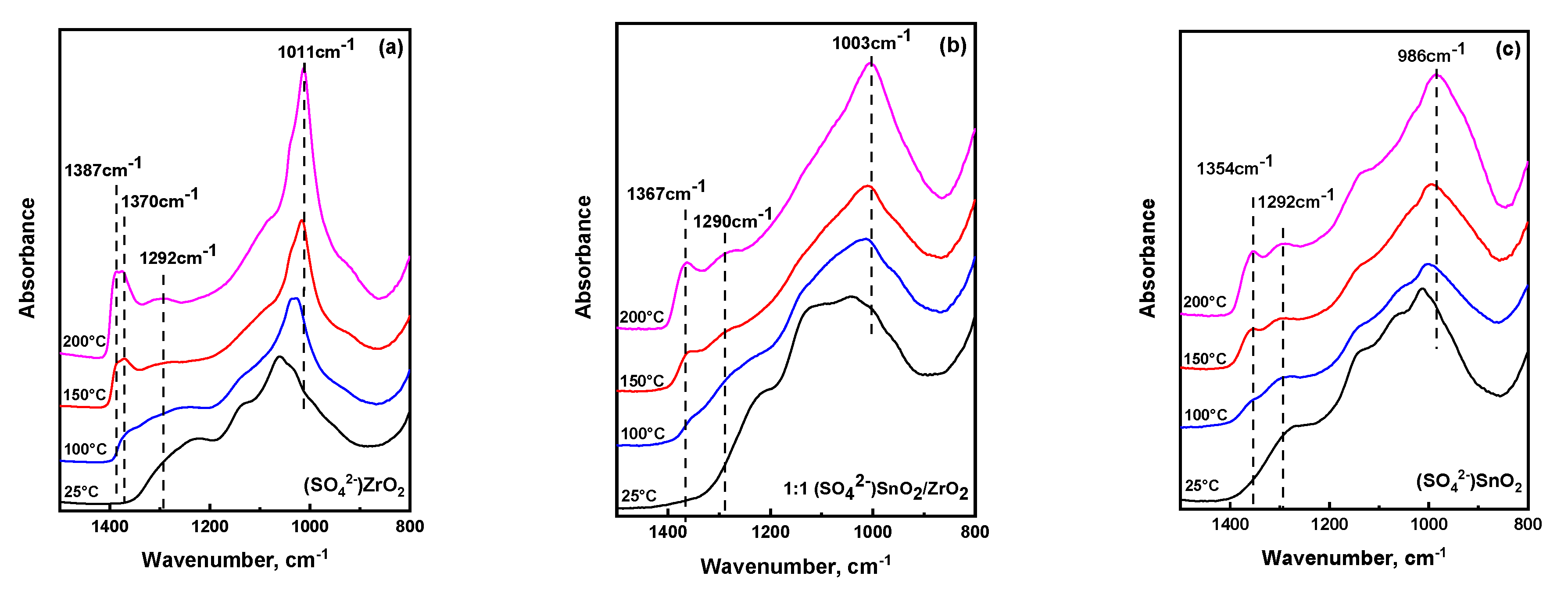
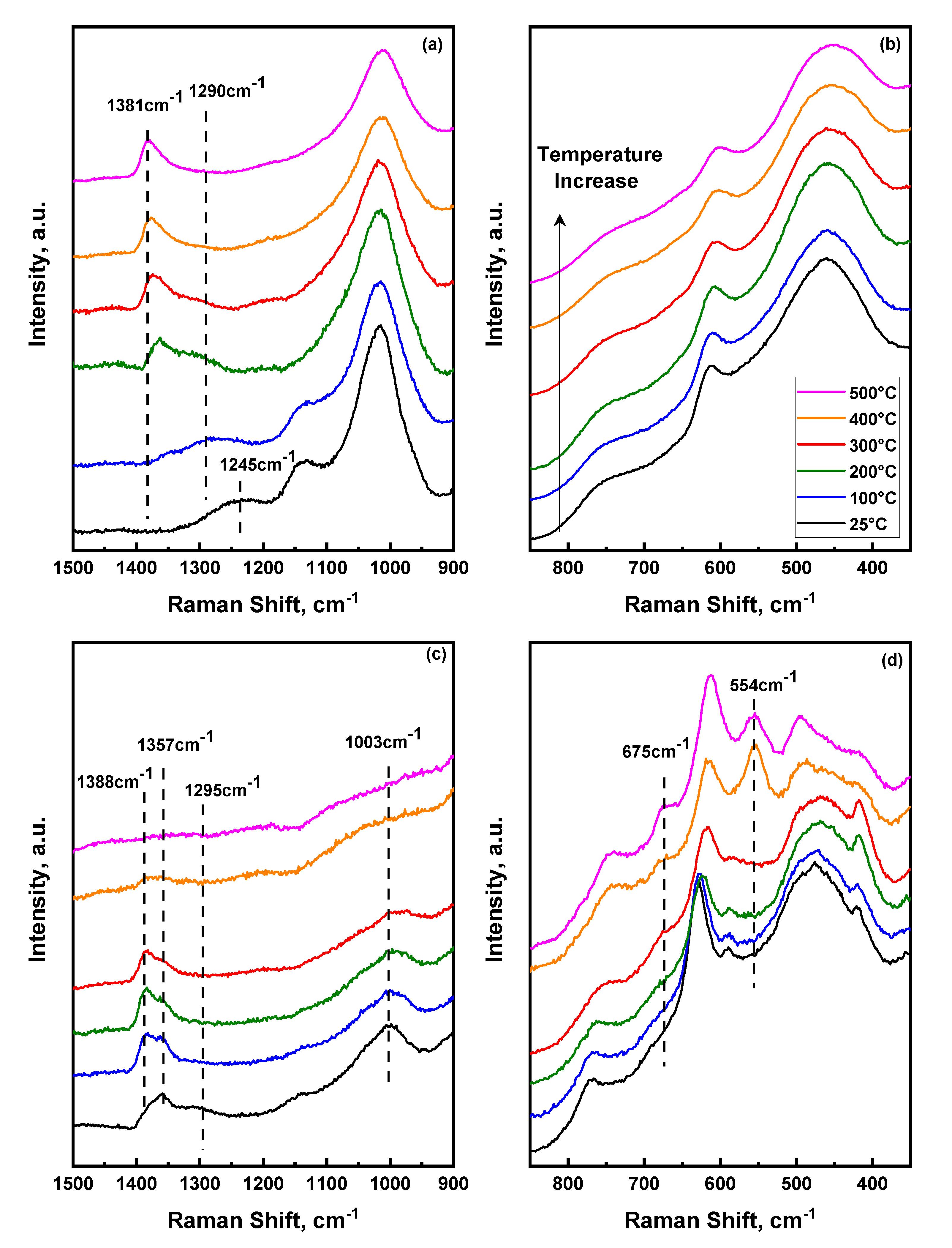
2.3. Temperature Dependent Evolution of Surface Sulfate Species on Mixed Oxides
2.4. Catalytic Activity of S-SnO2/ZrO2 on Tert-Butylation of Phenol
3. Materials and Methods
3.1. Materials and Synthesis of Catalysts
3.2. Physicochemical Characterization
3.3. Catalytic Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tanabe, K.; Holderich, W.F. Industrial application of solid acid-base catalysts. Appl. Catal. A Gen. 1999, 181, 399–434. [Google Scholar] [CrossRef]
- Okuhara, T. Water-tolerant solid acid catalysts. Chem. Rev. 2002, 102, 3641–3665. [Google Scholar] [CrossRef] [PubMed]
- Corma, A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614. [Google Scholar] [CrossRef]
- Arata, K.; Matsuhashi, H.; Hino, M.; Nakamura, H. Synthesis of solid superacids and their activities for reactions of alkanes. Catal. Today 2003, 81, 17–30. [Google Scholar] [CrossRef]
- Arata, K.; Hino, M. Preparation of Superacids by Metal-Oxides and Their Catalytic Action. Mater Chem. Phys. 1990, 26, 213–237. [Google Scholar] [CrossRef]
- Reddy, B.M.; Sreekanth, P.M.; Yamada, Y.; Xu, X.; Kobayashi, T. Surface characterization of sulfate, molybdate, and tungstate promoted TiO2-ZrO2 solid acid catalysts by XPS and other techniques. Appl. Catal. A Gen. 2002, 228, 269–278. [Google Scholar] [CrossRef]
- Morterra, C.; Cerrato, G.; Pinna, F.; Signoretto, M.; Strukul, G. On the Acid-Catalyzed Isomerization of Light Paraffins over a Zro2/So4 System: The Effect of Hydration. J. Catal. 1994, 149, 181–188. [Google Scholar] [CrossRef]
- Morterra, C.; Cerrato, G.; Signoretto, M. On the role of the calcination step in the preparation of active (superacid) sulfated zirconia catalysts. Catal. Lett. 1996, 41, 101–109. [Google Scholar] [CrossRef]
- Signoretto, M.; Pinna, F.; Strukul, G.; Cerrato, G.; Morterra, C. Platinum promoted zirconia-sulfate catalysts: One-pot preparation, physical properties and catalytic activity. Catal. Lett. 1996, 36, 129–133. [Google Scholar] [CrossRef]
- Wachs, I.E.; Routray, K. Catalysis Science of Bulk Mixed Oxides. ACS Catal. 2012, 2, 1235–1246. [Google Scholar] [CrossRef]
- Rosenberg, D.J.; Coloma, F.; Anderson, J.A. Modification of the acid properties of silica-zirconia aerogels by in situ and ex situ sulfation. J. Catal. 2002, 210, 218–228. [Google Scholar] [CrossRef]
- Bensitel, M.; Saur, O.; Lavalley, J.C.; Morrow, B.A. An Infrared Study of Sulfated Zirconia. Mater. Chem. Phys. 1988, 19, 147–156. [Google Scholar] [CrossRef]
- Jin, T.; Yamaguchi, T.; Tanabe, K. Mechanism of Acidity Generation on Sulfur-Promoted Metal-Oxides. J. Phys. Chem. 1986, 90, 4794–4796. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Jin, T.; Tanabe, K. Structure of Acid Sites on Sulfur-Promoted Iron-Oxide. J. Phys. Chem. 1986, 90, 3148–3152. [Google Scholar] [CrossRef]
- Babou, F.; Coudurier, G.; Vedrine, J.C. Acidic Properties of Sulfated Zirconia: An Infrared Spectroscopic Study. J. Catal. 1995, 152, 341–349. [Google Scholar] [CrossRef]
- Saur, O.; Bensitel, M.; Saad, A.B.M.; Lavalley, J.C.; Tripp, C.P.; Morrow, B.A. The Structure and Stability of Sulfated Alumina and Titania. J. Catal. 1986, 99, 104–110. [Google Scholar] [CrossRef]
- Ward, D.A.; Ko, E.I. One-Step Synthesis and Characterization of Zirconia-Sulfate Aerogels as Solid Superacids. J. Catal. 1994, 150, 18–33. [Google Scholar] [CrossRef]
- Tabora, J.E.; Davis, R.J. On the superacidity of sulfated zirconia catalysts for low-temperature isomerization of butane. J. Am. Chem. Soc. 1996, 118, 12240–12241. [Google Scholar] [CrossRef]
- Suzuki, T.; Yokoi, T.; Otomo, R.; Kondo, J.N.; Tatsumi, T. Dehydration of xylose over sulfated tin oxide catalyst: Influences of the preparation conditions on the structural properties and catalytic performance. Appl. Catal. A Gen. 2011, 408, 117–124. [Google Scholar] [CrossRef]
- Furuta, S.; Matsuhashi, H.; Arata, K. Biodiesel fuel production with solid superacid catalysis in fixed bed reactor under atmospheric pressure. Catal. Commun. 2004, 5, 721–723. [Google Scholar] [CrossRef]
- Furuta, S.; Matsuhashi, H.; Arata, K. Catalytic action of sulfated tin oxide for etherification and esterification in comparison with sulfated zirconia. Appl Catal. A Gen. 2004, 269, 187–191. [Google Scholar] [CrossRef]
- Varala, R.; Narayana, V.; Kulakarni, S.R.; Khan, M.; Alwarthan, A.; Adil, S.F. Sulfated tin oxide (STO)—Structural properties and application in catalysis: A review. Arab. J. Chem. 2016, 9, 550–573. [Google Scholar] [CrossRef]
- Ahmed, A.I.; El-Hakam, S.A.; Khder, A.S.; El-Yazeed, W.S.A. Nanostructure sulfated tin oxide as an efficient catalyst for the preparation of 7-hydroxy-4-methyl coumarin by Pechmann condensation reaction. J. Mol. Catal. A Chem. 2013, 366, 99–108. [Google Scholar] [CrossRef]
- Wang, G.W.; Hattori, H.; Tanabe, K. Acid-Base and Catalytic Properties of ZrO2-SnO2. Bull. Chem. Soc. Jpn. 1983, 56, 2407–2410. [Google Scholar] [CrossRef]
- Patel, A.; Coudurier, G.; Essayem, N.; Vedrine, J.C. Effect of the addition of Sn to zirconia on the acidic properties of the sulfated mixed oxide. J. Chem. Soc. Faraday Trans. 1997, 93, 347–353. [Google Scholar] [CrossRef]
- Chen, W.H.; Ko, H.H.; Sakthivel, A.; Huang, S.J.; Liu, S.H.; Lo, A.Y.; Tsai, T.C.; Liu, S.B. A solid-state NMR, FT-IR and TPD study on acid properties of sulfated and metal-promoted zirconia: Influence of promoter and sulfation treatment. Catal. Today 2006, 116, 111–120. [Google Scholar] [CrossRef]
- Stichert, W.; Schuth, F.; Kuba, S.; Knozinger, H. Monoclinic and tetragonal high surface area sulfated zirconias in butane isomerization: CO adsorption and catalytic results. J. Catal. 2001, 198, 277–285. [Google Scholar] [CrossRef]
- Occhiuzzi, M.; Cordischi, D.; Dragone, R. Manganese ions in the monoclinic, tetragonal and cubic phases of zirconia: An XRD and EPR study. Phys. Chem. Chem. Phys. 2003, 5, 4938–4945. [Google Scholar] [CrossRef]
- Jiang, L.H.; Sun, G.Q.; Zhou, Z.H.; Sun, S.G.; Wang, Q.; Yan, S.Y.; Li, H.Q.; Tian, J.; Guo, J.S.; Zhou, B.; et al. Size-controllable synthesis of monodispersed SnO2 nanoparticles and application in electrocatalysts. J. Phys. Chem. B 2005, 109, 8774–8778. [Google Scholar] [CrossRef]
- Wilson, G.; Glasser, F.P. Solid-Solution in the ZrO2-Sno2-TiO2 System. Br. Ceram. Trans. J. 1989, 88, 69–74. [Google Scholar]
- Zhou, W.; Soultanidis, N.; Xu, H.; Wong, M.S.; Neurock, M.; Kiely, C.J.; Wachs, I.E. Nature of Catalytically Active Sites in the Supported WO3/ZrO2 Solid Acid System: A Current Perspective. ACS Catal. 2017, 7, 2181–2198. [Google Scholar] [CrossRef]
- Nakamoto, K. Applications in Inorganic Chemistry. In Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A: Theory and Applications in Inorganic Chemistry; Wiley: Hoboken, NJ, USA, 2008; pp. 149–354. [Google Scholar] [CrossRef]
- Hug, S.J. In situ Fourier transform infrared measurements of sulfate adsorption on hematite in aqueous solutions. J. Colloid. Interface Sci. 1997, 188, 415–422. [Google Scholar] [CrossRef]
- Ben Mabrouk, K.; Kauffmann, T.H.; Aroui, H.; Fontana, M.D. Raman study of cation effect on sulfate vibration modes in solid state and in aqueous solutions. J. Raman Spectrosc. 2013, 44, 1603–1608. [Google Scholar] [CrossRef]
- Lefevre, G. In situ Fourier-transform infrared spectroscopy studies of inorganic ions adsorption on metal oxides and hydroxides. Adv. Colloid. Interface 2004, 107, 109–123. [Google Scholar] [CrossRef]
- Lavalley, J.C. Infrared spectrometric studies of the surface basicity of metal oxides and zeolites using adsorbed probe molecules. Catal. Today 1996, 27, 377–401. [Google Scholar] [CrossRef]
- Tabora, J.E.; Davis, R.J. Structure of Fe, Mn-Promoted Sulfated Zirconia Catalyst by X-ray and in Absorption Spectroscopies. J. Chem. Soc. Faraday Trans. 1995, 91, 1825–1833. [Google Scholar] [CrossRef]
- Muller, K.; Lefevre, G. Vibrational Characteristics of Outer-Sphere Surface Complexes: Example of Sulfate Ions Adsorbed onto Metal (Hydr)oxides. Langmuir 2011, 27, 6830–6835. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.X.; Wang, A.Q.; Wachs, I.E.; Baltrusaitis, J. Critical review on the active site structure of sulfated zirconia catalysts and prospects in fuel production. Appl. Catal. A Gen. 2019, 572, 210–225. [Google Scholar] [CrossRef]
- Haase, F.; Sauer, J. The surface structure of sulfated zirconia: Periodic ab initio study of sulfuric acid adsorbed on ZrO2(101) and ZrO2(001). J. Am. Chem. Soc. 1998, 120, 13503–13512. [Google Scholar] [CrossRef]
- Klose, B.S.; Jentoft, F.C.; Schlogl, R. In situ diffuse-reflectance infrared spectroscopic investigation of promoted sulfated zirconia catalysts during n-butane isomerization. J. Catal. 2005, 233, 68–80. [Google Scholar] [CrossRef]
- Kalampounias, A.G.; Tsilomelekis, G.; Berg, R.W.; Boghosian, S. Molybdenum(VI) Oxosulfato Complexes in MoO3-K2S2O7-K2SO4 Molten Mixtures: Stoichiometry, Vibrational Properties, and Molecular Structures. J. Phys. Chem. A 2012, 116, 8861–8872. [Google Scholar] [CrossRef] [PubMed]
- Kalampounias, A.G.; Tsilomelekis, G.; Boghosian, S. Liquid phase dynamics of molten M2S2O7 (M = K, Cs): A temperature dependent Raman spectroscopic study. Vib. Spectrosc. 2013, 65, 66–73. [Google Scholar] [CrossRef]
- Kalampounias, A.G.; Tsilomelekis, G.; Boghosian, S. Molten and glassy tellurium(IV) oxosulfato complexes in the TeO2-K2S2O7 system studied by Raman spectroscopy: Stoichiometry, vibrational properties and molecular structure. Vib. Spectrosc. 2018, 97, 85–90. [Google Scholar] [CrossRef]
- Dong, J.L.; Li, X.H.; Zhao, L.J.; Xiao, H.S.; Wang, F.; Guo, X.; Zhang, Y.H. Raman observation of the interactions between NH4+, SO42-, and H2O in supersaturated (NH4)(2)SO4 droplets. J. Phys. Chem. B 2007, 111, 12170–12176. [Google Scholar] [CrossRef]
- Andriopoulou, C.; Boghosian, S. Heterogeneity of deposited phases in supported transition metal oxide catalysts: Reversible temperature-dependent evolution of molecular structures and configurations. Phys. Chem. Chem. Phys. 2018, 20, 1742–1751. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Tribalis, A.; Kalampounias, A.G.; Boghosian, S.; Panagiotou, G.D.; Bourikas, K.; Kordulis, C.; Lycourghiotis, A. Temperature-dependent evolution of molecular configurations of oxomolybdenum species on MoO3/TiO2 catalysts monitored by in situ Raman spectroscopy. Stud. Surf. Sci. Catal. 2010, 175, 613–616. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Boghosian, S. On the configuration, molecular structure and vibrational properties of MoOx sites on alumina, zirconia, titania and silica. Catal. Sci. Technol. 2013, 3, 1869–1888. [Google Scholar] [CrossRef]
- Tribalis, A.; Panagiotou, G.D.; Tsilomelekis, G.; Kalampounias, A.G.; Bourikas, K.; Kordulis, C.; Boghosian, S.; Lycourghiotis, A. Temperature-Dependent Evolution of the Molecular Configuration of Oxo-Tungsten(VI) Species Deposited on the Surface of Titania. J. Phys. Chem. C 2014, 118, 11319–11332. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Panagiotou, G.D.; Stathi, P.; Kalampounias, A.G.; Bourikas, K.; Kordulis, C.; Deligiannakis, Y.; Boghosian, S.; Lycourghiotis, A. Molybdena deposited on titania by equilibrium deposition filtration: Structural evolution of oxo-molybdenum(VI) sites with temperature. Phys. Chem. Chem. Phys. 2016, 18, 23980–23989. [Google Scholar] [CrossRef]
- Anderson, B.G.; Dang, Z.; Morrow, B.A. Silica-Supported Zirconia. 2. Effect of Sulfation on the Surface-Acidity and Its Potential as a Catalyst for Methane-Olefin Coupling. J. Phys. Chem. 1995, 99, 14444–14449. [Google Scholar] [CrossRef]
- Platero, E.E.; Mentruit, M.P.; Arean, C.O.; Zecchina, A. FTIR studies on the acidity of sulfated zirconia prepared by thermolysis of zirconium sulfate. J. Catal. 1996, 162, 268–276. [Google Scholar] [CrossRef]
- Breitkopf, C.; Papp, H.; Li, X.; Olindo, R.; Lercher, J.A.; Lloyd, R.; Wrabetz, S.; Jentoft, F.C.; Meinel, K.; Forster, S.; et al. Activation and isomerization of n-butane on sulfated zirconia model systems—An integrated study across the materials and pressure gaps. Phys. Chem. Chem. Phys. 2007, 9, 3600–3618. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-F.; Yan, P.; Hao, X.-Y.; Wang, Z.-Z. Influences of introducing Al on the solid super acid SO42−/SnO2. Mater. Chem. Phys. 2008, 112, 1065–1068. [Google Scholar] [CrossRef]
- Matsuhashi, H.; Miyazaki, H.; Kawamura, Y.; Nakamura, H.; Arata, K. Preparation of a solid superacid of sulfated tin oxide with acidity higher than that of sulfated zirconia and its applications to aldol condensation and benzoylation. Chem. Mater. 2001, 13, 3038–3042. [Google Scholar] [CrossRef]
- Cui, X.; Ma, H.Z.; Wang, B.; Chen, H.W. Direct oxidation of n-heptane to ester over modified sulfated SnO2 catalysts under mild conditions. J. Hazard. Mater. 2007, 147, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, J.; Zhang, W.; Tan, Y.; Tang, T.; Wang, S.; Tang, B.; Zhao, J. SO42-/SnO2 as a new electrode for electrochemical supercapacitors. Ceram. Int. 2014, 40, 8925–8929. [Google Scholar] [CrossRef]
- Li, C.; Stair, P.C. Ultraviolet Raman spectroscopy characterization of sulfated zirconia catalysts: Fresh, deactivated and regenerated. Catal. Lett. 1996, 36, 119–123. [Google Scholar] [CrossRef]
- Due-Hansen, J.; Boghosian, S.; Kustov, A.; Fristrup, P.; Tsilomelekis, G.; Stahl, K.; Christensen, C.H.; Fehrmann, R. Vanadia-based SCR catalysts supported on tungstated and sulfated zirconia: Influence of doping with potassium. J. Catal. 2007, 251, 459–473. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Castro, R.H.R.; Zhou, W.; Navrotsky, A. Surface enthalpy and enthalpy of water adsorption of nanocrystalline tin dioxide: Thermodynamic insight on the sensing activity. J. Mater. Res. 2011, 26, 848–853. [Google Scholar] [CrossRef]
- Zuo, J.; Xu, C.Y.; Liu, X.M.; Wang, C.S.; Wang, C.Y.; Hu, Y.; Qian, Y.T. Study of the Raman-Spectrum of Nanometer SnO2. J. Appl. Phys. 1994, 75, 1835–1836. [Google Scholar] [CrossRef]
- Aragon, F.H.; Coaquira, J.A.H.; Hidalgo, P.; da Silva, S.W.; Brito, S.L.M.; Gouvea, D.; Moraisa, P.C. Evidences of the evolution from solid solution to surface segregation in Ni-doped SnO2 nanoparticles using Raman spectroscopy. J. Raman Spectrosc. 2011, 42, 1081–1086. [Google Scholar] [CrossRef]
- Wang, H.W.; Wesolowski, D.J.; Proffen, T.E.; Vlcek, L.; Wang, W.; Allard, L.F.; Kolesnikov, A.I.; Feygenson, M.; Anovitz, L.M.; Paul, R.L. Structure and Stability of SnO2 Nanocrystals and Surface-Bound Water Species. J. Am. Chem. Soc. 2013, 135, 6885–6895. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.J. A review of refinery complexity applications. Petrol Sci. 2017, 14, 167–194. [Google Scholar] [CrossRef]
- Ma, Q.S.; Chakraborty, D.; Faglioni, F.; Muller, R.P.; Goddard, W.A.; Harris, T.; Campbell, C.; Tang, Y.C. Alkylation of phenol: A mechanistic view. J. Phys. Chem. A 2006, 110, 2246–2252. [Google Scholar] [CrossRef] [PubMed]
- Colon-Ortiz, J.; Ramesh, P.; Tsilomelekis, G.; Neimark, A.V. Permeation dynamics of dimethyl methylphosphonate through polyelectrolyte composite membranes by in-situ Raman spectroscopy. J. Membr. Sci. 2020, 595. [Google Scholar] [CrossRef]
- Ramesh, P.; Kritikos, A.; Tsilomelekis, G. Effect of metal chlorides on glucose mutarotation and possible implications on humin formation. React. Chem. Eng. 2019, 4, 273–277. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Boghosian, S. In Situ Raman and FTIR Spectroscopy of Molybdenum(VI) Oxide Supported on Titania Combined with O-18/O-16 Exchange: Molecular Structure, Vibrational Properties, and Vibrational Isotope Effects. J. Phys. Chem. C 2011, 115, 2146–2154. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Josephson, T.R.; Nikolakis, V.; Caratzoulas, S. Origin of 5-Hydroxymethylfurfural Stability in Water/Dimethyl Sulfoxide Mixtures. ChemSusChem 2014, 7, 117–126. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Calcination Temperature | Concentration of H2SO4 | Major Crystalline Phases Detected | Surface Area (S.A.) (m2/g) | Literature (S.A.) (m2/g) |
|---|---|---|---|---|---|
| SnO2 | 500 °C | - | Tetragonal SnO2 | 43 | 30.5 [22] |
| S-SnO2 | 500 °C | 1M | Tetragonal SnO2 | 91 | 66–93 [22,23] |
| 10:1 SnO2/ZrO2 | 600 °C | - | Tetragonal SnO2 | 34 | 37.4 [24] |
| 10:1 S- SnO2/ZrO2 | 600 °C | 1M | Tetragonal SnO2 | 81 | N/A |
| 1:1 SnO2/ZrO2 | 600 °C | - | Tetragonal SnO2 Monoclinic ZrO2 | 54 | 31.4 [24] |
| 1:1 S-SnO2/ZrO2 | 600 °C | 1M | Tetragonal SnO2 Monoclinic ZrO2 | 74 | N/A |
| 1:10 SnO2/ZrO2 | 600 °C | - | Monoclinic ZrO2 | 55 | 35.8 [24] |
| 1:10 S-SnO2/ZrO2 | 600 °C | 1M | Tetragonal ZrO2 | 129 | 53 [25] |
| ZrO2 | 600 °C | - | Monoclinic ZrO2 | 45 | 30–40 [26,27] |
| S-ZrO2 | 600 °C | 1M | Tetragonal ZrO2 | 149 | 90–150 [17,26,27] |
| Conversion, % | Product Distribution, % | ||||||
|---|---|---|---|---|---|---|---|
| Catalyst | TBA | 2-TBP | 4-TBP | 2,4-TBP | 2,6-TBP | 2,4,6-TBP | TBPE |
| S-SnO2 | 89.9 | 62.0 | 33.1 | 4.4 | 0.3 | 0.1 | 0 |
| 10:1 S- SnO2/ZrO2 | 88.0 | 59.2 | 30.9 | 5.2 | 0.7 | 0.2 | 3.7 |
| 1:1 S-SnO2/ZrO2 | 47.6 | 34.9 | 19.6 | 0.3 | 0.0 | 0 | 45.1 |
| 1:10 S-SnO2/ZrO2 | 66.4 | 54.2 | 28.6 | 2.9 | 0.3 | 0.1 | 14.0 |
| S-ZrO2 | 55.2 | 45.2 | 25.5 | 0.9 | 0.0 | 0 | 28.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marlowe, J.; Acharya, S.; Zuber, A.; Tsilomelekis, G. Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol. Catalysts 2020, 10, 726. https://doi.org/10.3390/catal10070726
Marlowe J, Acharya S, Zuber A, Tsilomelekis G. Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol. Catalysts. 2020; 10(7):726. https://doi.org/10.3390/catal10070726
Chicago/Turabian StyleMarlowe, Justin, Shreyas Acharya, Adam Zuber, and George Tsilomelekis. 2020. "Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol" Catalysts 10, no. 7: 726. https://doi.org/10.3390/catal10070726
APA StyleMarlowe, J., Acharya, S., Zuber, A., & Tsilomelekis, G. (2020). Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol. Catalysts, 10(7), 726. https://doi.org/10.3390/catal10070726