Catalytic Foldamers: When the Structure Guides the Function


Abstract
1. Introduction
2. Principles Sustaining the Design of Catalytic Peptide Foldamers
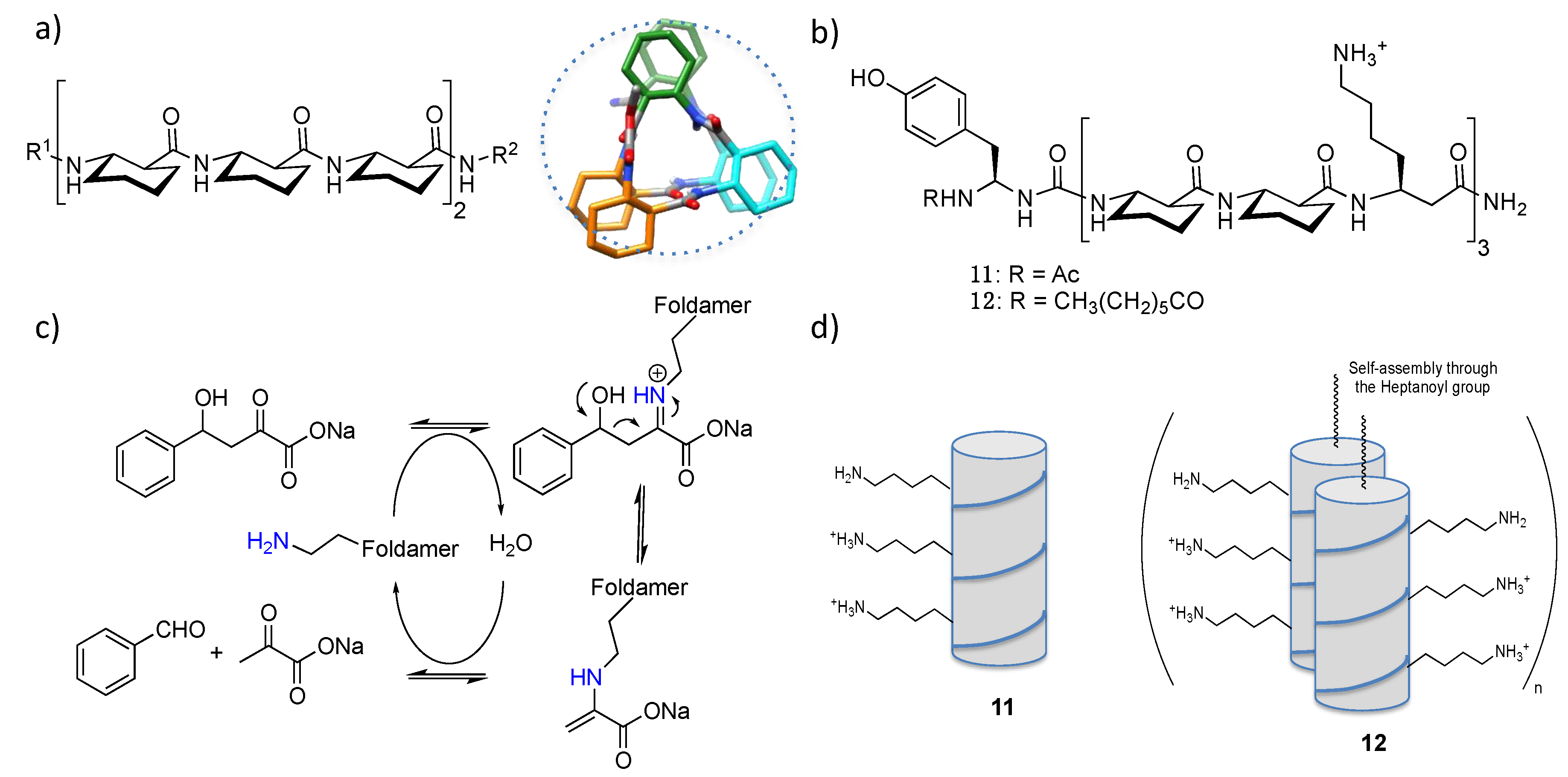
3. Installation of Catalytic Prosthetic Groups in Foldamers: The Peptoid Example
4. Active Sites Resulting from Spatially Preorganized Reactive Side Chains
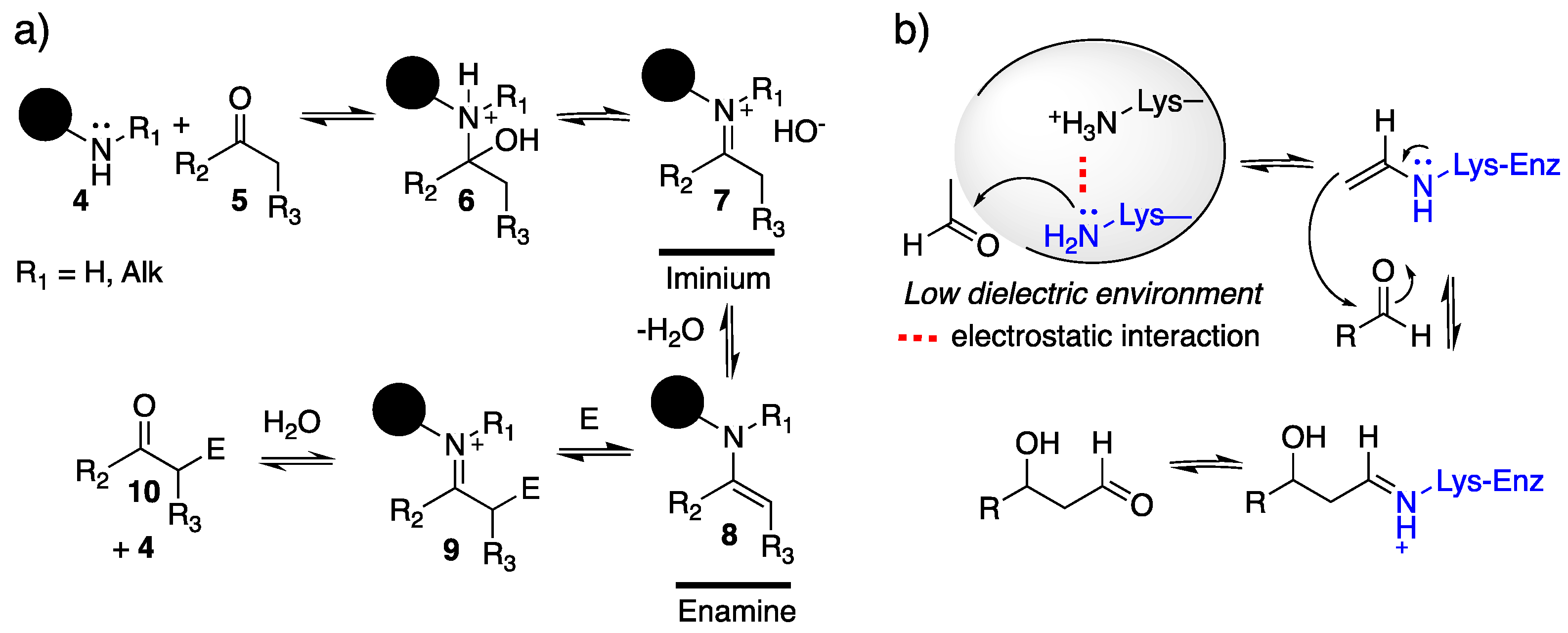
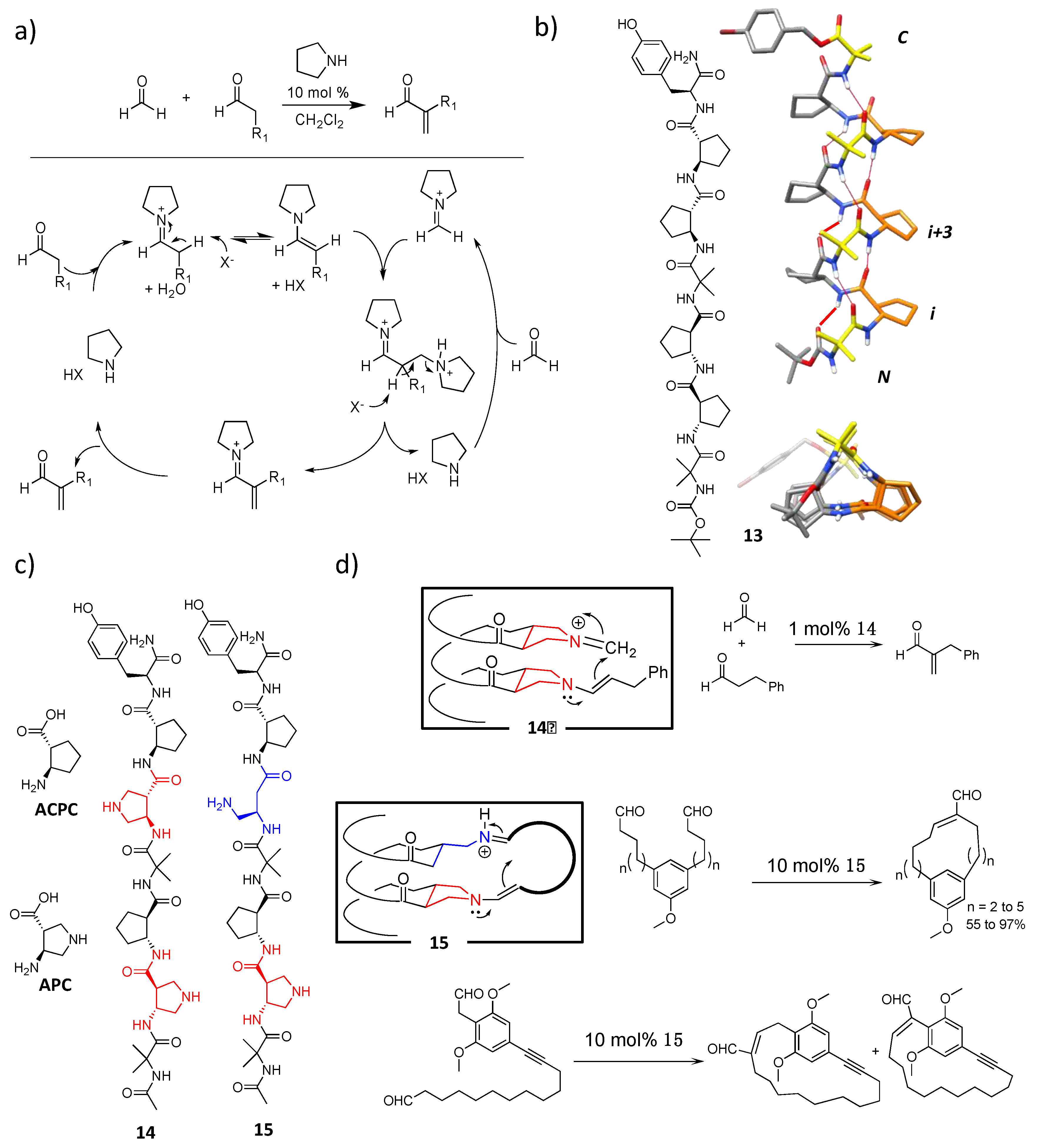
4.1. Enamine/Iminium Mediated Catalysis
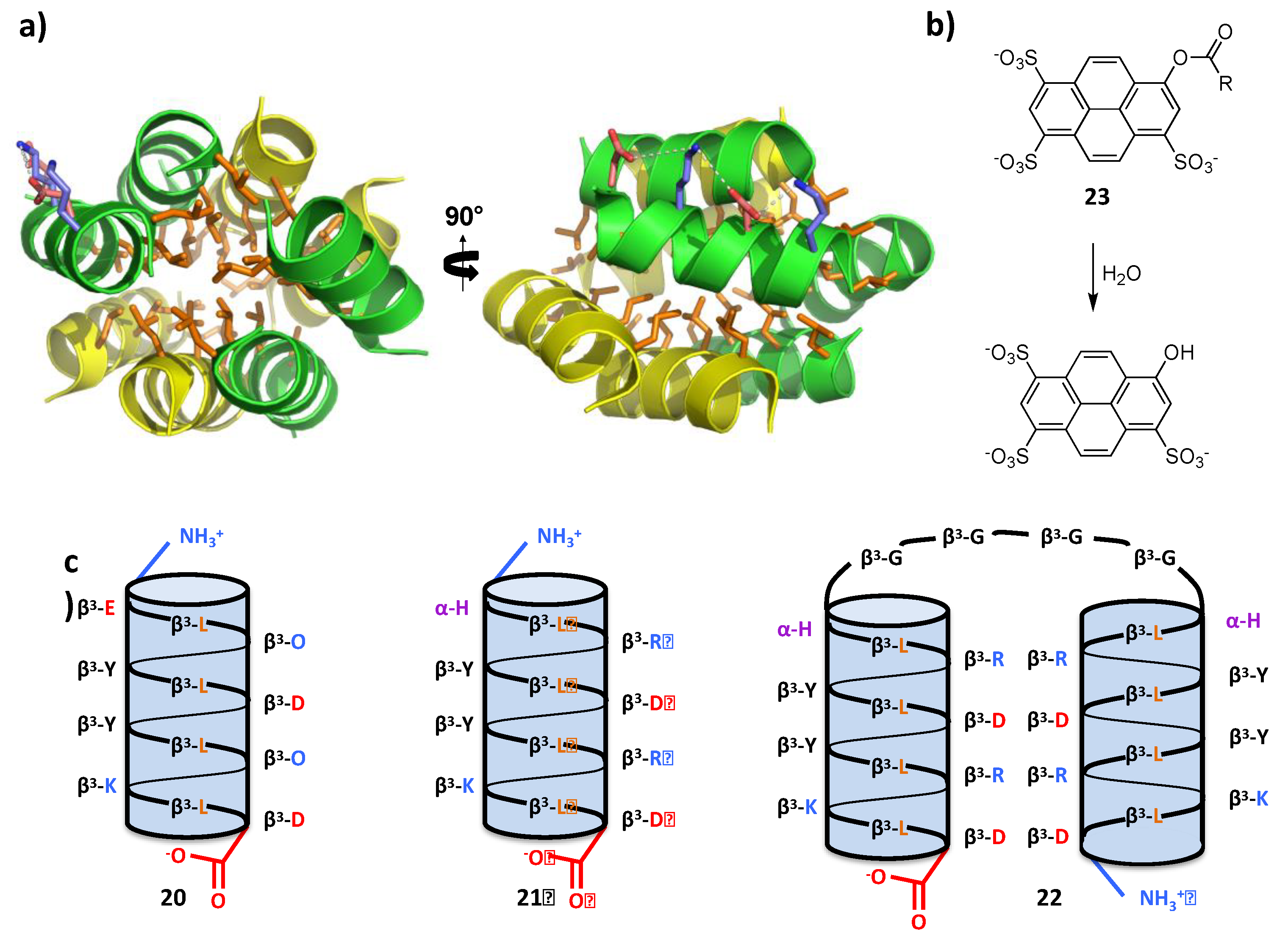
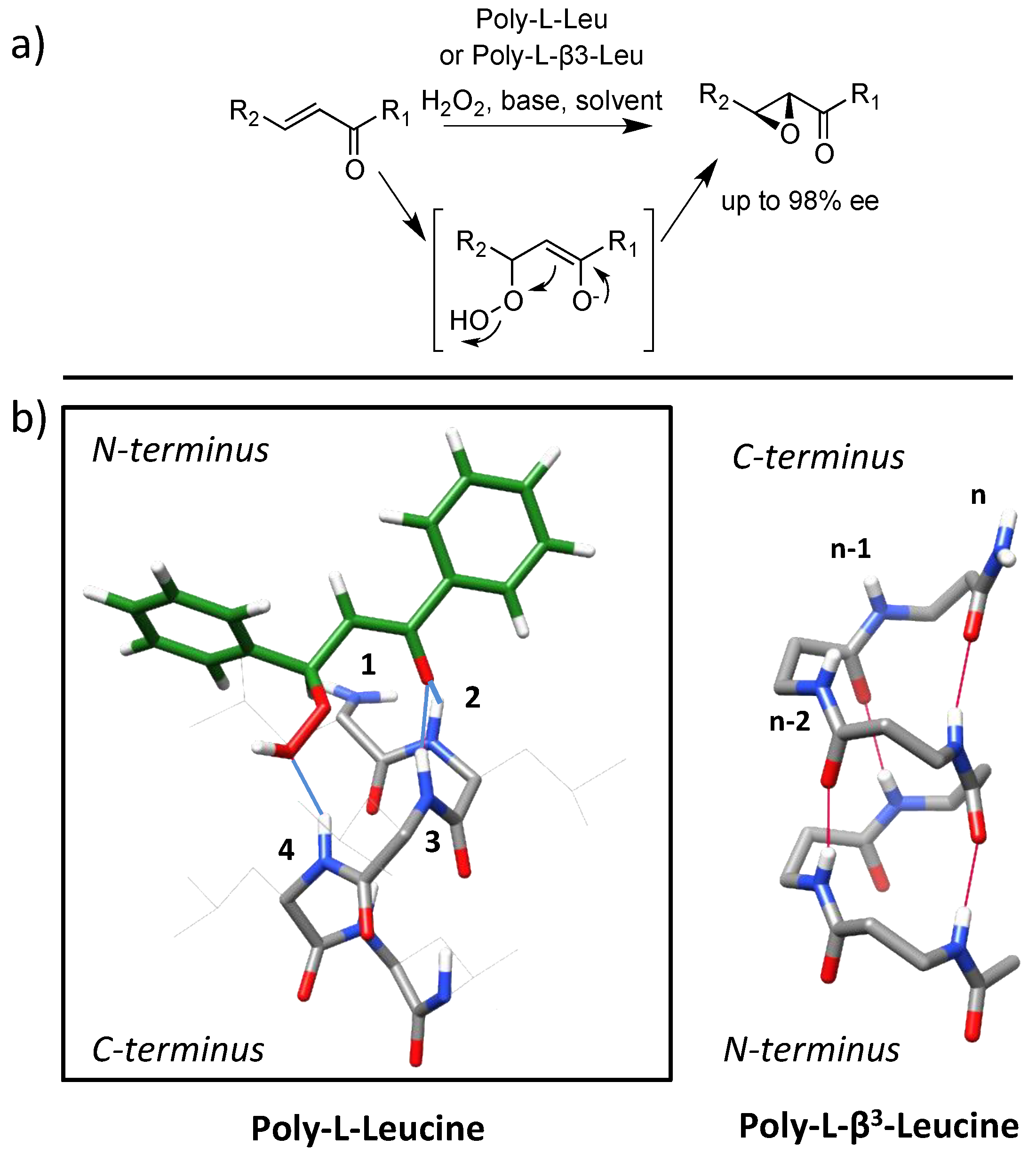
4.2. Bundle Foldamers as Artificial Esterase
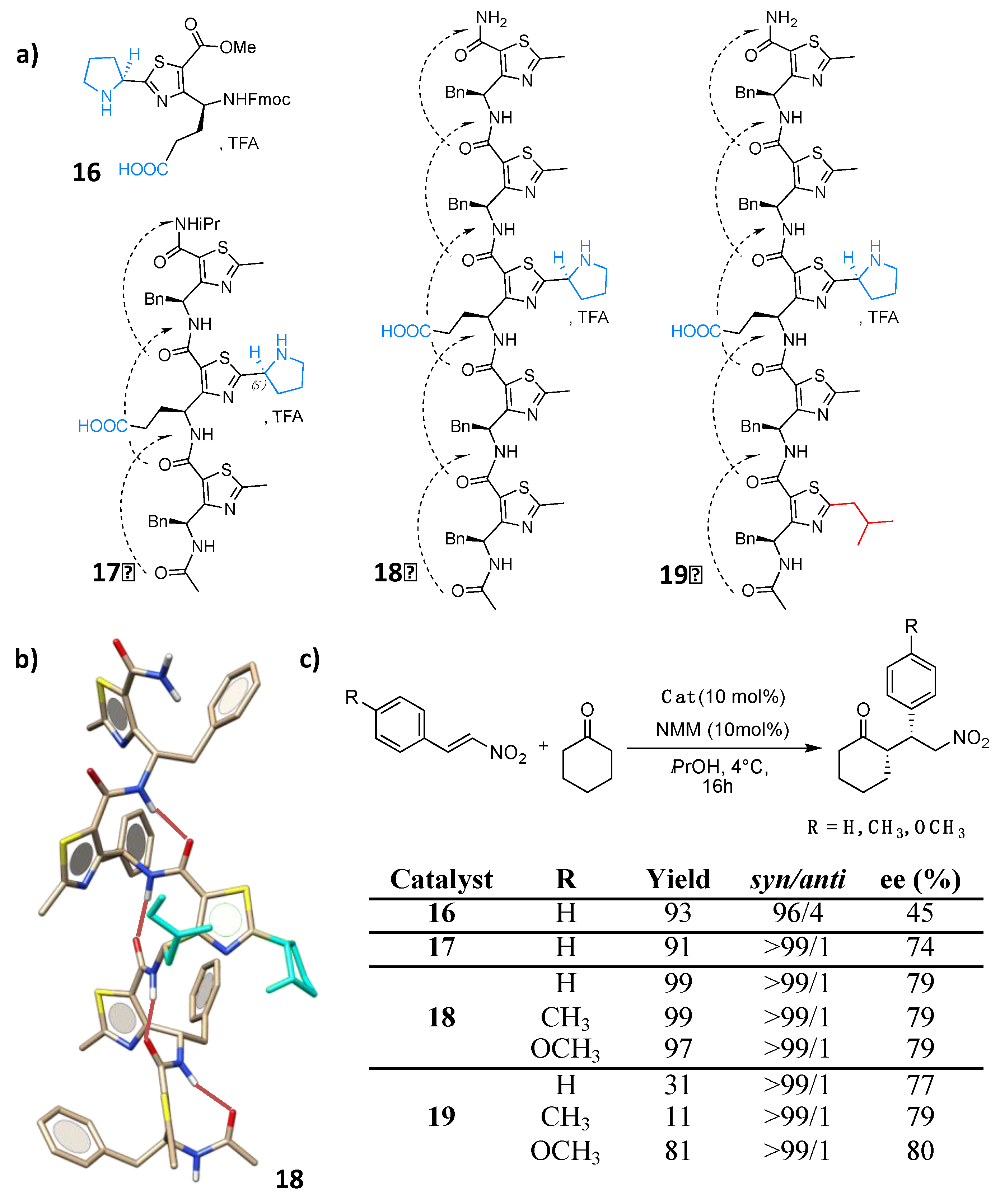
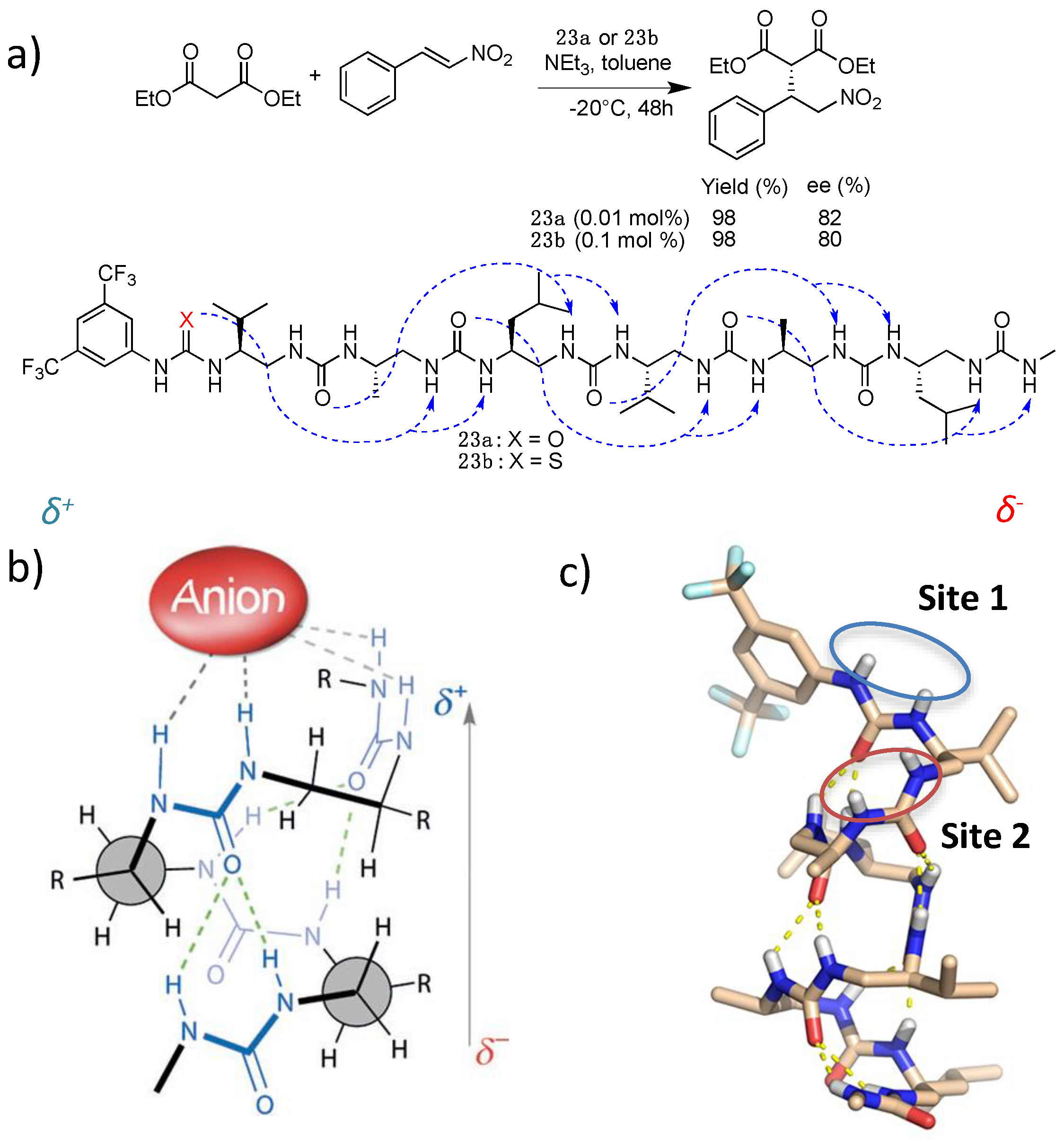
5. Hydrogen-Bond Catalysis Directed by the Main Chain Atoms
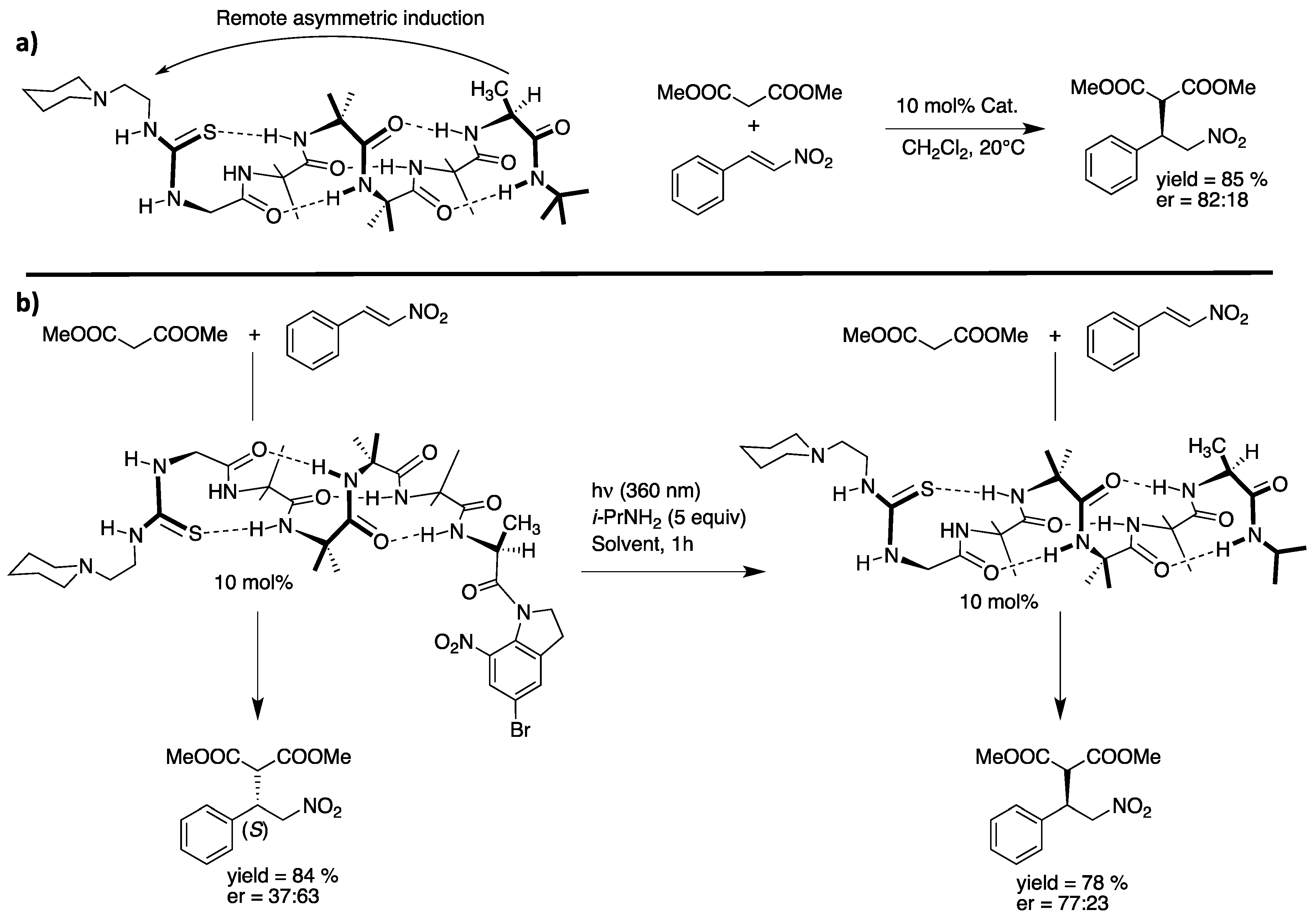
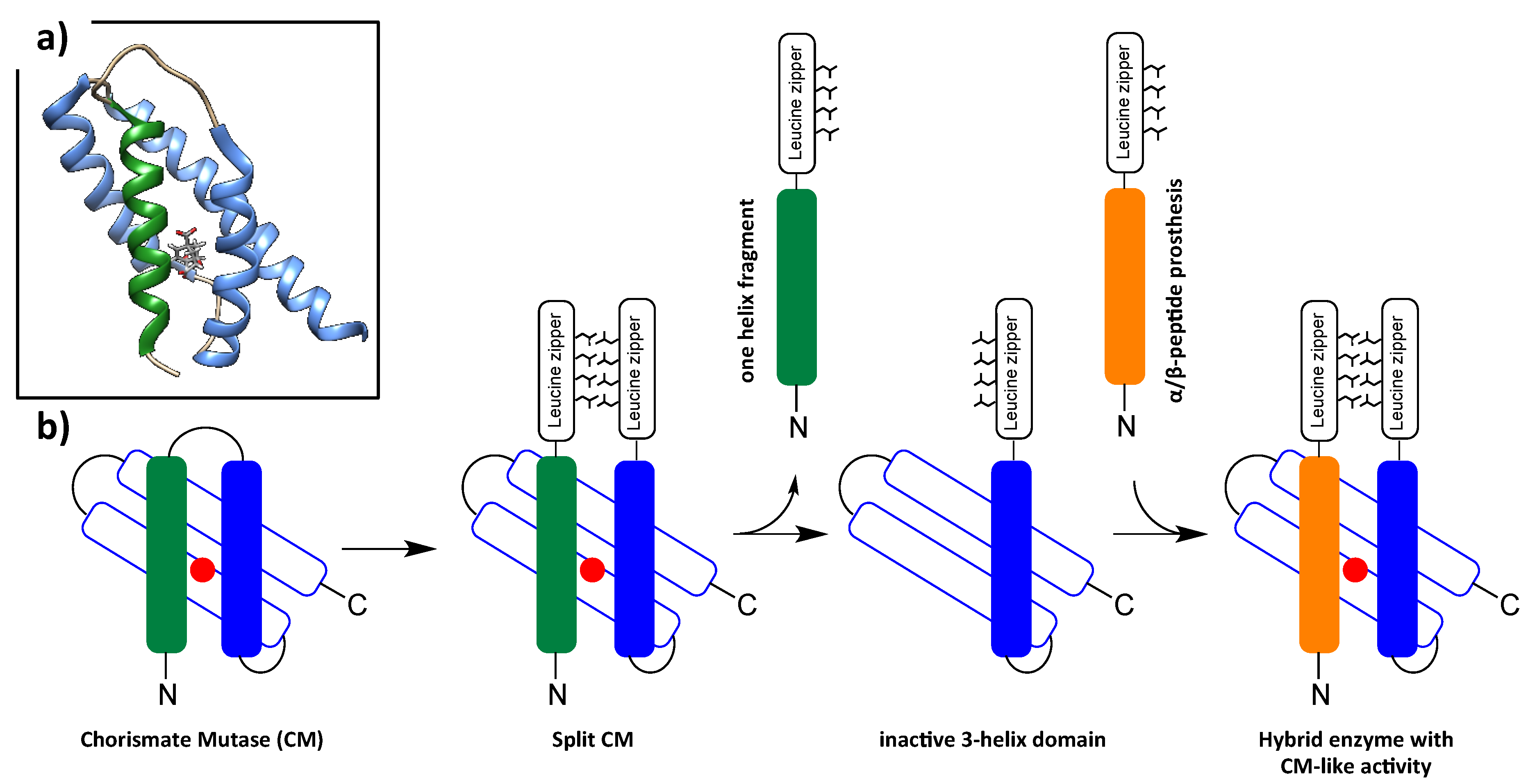
6. Foldamers as Protein Prosthesis
7. Conclusions
Funding
Conflicts of Interest
References
- Nestl, B.M.; Hammer, S.C.; Nebel, B.A.; Hauer, B. New generation of biocatalysts for organic synthesis. Angew. Chem. Int. Ed. Engl. 2014, 53, 3070–3095. [Google Scholar] [CrossRef] [PubMed]
- Voet, D.; Voet, J.G. Biochemistry, 3rd ed.; Pubisher: New York, NY, USA, 2004. [Google Scholar]
- Schomburg, I.; Chang, A.; Placzek, S.; Sohngen, C.; Rother, M.; Lang, M.; Munaretto, C.; Ulas, S.; Stelzer, M.; Grote, A.; et al. BRENDA in 2013: Integrated reactions, kinetic data, enzyme function data, improved disease classification: New options and contents in BRENDA. Nucleic Acids Res. 2013, 41, D764–D772. [Google Scholar] [CrossRef]
- Wulff, G. Enzyme-like catalysis by molecularly imprinted polymers. Chem. Rev. 2002, 102, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.S. Synthesis of polymeric enzyme-like catalysts. Synlett 2001, 9, 1343–1363. [Google Scholar] [CrossRef]
- Kirby, A.J. Enzyme mechanisms, models, and mimics. Angew. Chem. Int. Ed. Engl. 1996, 35, 707–724. [Google Scholar] [CrossRef]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef]
- Kuah, E.; Toh, S.; Yee, J.; Ma, Q.; Gao, Z. Enzyme mimics: Advances and applications. Chem. Eur. J. 2016, 22, 8404–8430. [Google Scholar] [CrossRef]
- Yin, Y.; Dong, Z.; Luo, Q.; Liu, J. Biomimetic catalysts designed on macromolecular scaffolds. Prog. Polym. Sci. 2012, 37, 1476–1509. [Google Scholar] [CrossRef]
- Song, W.; Zhao, B.; Wang, C.; Ozaki, Y.; Lu, X. Functional nanomaterials with unique enzyme-like characteristics for sensing applications. J. Mater. Chem. B 2019, 7, 850–875. [Google Scholar] [CrossRef]
- Stevenson, J.D.; Thomas, N.R. Catalytic antibodies and other biomimetic catalysts. Nat. Prod. Rep. 2000, 17, 535–577. [Google Scholar] [CrossRef]
- Qi, D.; Tann, C.M.; Haring, D.; Distefano, M.D. Generation of new enzymes via covalent modification of existing proteins. Chem. Rev. 2001, 101, 3081–3111. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, M. Engineering allosteric protein switches by domain insertion. Protein Eng. Des. Sel. 2005, 18, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Creus, M.; Ward, T.R. Designed evolution of artificial metalloenzymes:protein catalysts made to orde. Org. Biomol. Chem. 2007, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Davie, E.A.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
- Miller, S.J. In search of peptide-based catalysts for asymmetric organic synthesis. Acc. Chem. Res. 2004, 37, 601–610. [Google Scholar] [CrossRef]
- Mogharabi, M.; Rezaei, S.; Faramarzi, M.A. Peptide-catalysis in asymmetric organic synthesis. Trends Pept. Protein Sci. 2017, 1, 89–98. [Google Scholar] [CrossRef]
- Zozulia, O.; Dolan, M.A.; Korendovych, I.V. Catalytic peptide assemblies. Chem. Soc. Rev. 2018, 47, 3621–3639. [Google Scholar] [CrossRef]
- Akagawa, K. Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 513–564. [Google Scholar] [CrossRef]
- Wennemers, H. Asymmetric catalysis with peptides. Chem. Commun. 2011, 47, 12036–12041. [Google Scholar] [CrossRef]
- Redfern, O.C.; Dessailly, B.; Orengo, C.A. Exploring the structure and function paradigm. Curr. Opin. Struct. Biol. 2008, 18, 394–402. [Google Scholar] [CrossRef]
- Dessailly, B.H.; Redfern, O.C.; Cuff, A.; Orengo, C.A. Exploiting structural classifications for function prediction: Towards a domain grammar for protein function. Curr. Opin. Struct. Biol. 2009, 19, 349–356. [Google Scholar] [CrossRef]
- Freiberger, M.I.; Guzovsky, A.B.; Wolynes, P.G.; Parra, R.G.; Ferreiro, D.U. Local frustration around enzyme active sites. Proc. Natl. Acad. Sci. USA 2019, 116, 4037–4043. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.J.; Mio, M.J.; Prince, R.B.; Hughes, T.S.; Moore, J.S. A field guide to foldamers. Chem. Rev. 2001, 101, 3893–4012. [Google Scholar] [CrossRef] [PubMed]
- Guichard, G.; Huc, I. Synthetic foldamers. Chem. Commun. 2011, 47, 5933–5941. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.; Huc, I. Foldamers: Structure, Properties And Applications; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2007; p. 425. [Google Scholar] [CrossRef]
- Roy, A.; Prabhakaran, P.; Baruah, P.K.; Sanjayan, G.J. Diversifying the structural architecture of synthetic oligomers: The hetero foldamer approach. Chem. Commun. 2011, 47, 11593–11611. [Google Scholar] [CrossRef]
- Martinek, T.A.; Fulop, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.M.; Choi, S.; Shandler, S.; DeGrado, W.F. Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007, 3, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, B.; Godde, F.; Huc, I. How can folded biopolymers and synthetic foldamersrecognize each other. Chembiochem 2009, 10, 1765–1767. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, R.; Frolov, A.I.; Knerr, L.; Drury, W.J., III; Valeur, E. Therapeutic potential of foldamers: From chemical biology tools to drug candidates. J. Med. Chem. 2016, 59, 9599–9621. [Google Scholar] [CrossRef]
- Checco, J.W.; Gellman, S.H. Targeting recognition surfaces on natural proteins with peptidic foldamers. Curr. Opin. Struct. Biol. 2016, 39, 96–105. [Google Scholar] [CrossRef]
- Kulkarni, K.; Habila, N.; Del Borgo, M.P.; Aguilar, M.I. Novel materials from the supramolecular self-assembly of short helical beta(3)-peptide foldamers. Front. Chem. 2019, 7, 70. [Google Scholar] [CrossRef]
- Wang, T.; Fan, X.; Hou, C.; Liu, J. Design of artificial enzymes by supramolecular strategies. Curr. Opin. Struct. Biol. 2018, 51, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Dugas, H. Bioorganic Chemistry A Chemical Approach to Enzyme Action, Chapter 4; Springer: New York, NY, USA, 1996; pp. 159–251. [Google Scholar]
- Silverman, R.B. Organic Chemistry of Enzyme-Catalyzed Reactions, Chapter 1; Academic Press: San Diego, CA, USA, 2002. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Cleland, W.W.; Hengge, A.C. Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 2006, 106, 3252–3278. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R. Bifunctional acid-base catalysis by imidazole groups in enzyme mimics. J. Mol. Cat. 1994, 91, 161–174. [Google Scholar] [CrossRef]
- Peters, R.E.A. Cooperative Catalysis Designing Efficient Catalysts for Synthesis; Wiley-VCH Verlag GmbH &, Co.: Weinheim, Germany, 2015; p. 456. [Google Scholar]
- Wende, R.C.; Schreiner, P.R. Evolution of asymmetric organocatalysis: Multi- and retrocatalysis. Green Chem. 2012, 14, 1821–1849. [Google Scholar] [CrossRef]
- Zhou, J. Multi-Catalyst System in Asymmetric Catalysis; Wiley: Hoboken, NJ, USA, 2014; p. 1. [Google Scholar]
- Liu, F. The upside of downsizing: Asymmetric trifunctional organocatalysts as small enzyme mimics for cooperative enhancement of both rate and enantioselectivity with regulation. Chirality 2013, 25, 675–683. [Google Scholar] [CrossRef]
- Maayan, G.; Ward, M.D.; Kirshenbaum, K. Folded biomimetic oligomers for enantioselective catalysis. Proc. Natl. Acad. Sci. USA 2009, 106, 13679–13684. [Google Scholar] [CrossRef]
- Kirshenbaum, K.; Barron, A.E.; Goldsmith, R.A.; Armand, P.; Bradley, E.K.; Truong, K.T.; Dill, K.A.; Cohen, F.E.; Zuckermann, R.N. Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. USA 1998, 95, 4303–4308. [Google Scholar] [CrossRef]
- Wu, C.W.; Sanborn, T.J.; Huang, K.; Zuckermann, R.N.; Barron, A.E. Peptoid oligomers with alpha-chiral, aromatic side chains: Sequence requirements for the formation of stable peptoid helices. J. Am. Chem. Soc. 2001, 123, 6778–6784. [Google Scholar] [CrossRef]
- Anelli, P.L.; Biffi, C.; Montanari, F.; Quici, S. Fast and selective oxidation of primary alcohols to aldehydes or to carboxylic acids and of secondary alcohols to ketones mediated by oxoammonium salts under two-phase conditions. J. Org. Chem. 1987, 52, 2559–2562. [Google Scholar] [CrossRef]
- Anelli, P.L.; Banfi, S.; Montanari, F.; Quici, S. Oxidation of diols with alkali hypochlorites catalyzed by oxammonium salts under two-phase conditions. J. Org. Chem. 1989, 54, 2970–2972. [Google Scholar] [CrossRef]
- List, B. Enamine catalysis is a powerful strategy for the catalytic generation and use of carbanion equivalents. Acc. Chem. Res. 2004, 37, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Pihko, P.M.; Majander, I.; Erkkilä, A. Enamine Catalysis. In Asymmetric Organocatalysis. Topics in Current Chemistry; List, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 291. [Google Scholar]
- Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Noto, R. Low-loading asymmetric organocatalysis. Chem. Soc. Rev. 2012, 41, 2406–2447. [Google Scholar] [CrossRef] [PubMed]
- Notz, W.; Tanaka, F.; Barbas, C.F., III. Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric Aldol, Mannich, Michael, and Diels-alder reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef]
- Hamilton, G.A.; Westheimer, F.H. On the mechanism of the enzymatic decarboxylation of acetoacetate. J. Am. Chem. Soc. 1959, 81, 6332–6333. [Google Scholar] [CrossRef]
- Laursen, R.A.; Westheimer, F.H. The active site of acetoacetate decarboxylase. J. Am. Chem. Soc. 1966, 88, 3426–3430. [Google Scholar] [CrossRef]
- Grazi, E.; Rowley, P.T.; Cheng, T.; Tchola, O.; Horecker, B.L. The mechanism of action of aldolases. III. Schiff base formation with lysine. Biochem. Biophys. Res. Commun. 1962, 9, 38–43. [Google Scholar] [CrossRef]
- Lai, C.Y.; Nakai, N.; Chang, D. Amino acid sequence of rabbit muscle aldolase and the structure of the active center. Science 1974, 183, 1204–1206. [Google Scholar] [CrossRef]
- Westheimer, F.H. Coincidences, decarboxylation, and electrostatic effects. Tetrahedron 1995, 51, 3–20. [Google Scholar] [CrossRef]
- Heine, A.; Luz, J.G.; Wong, C.H.; Wilson, I.A. Analysis of the class I aldolase binding site architecture based on the crystal structure of 2-deoxyribose-5-phosphate aldolase at 0.99A resolution. J. Mol. Biol. 2004, 343, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, K.; Allemann, R.K.; Widmer, H.; Benner, S.A. Synthesis, structure and activity of artificial, rationally designed catalytic polypeptides. Nature 1993, 365, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Weston, C.J.; Cureton, C.H.; Calvert, M.J.; Smart, O.S.; Allemann, R.K. A stable miniature protein with oxaloacetate decarboxylase activity. ChemBioChem 2004, 5, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.M.; Windsor, M.A.; Pomerantz, W.C.; Gellman, S.H.; Hilvert, D. A rationally designed aldolase foldamer. Angew. Chem. Int. Ed. Engl. 2009, 48, 922–925. [Google Scholar] [CrossRef]
- Appella, D.H.; Christianson, L.A.; Klein, D.A.; Powell, D.R.; Huang, X.; Barchi, J.J., Jr.; Gellman, S.H. Residue-based control of helix shape in beta-peptide oligomers. Nature 1997, 387, 381–384. [Google Scholar] [CrossRef]
- Raguse, T.L.; Lai, J.R.; Gellman, S.H. Environment-independent 14-helix formation in short beta-peptides: Striking a balance between shape control and functional diversity. J. Am. Chem. Soc. 2003, 125, 5592–5593. [Google Scholar] [CrossRef]
- Erkkilä, A.; Pihko, P.M. Rapid organocatalytic aldehyde-aldehyde condensation reactions. Eur. J. Org. Chem. 2007, 4205–4216. [Google Scholar] [CrossRef]
- Girvin, Z.C.; Gellman, S.H. Exploration of diverse reactive diad geometries for bifunctional catalysis via foldamer backbone variation. J. Am. Chem. Soc. 2018, 140, 12476–12483. [Google Scholar] [CrossRef]
- Girvin, Z.C.; Andrews, M.K.; Liu, X.; Gellman, S.H. Foldamer-templated catalysis of macrocycle formation. Science 2019, 366, 1528–1531. [Google Scholar] [CrossRef]
- Mathieu, L.; Legrand, B.; Deng, C.; Vezenkov, L.; Wenger, E.; Didierjean, C.; Amblard, M.; Averlant-Petit, M.C.; Masurier, N.; Lisowski, V.; et al. Helical oligomers of thiazole-based gamma-amino acids: Synthesis and structural studies. Angew. Chem. Int. Ed. Engl. 2013, 52, 6006–6010. [Google Scholar] [CrossRef]
- Bonnel, C.; Legrand, B.; Bantignies, J.L.; Petitjean, H.; Martinez, J.; Masurier, N.; Maillard, L.T. FT-IR and NMR structural markers for thiazole-based gamma-peptide foldamers. Org. Biomol. Chem. 2016, 14, 8664–8669. [Google Scholar] [CrossRef]
- Mathieu, L.; Bonnel, C.; Masurier, N.; Maillard, L.T.; Martinez, J.; Lisowski, V. Cross-Claisen condensation of n-fmoc-amino acids—A short route to heterocyclic γ-amino acids. Eur. J. Org. Chem. 2015, 2015, 2262–2270. [Google Scholar] [CrossRef]
- Ali, L.M.A.; Simon, M.; El Cheikh, K.; Aguesseau-Kondrotas, J.; Godefroy, A.; Nguyen, C.; Garcia, M.; Morere, A.; Gary-Bobo, M.; Maillard, L. Topological requirements for CI-M6PR-Mediated cell uptake. Bioconjug. Chem. 2019, 30, 2533–2538. [Google Scholar] [CrossRef]
- Simon, M.; Ali, L.M.A.; Cheikh, K.E.; Aguesseau, J.; Gary-Bobo, M.; Garcia, M.; Morère, A.; Maillard, L.T. Can heterocyclic γ-peptides provide polyfunctional platforms for synthetic glycocluster construction. Chem. Eur. J. 2018, 24, 11426–11432. [Google Scholar] [CrossRef] [PubMed]
- Aguesseau-Kondrotas, J.; Simon, M.; Legrand, B.; Bantignies, J.L.; Kang, Y.K.; Dumitrescu, D.; Van Der Lee, A.; Campagne, J.M.; de Figueiredo, R.M.; Maillard, L.T. Prospect of Thiazole-based gamma-peptide foldamers in enamine catalysis: Exploration of the Nitro-michael addition. Chem. Eur. J. 2019, 25, 7396–7401. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, M.; Revell, J.D.; Tonazzi, S.; Wennemers, H. Peptide catalyzed asymmetric conjugate addition reactions of aldehydes to nitroethylene—A convenient entry into gamma2-amino acids. J. Am. Chem. Soc. 2008, 130, 5610–5611. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, M.; Revell, J.D.; Wennemers, H. Tripeptides as efficient asymmetric catalysts for 1,4-addition reactions of aldehydes to nitroolefins—A rational approach. Angew. Chem. Int. Ed. Engl. 2008, 47, 1871–1874. [Google Scholar] [CrossRef]
- Wiesner, M.; Neuburger, M.; Wennemers, H. Tripeptides of the type H-D-Pro-Pro-Xaa-NH2 as catalysts for asymmetric 1,4-addition reactions: Structural requirements for high catalytic efficiency. Chem. Eur. J. 2009, 15, 10103–10109. [Google Scholar] [CrossRef]
- Wiesner, M.; Upert, G.; Angelici, G.; Wennemers, H. Enamine catalysis with low catalyst loadings--high efficiency via kinetic studies. J. Am. Chem. Soc. 2010, 132, 6–7. [Google Scholar] [CrossRef]
- Moffet, D.A.; Hecht, M.H. De novo proteins from combinatorial libraries. Chem. Rev. 2001, 101, 3191–3203. [Google Scholar] [CrossRef]
- Hecht, M.H.; Das, A.; Go, A.; Bradley, L.H.; Wei, Y. De novo proteins from designed combinatorial libraries. Protein Sci. 2004, 13, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, P.; Stetefeld, J.; Strelkov, S.V. Coiled coils: A highly versatile protein folding motif. Trends Cell Biol. 2001, 11, 82–88. [Google Scholar] [CrossRef]
- Woolfson, D.N. The design of coiled-coil structures and assemblies. Adv. Protein Chem. 2005, 70, 79–112. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, M.W.; Horne, W.S.; Gellman, S.H. An alpha/beta-peptide helix bundle with a pure beta3-amino acid core and a distinctive quaternary structure. J. Am. Chem. Soc. 2009, 131, 9860–9861. [Google Scholar] [CrossRef]
- Horne, W.S.; Price, J.L.; Gellman, S.H. Interplay among side chain sequence, backbone composition, and residue rigidification in polypeptide folding and assembly. Proc. Natl. Acad. Sci. USA 2008, 105, 9151–9156. [Google Scholar] [CrossRef]
- Daniels, D.S.; Petersson, E.J.; Qiu, J.X.; Schepartz, A. High-resolution structure of a beta-peptide bundle. J. Am. Chem. Soc. 2007, 129, 1532–1533. [Google Scholar] [CrossRef]
- Wang, P.S.; Schepartz, A. Beta-peptide bundles: Design. Build. Analyze. Biosynthesize. Chem. Commun. 2016, 52, 7420–7432. [Google Scholar] [CrossRef]
- Collie, G.W.; Bailly, R.; Pulka-Ziach, K.; Lombardo, C.M.; Mauran, L.; Taib-Maamar, N.; Dessolin, J.; Mackereth, C.D.; Guichard, G. Molecular recognition within the cavity of a foldamer helix bundle: Encapsulation of primary alcohols in aqueous conditions. J. Am. Chem. Soc. 2017, 139, 6128–6137. [Google Scholar] [CrossRef]
- Lombardo, C.M.; Collie, G.W.; Pulka-Ziach, K.; Rosu, F.; Gabelica, V.; Mackereth, C.D.; Guichard, G. Anatomy of an oligourea six-helix bundle. J. Am. Chem. Soc. 2016, 138, 10522–10530. [Google Scholar] [CrossRef]
- Collie, G.W.; Pulka-Ziach, K.; Lombardo, C.M.; Fremaux, J.; Rosu, F.; Decossas, M.; Mauran, L.; Lambert, O.; Gabelica, V.; Mackereth, C.D.; et al. Shaping quaternary assemblies of water-soluble non-peptide helical foldamers by sequence manipulation. Nat. Chem. 2015, 7, 871–878. [Google Scholar] [CrossRef]
- Craig, C.J.; Goodman, J.L.; Schepartz, A. Enhancing beta3 -peptide bundle stability by design. ChemBioChem 2011, 12, 1035–1038. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.S.; Nguyen, J.B.; Schepartz, A. Design and high-resolution structure of a beta(3)-peptide bundle catalyst. J. Am. Chem. Soc. 2014, 136, 6810–6813. [Google Scholar] [CrossRef] [PubMed]
- Pihko, P.M.; Rapakko, S.; Wierenga, R.K. Hydrogen Bonding in Organic Synthesis; Wiley-VCH Verlag GmbH: Mörlenbach, Germany, 2009. [Google Scholar]
- Kamerlin, S.C.; Chu, Z.T.; Warshel, A. On catalytic preorganization in oxyanion holes: Highlighting the problems with the gas-phase modeling of oxyanion holes and illustrating the need for complete enzyme models. J. Org. Chem. 2010, 75, 6391–6401. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. Engl. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Zhang, Z.; Schreiner, P.R. (Thio)Urea organocatalysis—What can be learnt from anion recognition. Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef]
- Juliá, S.; Masana, J.; Vega, J.C. “Synthetic enzymes”. Highly stereoselective epoxidation of chalcone in a triphasic toluene-water-poly[(S)-alanine] system. Angew. Chem. Int. Ed. Engl. 1980, 19, 929–931. [Google Scholar]
- Juliá, S.; Guixer, J.; Masana, J.; Rocas, J.; Colonna, S.; Annuziata, R.; Molinari, H. Synthetic enzymes. Part 2. Catalytic asymmetric epoxidation by means of polyamino-acids in a triphase system. J. Chem. Soc. Perkin Trans. 1982, 1, 1317–1324. [Google Scholar] [CrossRef]
- Allen, J.V.; Drauz, K.H.; Flood, R.W.; Roberts, S.M.; Skidmore, J. Polyamino acid-catalysed asymmetric epoxidation: Sodium percarbonate as a source of base and oxidant. Tet. Lett. 1999, 40, 5417–5420. [Google Scholar] [CrossRef]
- Carrea, G.; Colonna, S.; Meek, A.D.; Ottolina, G.; Roberts, S.M. Kinetics of chalcone oxidation by peroxide anion catalysed by poly-l-leucine. Chem. Commun. 2004, 1412–1413. [Google Scholar] [CrossRef]
- Carrea, G.; Colonna, S.; Kelly, D.R.; Lazcano, A.; Ottolina, G.; Roberts, S.M. Polyamino acids as synthetic enzymes: Mechanism, applications and relevance to prebiotic catalysis. Trends Biotechnol. 2005, 23, 507–513. [Google Scholar] [CrossRef]
- Berkessel, A.; Gasch, N.; Glaubitz, K.; Koch, C. Highly enantioselective enoneepoxidation catalyzed by short solidphase-bound peptides: Dominant role of peptide helicity. Org. Lett. 2001, 3, 3839–3842. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.R.; Roberts, S.M. The mechanism of polyleucine catalysed asymmetric epoxidation. Chem. Commun. 2004, 2018–2020. [Google Scholar] [CrossRef]
- Coffey, P.E.; Drauz, K.H.; Roberts, S.M.; Skidmore, J.; Smith, J.A. beta-peptides as catalysts: Poly-beta-leucine as a catalyst for the Julia-Colonna asymmetric epoxidation of enones. Chem. Commun. 2001, 2330–2331. [Google Scholar] [CrossRef]
- Ueda, A.; Umeno, T.; Doi, M.; Akagawa, K.; Kudo, K.; Tanaka, M. Helical-peptide-catalyzed enantioselective michael addition reactions and their mechanistic insights. J. Org. Chem. 2016, 81, 6343–6356. [Google Scholar] [CrossRef]
- Umeno, T.; Ueda, A.; Doi, M.; Kato, T.; Oba, M.; Tanaka, M. Helical foldamer-catalyzed enantioselective 1,4-addition reaction of dialkyl malonates to cyclic enones. Tet. Lett. 2019, 60, 151301. [Google Scholar] [CrossRef]
- Becart, D.; Diemer, V.; Salaun, A.; Oiarbide, M.; Nelli, Y.R.; Kauffmann, B.; Fischer, L.; Palomo, C.; Guichard, G. Helical oligourea foldamers as powerful hydrogen bonding catalysts for enantioselective C-C bond-forming reactions. J. Am. Chem. Soc. 2017, 139, 12524–12532. [Google Scholar] [CrossRef] [PubMed]
- Burgess, K.; Ibarzo, J.; Linthicum, D.S.; Russell, D.H.; Shin, H.; Shitangkoon, A.; Totani, R.; Zhang, A.J. Solid phase syntheses of oligoureas. J. Am. Chem. Soc. 1997, 119, 1556–1564. [Google Scholar] [CrossRef]
- Semetey, V.; Rognan, D.; Hemmerlin, C.; Graff, R.; Briand, J.-P.; Marraud, M.; Guichard, G. Stable helical secondary structure in short-chain N,N’-linked oligoureas bearing proteinogenic side chains. Angew. Chem. Int. Ed. Engl. 2002, 41, 1893–1895. [Google Scholar] [CrossRef]
- Fischer, L.; Claudon, P.; Pendem, N.; Miclet, E.; Didierjean, C.; Ennifar, E.; Guichard, G. The canonical helix of urea oligomers at atomic resolution: Insights into folding-induced axial organization. Angew. Chem. Int. Ed. Engl. 2010, 122, 1085–1088. [Google Scholar] [CrossRef]
- Diemer, V.; Fischer, L.; Kauffmann, B.; Guichard, G. Anion recognition by aliphatic helical oligoureas. Chem. Eur. J. 2016, 22, 15684–15692. [Google Scholar] [CrossRef]
- Pihko, P.M. Activation of carbonyl compounds by double hydrogen bonding: An emerging tool in asymmetric catalysis. Angew. Chem. Int. Ed. Engl. 2004, 43, 2062–2064. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, C.J. Recent advances in asymmetric organocatalysis mediated by bifunctional amine-thioureas bearing multiple hydrogen-bonding donors. Chem. Commun. 2015, 51, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, K. Science of Synthesis: Asymmetric Organocatalysis Vol. 2: Brønsted Base and Acid Catalysts, and Additional Topics; Thieme Publishing Group: Stuttgart, Germany, 2012. [Google Scholar]
- Jones, C.R.; Dan Pantos, G.; Morrison, A.J.; Smith, M.D. Plagiarizing proteins: Enhancing efficiency in asymmetric hydrogen-bonding catalysis through positive cooperativity. Angew. Chem. Int. Ed. Engl. 2009, 48, 7391–7394. [Google Scholar] [CrossRef]
- Probst, N.; Madarasz, A.; Valkonen, A.; Papai, I.; Rissanen, K.; Neuvonen, A.; Pihko, P.M. Cooperative assistance in bifunctional organocatalysis: Enantioselective Mannich reactions with aliphatic and aromatic imines. Angew. Chem. Int. Ed. Engl. 2012, 51, 8495–8499. [Google Scholar] [CrossRef]
- Pengo, B.; Formaggio, F.; Crisma, M.; Toniolo, C.; Bonora, G.M.; Broxterman, Q.B.; Kamphuis, J.; Saviano, M.; Lacovino, R.; Rossi, F.; et al. Linear oligopeptides. Part 406.1 Helical screw sense of peptide molecules: The pentapeptide system (Aib)4/L-Val[L-(αMe)Val] in solution. J. Chem. Soc. Perkin Trans. 2 1998, 1651–1658. [Google Scholar] [CrossRef]
- Brown, R.A.; Marcelli, T.; De Poli, M.; Sola, J.; Clayden, J. Induction of unexpected left-handed helicity by an N-terminal L-amino acid in an otherwise achiral peptide chain. Angew. Chem. Int. Ed. Engl. 2012, 51, 1395–1399. [Google Scholar] [CrossRef]
- De Poli, M.; Byrne, L.; Brown, R.A.; Sola, J.; Castellanos, A.; Boddaert, T.; Wechsel, R.; Beadle, J.D.; Clayden, J. Engineering the structure of an N-terminal beta-turn to maximize screw-sense preference in achiral helical peptide chains. J. Org. Chem. 2014, 79, 4659–4675. [Google Scholar] [CrossRef]
- Le Bailly, B.A.; Clayden, J. Controlling the sign and magnitude of screw-sense preference from the C-terminus of an achiral helical foldamer. Chem. Commun. 2014, 50, 7949–7952. [Google Scholar] [CrossRef]
- Le Bailly, B.A.; Byrne, L.; Clayden, J. Refoldable foldamers: Global conformational switching by deletion or insertion of a single hydrogen bond. Angew. Chem. Int. Ed. Engl. 2016, 55, 2132–2136. [Google Scholar] [CrossRef]
- David, R.; Gunther, R.; Baumann, L.; Luhmann, T.; Seebach, D.; Hofmann, H.J.; Beck-Sickinger, A.G. Artificial chemokines: Combining chemistry and molecular biology for the elucidation of interleukin-8 functionality. J. Am. Chem. Soc. 2008, 130, 15311–15317. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Zuckermann, R.N. Protein side-chain translocation mutagenesis via incorporation of peptoid residues. ACS Chem. Biol. 2011, 6, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Craven, T.W.; Bonneau, R.; Kirshenbaum, K. PPII helical peptidomimetics templated by cation-pi interactions. Chembiochem 2016, 17, 1824–1828. [Google Scholar] [CrossRef]
- Mayer, C.; Muller, M.M.; Gellman, S.H.; Hilvert, D. Building proficient enzymes with foldamer prostheses. Angew. Chem. Int. Ed. Engl. 2014, 53, 6978–6981. [Google Scholar] [CrossRef]
- Hegedus, Z.; Grison, C.M.; Miles, J.A.; Rodriguez-Marin, S.; Warriner, S.L.; Webb, M.E.; Wilson, A.J. A catalytic protein-proteomimetic complex: Using aromatic oligoamide foldamers as activators of RNase S. Chem. Sci. 2019, 10, 3956–3962. [Google Scholar] [CrossRef]
- Muller, M.M.; Kries, H.; Csuhai, E.; Kast, P.; Hilvert, D. Design, selection, and characterization of a split chorismate mutase. Protein Sci. 2010, 19, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Vamvaca, K.; Vogeli, B.; Hilvert, D. Structure and dynamics of a molten globular enzyme. Nat. Struct. Mol. Biol. 2007, 14, 1202–1206. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Catalytic System | Conversion a (%) | Selectivity (%) | ee (%) | |
| 1 |  | 22 | none | none |
| 2 |  | 86 | none | none |
| 3 |  | 84 | 60 (S) | >99 (R) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Legrand, B.; Aguesseau-Kondrotas, J.; Simon, M.; Maillard, L. Catalytic Foldamers: When the Structure Guides the Function. Catalysts 2020, 10, 700. https://doi.org/10.3390/catal10060700
Legrand B, Aguesseau-Kondrotas J, Simon M, Maillard L. Catalytic Foldamers: When the Structure Guides the Function. Catalysts. 2020; 10(6):700. https://doi.org/10.3390/catal10060700
Chicago/Turabian StyleLegrand, Baptiste, Julie Aguesseau-Kondrotas, Matthieu Simon, and Ludovic Maillard. 2020. "Catalytic Foldamers: When the Structure Guides the Function" Catalysts 10, no. 6: 700. https://doi.org/10.3390/catal10060700
APA StyleLegrand, B., Aguesseau-Kondrotas, J., Simon, M., & Maillard, L. (2020). Catalytic Foldamers: When the Structure Guides the Function. Catalysts, 10(6), 700. https://doi.org/10.3390/catal10060700