Abstract
The lack of supported versions of the tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) ligand, suitable for flow-chemistry applications at scale, prompted us to develop a new route for the immobilization of such tris-triazole chelating units on highly cross-linked polystyrene resins. With this aim, the preparation of the known TBTA-type monomer 3 was optimized to develop a high-yield synthetic sequence, devoid of chromatographic purifications at any stage. Then, bead-type (P7) and monolithic (M7) functional resins were obtained by the easy and scalable suspension- or mold-copolymerization of 3 with divinylbenzene. Both types of materials were found to possess a highly porous morphology and specific surface area in the dry state and could be charged with substantial amounts of Cu(I) or Cu(II) salts. After treatment of the latter with a proper reducing agent, the corresponding supported Cu(I) complexes were tested in the copper-catalyzed alkyne-azide cycloaddition reaction (CuAAC). The immobilized catalysts proved active at room temperature and, in batch and with catalyst loadings as low as 0.6 mol%, afforded quantitative conversions within 20 h. Independent of the alkyne structure, extended use of the supported catalyst in flow was also possible. In the reaction of benzylazide and propargyl alcohol, this allowed a total turnover number larger than 400 to be reached.
1. Introduction
Independently disclosed in 2002 by the groups of Meldal and of Fokin and Sharpless [1,2], the copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC) has become an essential component of the click-chemistry toolbox [3,4,5]. Driven by many applications [6,7], the strong interest in CuAAC stimulated an intense search for improved catalytic systems [3,4,5,8]. The main goals of these efforts were the stabilization of the Cu(I) active form and reduction of catalyst loading [9,10,11], the suppression of side-reactions [12], and the lowering of metal contamination in 1,2,3-triazole products. Concerning the last aim, the immobilization of copper species on insoluble supports is one of the most actively investigated options [13,14,15,16]. Broadly speaking, this task has been pursued by either preparing nanostructured Cu(0), Cu(I), or Cu(II) (pre)catalysts on insoluble materials, or by anchoring a discrete Cu(I) or Cu(I) complex to a suitable support.
In principle, with the second choice, an advantage can be produced by the capacity of some ligands, such as tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, 1 in Figure 1), to stabilize the catalytically active Cu(I) form and to increase the CuAAC reaction rates [9,10]. In fact, a supported version of 1 on TentaGelTM resin was developed in 2007 by Chan and Fokin (P2) [17], and made commercially available a few years later, albeit at a relatively high price (€133.00 for 250 mg with a 0.17 mmol g−1 content of TBTA units) [18]. After loading with [Cu(MeCN)4]PF6, the resulting Cu(I) complex proved active in the CuAAC of model substrates [19], with a remarkable capacity to work in both protic and aprotic reaction media and a good stability upon recycling [17].
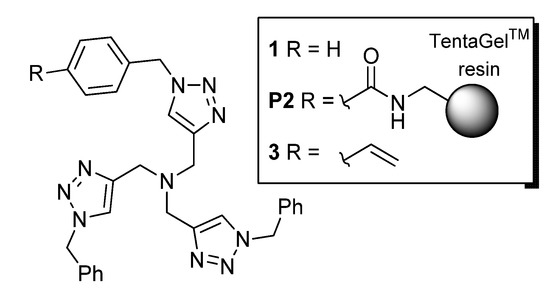
Figure 1.
Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) ligand (1), commercial supported variant (P2), and TBTA styryl monomer (3).
Variants of P2 were reported in the following years to include soluble macromolecular analogs [20,21,22] and surface-immobilized TBTA units in microfluidic devices [23]. However, to the best of our knowledge, no supported TBTA material suitable for continuous-flow chemistry at a scale of truly preparative interest has been described to date (for recent examples of CuAAC in flow with other supported catalytic systems, see [13,14,15,16,24,25,26,27,28,29,30,31,32]).
For this reason, due to our continuing interest in immobilized copper catalysts for selective transformations [33,34,35,36,37,38], we recently tackled the problem of designing a new polymeric TBTA ligand fit for use in mini- or meso-fluidic reactors.
Eventually, these efforts allowed a route for the convenient synthesis of the known TBTA monomer 3, as well as the conditions for its immobilization in bead-type or monolithic macroporous polystyrene resins, to be established. After charging with Cu(I) or Cu(II) salts, the resulting materials proved to be competent catalysts for batch and continuous-flow CuAAC reactions.
2. Results
2.1. Preparation of the Functional Monomer 3
The synthesis of the styrene monomer 3 was carried out according to Scheme 1. In brief, dipropargylamine (5) was subjected to CuAAC with benzylazide to provide 6, which was subsequently alkylated with propargyl bromide to give 4. A second CuAAC step, in this case, with 4-azidomethylstyrene, led to the TBTA monomer 3 in a high overall yield and without the need for chromatographic purification in any stage of the synthetic sequence.
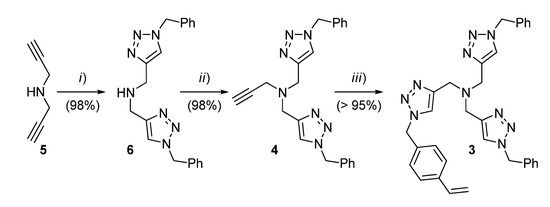
Scheme 1.
Preparation of the TBTA monomer 3. Conditions: (i) benzylazide (2 equiv.), CuSO4 (7 mol%), sodium l-ascorbate (20 mol%), H2O:CH2Cl2 (1:1), r.t., 7 h; (ii) NaH (1.3 equiv.), THF, then propargyl bromide in toluene (1.05 equiv.), r.t., 19 h; (iii) 4-azidomethylstyrene (1.0 equiv.), CuSO4 (20 mol%), sodium l-ascorbate (60 mol%), H2O:CH2Cl2 (1:1), r.t., 19 h.
2.2. Preparation of the Bead-Type Supported Catalysts
For the conversion of 3 into the corresponding functional resin P7, typical suspension polymerization conditions were adopted (Scheme 2). Gratifyingly, this step also took place with a high efficiency to give opaque polymer beads ranging from 100 to 300 μm in diameter. After extensive washing and drying, the material was characterized by elemental analysis, scanning electron microscopy (SEM), FT-IR, and N2 adsorption measurements (Supporting Information).
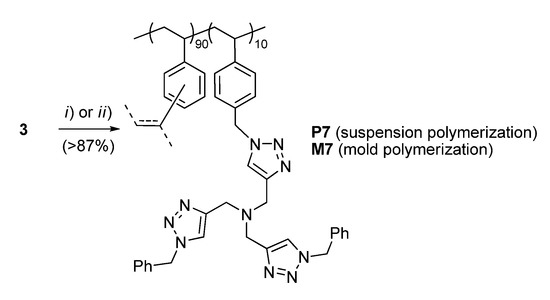
Scheme 2.
Preparation of the supported TBTA ligands P7 and M7. Conditions: (i) for P7, technical DVB (9 equiv.), AIBN (2 wt %), 1-dodecanol/toluene (1:9 v/v), 0.4% poly(vinyl alcohol) in water, 70 °C, 20 h; (ii) for M7, technical DVB (9 equiv.), AIBN (2 wt %), 1-dodecanol/toluene (2:1 v/v), 70 °C, 24 h.
The determination of nitrogen content by combustion analysis confirmed that >87% of the TBTA monomer had been incorporated into the resin, to give an actual loading of 0.50 mmol g−1 (28% by weight). As for other polymeric ligands from this laboratory [34], in spite of its seemingly compact surface exhibited by SEM analysis (Figure S2), P7 was found to possess a relatively large specific area in the dry state (SBET = 273 m2 g−1, Figure S4).
Additionally, given the moderate swelling in various aprotic solvents (e.g., 50% in THF), P7 proved to be an effective absorbent for copper salts. The largest loading (LCu ~ 0.5 mmol g−1) was recorded with CuCl or Cu(OTf)2 dissolved in MeCN and THF, respectively; nonetheless, a useful result (LCu = 0.17 mmol g−1) was also achieved with the cheaper CuSO4 in THF-H2O (2:1).
2.3. Preparation of the Monolithic Reactors
In order to immobilize TBTA units within monolithic continuous-flow reactors (MCFRs), three stainless-steel tubular molds (10 or 15 cm length × 4.6 mm internal diameter) were completely filled with a solution containing 3, DVB (3:DVB = 1:9 mole ratio), and AIBN (2 wt % with respect to the monomers) in 1-dodecanol/toluene (1:2 v/v). The molds were sealed and kept in an oil bath heated to 70 °C for 24 h, to give compact white monolithic resins M7 inside the metal housings. Then, the devices were flushed with dry THF for 24 h, whereupon they provided a P ~ 2.5 Kg cm−2 back-pressure at a ф = 35 μL min−1 flow-rate. Evaporation of the collected eluates under a reduced pressure gave oily residues that contained only 1-dodecanol and AIBN-related products, as shown by 1H NMR.
For the sake of characterization, one of the resin monoliths was removed from the housing and subjected to FT-IR, SEM, and N2 adsorption analyses (Supporting Information). While FT-IR did not show major differences with respect to the analogous resin in beads (Figure S3), M7 was found to possess a larger specific surface area in the dry state (SBET = 435 m2 g−1, Figure S5) and to swell to a lesser extent (≤30% in THF or CH2Cl2 ) than P7. In addition, the SEM images of M7 (Figure S2) showed a more rugged surface, with micrometric-sized groves decorated with ≤0.1 μm resin globules.
Loading of the resin monoliths with the copper salt was performed in flow, by pumping a solution of Cu(OTf)2 in THF through the remaining devices. After extensive washing with the same solvent, the iodometric titration of Cu(II) recovered in the eluates confirmed the capacity of the M7 material to retain an appreciable amount of the metal salt (LCu = 0.18–0.25 mmol g−1). Additionally, at this stage, the resin contained in one of the devices was expelled from the metal housing to give monolithic chunks of uniform turquoise shade (Figure 2a). Besides the obvious discoloring, the presence of Cu(OTf)2 in the recovered material was confirmed by the characteristic Lα,β and Kα lines of Cu and the Kα line of S in the Energy-Dispersive X-Ray Spectroscopy (EDS) analysis (Supporting Information). Moreover, mapping the metal content along a cross-section of the monolith with the same technique (Figure 2b and Figure S7) verified the conclusion drawn above regarding the uniform distribution of copper within the resin body. The further characterization of M7∙Cu(OTf)2 by SEM and N2 adsorption (SBET = 473 m2 g−1, Figure S6) revealed no major changes with respect to the bare M7 resin.
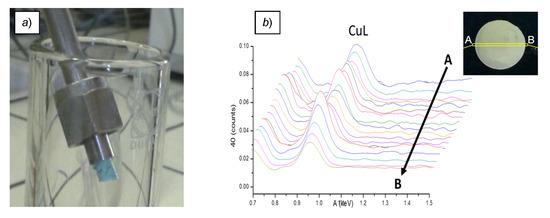
Figure 2.
Appearance of the M7∙Cu(OTf)2 monolith (a) and relative intensity of the Lα,β Cu lines, collected by SEM-EDS analysis along the scanning path A-B shown in the inset (b).
2.4. CuAAC Reactions in Batch
The catalytic properties of supported complexes were initially tested in batch CuAAC runs, carried out with the bead-type materials P7∙CuX1-2 (Table 1).

Table 1.
Copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC) runs in batch with P7 loaded with Cu(I) or Cu(II) salts.
In the case of P7∙CuCl in THF, the material was active in its pristine form and led to quantitative consumption of the benzylazide substrate in 20 h at room temperature at the substrate to catalyst ratio of S/C = 10 (Table 1, entry 1). Direct reuse of the resin recovered each time by filtration allowed four recycle runs, carried out with a progressive increase of S/C up to 50, to be successfully performed (Table 1, entries 2–5).
By contrast, the Cu(II)-supported complexes P7∙Cu(OTf)2 and P7∙CuSO4 had to be subjected to reduction before being used in CuAAC runs. Initial tests, aimed to optimized this stage, showed that phenylhydrazine in CH2Cl2 and hydroquinone in THF or MeOH were poorly effective in the activation of P7∙Cu(OTf)2 (Table 1, entries 6–8). On the contrary, the exposure of the same supported complex to sodium l-ascorbate in MeOH/H2O (4:1) afforded a brownish-yellow material with high activity in a series of consecutive CuAAC runs between benzylazide and propargyl alcohol (Table 1, entries 9–12).
As long as the same benchmark CuAAC reaction is concerned, P7∙CuSO4 reduced with sodium l-ascorbate in aqueous MeOH also proved to be a remarkably effective catalyst (Table 1, entry 13). However, when the recovered supported complex was employed for promoting the CuAAC between benzylazide and 1-hexyne, an appreciably slower reaction was observed (Table 1, entry 14).
Repetition of the last two runs above, by replacing P7 with the soluble TBTA ligand 1 (Table 1, entries 15 and 16), showed that the catalytic activity of the homogeneous system was higher than reduced P7∙CuSO4 under otherwise identical conditions Even in these experiments, however, 1-hexyne was found to react at a substantially slower rate than propargyl alcohol.
2.5. CuAAC Reactions in Flow
Initial continuous-flow CuAAC experiments (Table 2) focused on benchmarking the devices based on the M7∙Cu (OTf)2 material. Besides two monolithic reactors of different lengths (10 and 15 cm, respectively), this part of the study included the analogous packed-bed continuous flow reactor (PCFR). The latter was assembled by filling a stainless steel column (15 cm length × 4.6 mm internal diameter) with powder obtained by grinding chunks of the M7∙Cu(OTf)2 monolith involved in the characterization measurements discussed above.
Table 2.
Continuous-flow CuAAC runs in the monolithic (MCFR) and packed-bed (PCFR) devices based on reduced M7∙Cu(OTf)2.
The CuAAC runs were carried out by employing a syringe pump, which was used for flushing the reactor with a solution of the selected reducing agent and then with a solution of internal standard and equimolar propargyl alcohol and benzylazide.
The examination of different reductants for the Cu(II) metal centers in the MCFR (15 cm) or PCFR afforded results that closely mirror those already recorded in batch. Indeed, hydroquinone in THF turned out to be completely useless (Table 2, entries 1 and 8), while the addition of phenylhydrazine to the reactant stream proved moderately effective (Table 2, entries 2 and 9). In this latter case, however, the reaction progress at the reactor outlet was largely incomplete, in spite of the rather low flow-rate (ф = 0.4 mL h−1) adopted in the run.
On the contrary, good levels of conversion of the azide were attained if sodium l-ascorbate was used in the preliminary reduction step and then employed for saturating the stream of reactants in the subsequent CuAAC runs. Under these conditions, the complete consumption of benzylazide was achieved in a relatively long experiment (S/C = 67) carried out at ф = 1.5 mL h−1 in the MCFR (10 cm) (Table 2, entry 3). The increase of the flow-rate to 12 mL h−1, followed by its progressive decrease to the value of the initial run, led to conversion degrees that reflect the average residence time of the reactants through the catalyst bed (Table 2, entries 3–7).
Similarly, the use of sodium l-ascorbate for reducing the powdered material in the PCFR considerably improved its catalytic activity with respect to the use of hydroquinone or phenylhydrazine (Table 2, entries 8 and 9). This allowed 57% conversion to be attained at room temperature and with a flow-rate as high as ф = 6 mL h−1 (Table 2, entry 10). Further experiments with the PCFR were carried out at 60 °C and with ф = 3 mL h−1. Under these conditions, good to high conversion levels were recorded in a sequence of five runs (Table 2, entries 11–15).
The next stage of the investigation focused on PCFRs containing the bead-type complex P7∙CuSO4 reduced with sodium l-ascorbate. With this aim, sections of PTFE tubing (18 cm × 1.5 mm inner diameter) were packed with the resin beads (80–85 mg, approx. 14 μmol of copper), to give a set of identical flow reactors R1–R5. In this case, pumping was effected by a gas-tight syringe, used for the preliminary flushing of devices with 0.7% sodium l-ascorbate in aqueous MeOH and, after turning of the catalyst bed to pale-yellow, of the solution of reactants and internal standards.
The testing of R1 and R2 in the benchmark reaction was performed in MeOH-THF (4:1) and MeOH-H2O (4:1), respectively, with the devices being placed in a glove-bag filled with N2. The two rather long runs (S/C ~ 350 each) were carried out in parallel with a dual channel syringe pump, with simultaneous variation of the flow-rates in the range ф = 0.6-1.8 mL h−1 (Table 3, entries 1 and 3). Because the former solvent mixture turned-out to lead to higher conversion degrees, all of the subsequent experiments made use of mixtures of MeOH and THF as the reaction medium. Moreover, instead of keeping the reactors inside the glove-bag, sodium l-ascorbate was added to the reactant feed, which was pumped at the fixed flow-rate of ф = 0.6 mL h−1.
Table 3.
Continuous-flow CuAAC reactions in the PCFRs containing P7∙CuSO4 reduced with sodium l-ascorbate.
The reuse of R1 and R2 with the said experimental arrangement showed that both devices were still active and could promote the CuAAC reaction of benzylazide with propargyl alcohol (Table 3, entry 2) and 1-hexyne (Table 3, entry 4), respectively, albeit with noticeably reduced activity.
The substrate scope of the continuous-flow CuAAC reaction was explored with the fresh reactors R3, R4, and R5. In general, the alkyne substrates endowed with an alcohol or amine functional group (Table 3, entries 5–9 and 11–14) were found to provide much better results than the purely hydrocarbon derivative 1,7-octadiyne (Table 3, entry 10). Provided that the solvent composition was adjusted to prevent the precipitation of the corresponding triazole product inside the PCFR, total conversion and an excellent isolated yield could also be obtained in the case of phenyl-acetylene (Table 3, entries 7 and 8).
In order to evaluate the metal contamination in the CuAAC products, selected dried samples of the crude triazoles were analyzed by inductively-coupled plasma optical spectroscopy (ICP-OES). As shown in Table 3, significantly larger amounts of copper were found in the bis-triazole derivative 6 (e.g., 853 ppm, Table 3, entry 13) than in the mono-triazoles from the CuAAC of benzylazide with propargyl alcohol, 1-hexyne, or phenylacetylene (138–175 ppm, Table 3, entries 4, 5, and 9).
3. Discussion
3.1. Preparation of the TBTA Monomer and Supported Catalysts
Because neither the mechanical properties of the commercial resin P2 (large swelling in most solvents [17]) nor the published synthetic route for its preparation appeared to satisfy the requirements of continuous-flow large-scale applications, both aspects had to be addressed in the present study.
As for the polymer support, a highly cross-linked architecture was selected in order to minimize clogging problems in flow, due to excessive swelling of the support. Moreover, cost considerations led to disregard routes based on the grafting of TBTA units on commercial macroporous resins, in favor of the co-polymerization of the TBTA monomer 3 with technical divinylbenzene (DVB) as a cheap cross-linking agent.
Even though the preparation of 3 has been reported in the literature [20,21], at the beginning of this work, we set out to find a more convenient and scalable route for its synthesis. In particular, the goal was to avoid the use of expensive reagents and multiple chromatographic purification steps required by the published preparations of the TBTA monomer 3, or its direct precursor 4 [17,20,21,22,23].
After exploring alternative routes, a straightforward procedure was identified for converting dipropargylamine (5) into 3, in three high-yielding steps (Scheme 1). The key point of the whole sequence was the alkylation of the intermediate 6 with propargyl bromide that, after some optimization work, was found to proceed much better than anticipated [39]. This allowed 4 to be obtained in a nearly quantitative yield by simple precipitation of the crude product with water and subsequent recrystallization from AcOEt. Thanks to the high yield in the initial CuAAC reaction between 5 and benzylazide, the whole reaction sequence could be scaled-up in an effortless manner to provide 11.5 g of pure 4.
The subsequent CuAAC reaction between 4 and 4-azidomethylstyrene, followed by the removal of copper species through aqueous ammonia work-up, afforded the monomer 3 in an essentially quantitative yield. Because the NMR purity of 3 appeared to be high, the freshly prepared monomer was used directly in the crude form.
Having achieved the preparation of 3 through a high-yielding and chromatography-free synthetic sequence, its immobilization in styrene-type resins could be explored. This aim was pursued by following two alternative routes: (a) The co-polymerization of 3 and DVB in a poly(vinyl alcohol)-stabilized aqueous suspension, to give resin beads P7, and (b) Fréchet-type co-polymerization of the same monomers inside stainless-steel tubular molds [40,41,42,43], which afforded the monolithic resins M7.
As confirmed by the evidence detailed in the Results section, both approaches proved effective (>87% yield) towards the goal of high incorporation of the functional monomer in the cross-linked polymeric network. At the same time, and in spite of the identical co-monomer composition (3:DVB = 10:90), differences were observed in the swelling and morphological properties of the various resins and in their ability to adsorb Cu(I) or Cu(II) salts. Overall, the swelling ratios of M7 and P7 in THF (sr = 1.3 and sr = 1.5, respectively) and the SEM images suggest a more rigid polymer architecture for the monoliths than for the resin beads.
Concerning the specific surface area deduced from N2 adsorption measurements (M7: SBET = 435 m2 g−1; P7: SBET = 273 m2 g−1), the results look somewhat counterintuitive in view of the use of a larger amount of 1-dodecanol under the mold polymerization conditions. In fact, being a bad solvent for the growing polystyrene chains, this porogenic agent was expected to reduce SBET as a consequence of the shift of the porosity distribution towards the macropore domain [44]. Therefore, the observation of an experimental trend, which is the opposite of the predicted one, points to the occurrence of additional phenomena, possibly linked to the presence of the aqueous dispersing phase during the suspension polymerization process.
Both P7 and M7 demonstrated a good to excellent ability to adsorb CuCl or CuX2 salts (X = OTf or 1/2 SO4) dissolved in suitable solvents. The largest capacity (up to the theoretical value LCu ~ 0.5 mmol g−1 for a Cu(I/II):TBTA=1:1 stoichiometry [17]) was obtained in the uptake of CuCl or Cu(OTf)2 by P7, from polar aprotic media (MeCN and THF, respectively). Nevertheless, the same material could be used for adsorbing the cheaper CuSO4 salt, even from a protic THF-H2O (2:1) solution, albeit with some decrease in the metal uptake (LCu = 0.18 mmol g−1).
Flushing different M7 monoliths with Cu(OTf)2 in THF proved less effective, with a measured metal adsorption no higher than 50% of the calculated content of TBTA units. By comparing these results with those obtained with P7, it seems possible to conclude that the swelling extent of the resin has a more significant impact on the metal uptake than the bare surface area of the material in the dry state (see the discussion above).
3.2. Catalysis in Batch and in Flow
Initial runs in batch, with the bead-type resin P7 charged with CuCl, Cu(OTf)2, or CuSO4, confirmed that the supported complexes were competent catalysts (or pre-catalysts) for the CuAAC. In particular, a sample of P7∙CuCl was able to provide ≥83% final conversion in a sequence of five benchmark reaction cycles in CH2Cl2 (Table 1, entries 1–5), carried out with a progressive decrease of the catalyst loading from 10 to 2 mol% (calculated with respect to the initial copper content). In this regard, it is worth noting that, while the last three runs did not reach full conversion of the azide substrate, they did not evidence any appreciable drop of the activity of the supported complex.
Because Cu(II) salts are inactive in CuAAC [3], P7∙Cu(OTf)2 and P7∙CuSO4 were subjected to reduction before testing. Considering its effectiveness in our previous work with nanostructured CuAAC catalysts [37], as well as for other Cu(I)-supported systems [35], the use of phenylhydrazine in CH2Cl2 was initially selected for this purpose. Unfortunately, despite the apparently fast reduction of the supported Cu(II) sites (as judged from the fading of the azure color of P7∙Cu(OTf)2), the resulting material displayed unsatisfactory activity (Table 1, entries 6 and 7).
Given its successful use in the literature for the reduction of P2∙CuCl2 and P2∙CuSO4 [17], it came as a surprise to discover that hydroquinone in THF or MeOH was completely ineffective towards P7∙Cu(OTf)2. In this case, no change of the azure color of the resin beads was observed and the hydroquinone-treated material did not display any significant CuAAC activity (Table 1, entry 8).
Much to our delight, switching to the use of sodium l-ascorbate in aqueous MeOH eventually afforded excellent results in a row of four benchmark runs performed with reduced P7∙Cu(OTf)2 at S/C = 20 or S/C = 100 (Table 1, entries 9–12). Although, in this case, some loss of catalytic activity towards the end of the series is suggested by the conversion values at the intermediate reaction time of 2 h, such a trend did not preclude the full conversion of azide at the standard 20 h end time of each run.
Sodium l-ascorbate in MeOH was also effective for the reduction of P7∙CuSO4 and provided a supported system endowed with high catalytic activity in the benchmark reaction (Table 1, entry 13). Nevertheless, recovery of the supported catalyst by filtration and reuse for promoting the CuAAC between benzylazide and 1-hexyne resulted in a surprisingly sluggish reaction (Table 1, entry 14). Since control experiments ruled out major oxidative phenomena of Cu(I) or extensive metal leaching from the resin (vide infra), these findings prompted us to examine the CuAAC of propargyl alcohol and 1-hexyne in the presence of the soluble catalyst from the TBTA ligand 1 (Table 1, entries 15 and 16). As expected, both homogeneous-phase reactions proceeded at a higher rate than the corresponding heterogeneous-phase ones. At the same time, however, a marked influence of the alkyne structure was observed, even in this case, with 1-hexyne, which was found to react at least six times slower than propargyl alcohol. By taking into account some literature precedents [39,45], it seems reasonable to conclude that the hydroxyl group in the latter substrate exerts an accelerating effect on the CuAAC reaction, possibly through metal-chelation or by the increase of the acidity of the acetylenic proton.
Overall, the results in batch confirmed that the highly cross-linked polystyrene architecture of P7 was fit for the preparation of TBTA-Cu(I)-supported catalytic systems for the CuAAC. Despite the moderate swelling and lack of polyethylene side-chains, as in the TentaGelTM-based commercial ligand P2, the resin beads P7 could be charged with Cu(I) or Cu(II) salts dissolved in either polar aprotic solvents or mixtures featuring a high volume fraction of protic solvents. Lastly, after reduction (when necessary) with sodium l-ascorbate, the corresponding P7∙Cu(I) materials were demonstrated to be competent catalysts for CuAAC reactions in media of widely different polarities (from CH2Cl2 to nearly pure MeOH, see Table 1). Considering that all the CuAAC runs were carried out for 20 h at room temperature, the specific activity of the various P7∙Cu(I) catalysts compares well with that described for P2∙Cu(I) [17].
With the information above in hand, we could turn to the use of supported catalysts in the continuous-flow reactors. This was initially done for the MCFRs M7∙Cu(OTf)2, whose high specific surface area (SBET = 473 m2 g−1) was deemed advantageous for the intended catalytic applications in-flow. In this regard, it is worth noting that, especially with the 15 cm-long MFCRs, the flow-rate was limited to ф ≤ 50–100 μL min−1 by the ensuing back-pressure. Moreover, the longer devices showed a tendency to occasionally clog. In spite of this, the ability to flush the various reactors with a syringe pump confirmed that the monolithic bodies obtained under the conditions described in this work were endowed with a sufficiently large fraction of connected macropores to support the convective flow of the liquid stream.
Notwithstanding the limitations above, excellent results were obtained with properly reduced M7∙Cu(OTf)2. In fact, while phenylhydrazine or hydroquinone turned out to be essentially useless in this case (Table 2, entries 1 and 2), resorting to the use of sodium l-ascorbate caused the expected boost of catalytic activity. Such a reducing agent was employed both as a preliminary flush, in aqueous MeOH, and then at a very small [46], yet undetermined, saturating concentration, in the MeOH/THF (4:1) stream of the reactants. Under these conditions, quantitative consumption of the azide at the reactor outlet was obtained in a relatively long benchmark run (S/C = 67) at ф = 1.5 mL h−1 (Table 2, entry 3). Interestingly, the flow-rate adopted in the experiment corresponds to an average residence time (τ) of the reactants in the catalytic monolith body (as estimated by pycnometry) of less than 50 min.
Subsequent tests at higher flow-rates (Table 2, entries 4–6) led to a decrease of the conversion extent, with a trend that has an obvious relationship with the progressive shortening of τ (Figure 3). This conclusion is further supported by a final experiment under the initial conditions (S/C = 77 and ф = 1.5 mL h−1; Table 2, entry 7), where the MCFR proved to still be capable of fully converting the azide substrate, in spite of its extended usage, up to a total (accumulated) turnover number of TTON = 188.
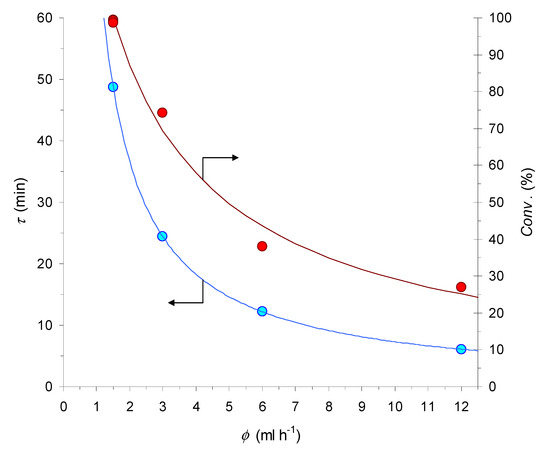
Figure 3.
Graphical representation of the relationship between the flow-rate (ф), estimated residence time (τ), and conversion degree (Conv. %) for the benchmark CuAAC reaction in the 10 cm-long MCFR (for the conditions see Table 2, entries 3–7).
Similar results were obtained with the reactor packed with the crushed M7∙Cu(OTf)2 monolith. In this case, hydroquinone or phenylhydrazine (Table 2, entries 8 and 9) were less effective in activating the Cu(II) pre-catalyst than sodium l-ascorbate (Table 2, entry 10). However, even in the latter case, the benchmark CuAAC reaction was largely incomplete at the high flow-rate adopted in the run (57% conversion at ф = 6 mL h−1). In order to improve this outcome, the influence of the reaction temperature was briefly investigated in a series of experiments carried out at 60 °C and with ф = 3 mL h−1 (Table 2, entries 11–15). As expected, the combined increase of the temperature and reduction of the flow-rate succeeded in improving the conversion degree. At the same time, however, these conditions led to some decrease of catalytic activity towards the latest runs, which discouraged further pursuit of the approach.
In this regard, it is worth pointing out that a cursory comparison of the entries 5 and 10 in Table 2 might give the feeling that the MCFR device (38% conversion and space-time yield of STY = 52 g L−1 h−1) was provided with a lower catalytic activity than the PCFR one (57% conversion and STY = 70 g L−1 h−1, under otherwise identical conditions). However, when the relative copper contents and estimated residence times in the two reactors are taken into account (approx. 64 μmol vs. 170 μmol and τ = 12 min vs. τ = 19 min, respectively), it is possible to conclude that, similar to other studies [47], the monolithic device was actually endowed with higher specific activity than the packed-bed one.
The last part of this investigation involved the PCFRs R1–R5, assembled by dry-packing P7∙CuSO4 (80–85 mg, approx. 14 μmol of copper) into PTFE tubing sections. Thanks to the translucent properties of PTFE, in this case, the reduction of the Cu(II) sites could be followed visually to confirm an essentially instantaneous process when the flush of sodium l-ascorbate in aqueous MeOH reached successive portions of the resin beads.
Initial experiments were designed to benchmark the properties of reduced P7∙CuSO4 in flow. With this aim, two PCFRs (R1 and R2; Table 3, entries 1 and 3) were fit to a dual-channel syringe pump and the whole apparatus was placed in a glove-bag under N2 to prevent the oxidation of Cu(I) sites by atmospheric O2. While the run in R1 involved the MeOH/THF (4:1) reaction medium, already employed in batch (see Table 1, entries 9–12), for the reaction in R2, the use of a fully protic MeOH/H2O (4:1) solvent mixture was explored.
With the adopted set-up, the influence of the flow-rate could be investigated simultaneously in the two PCFRs (Figure 4). Complete conversion of the azide at the outlet of both devices was observed for about 4.5 h at ф = 1.2 mL h−1 (around 17 column volumes of feed stream). After that time, the flow-rate was increased to ф = 1.8 mL h−1, with the applied change, which led to a progressive decrease of the conversions in the following 3.5 h. Besides the anticipated effect of the shorter residence time at higher ф, the observed trend was likely to involve some catalyst deactivation phenomena. This conclusion is substantiated by the observation that switching back to the ф = 1.2 mL h−1 setting caused a significant improvement of the reaction completeness, but did not restore the initial full conversions. Moreover, in the following 6 h at ф = 1.2 mL h−1, a further loss of catalytic activity was recorded, which is a problem that could be partially compensated for by a reduction of the flow-rate to ф = 0.6 mL h−1 in the final part of the run. Eventually, the trends in the instantaneous output of the reactors led to an 88% and 68% average degree of azide conversion in the cumulatively collected effluents from R1 and R2, respectively.
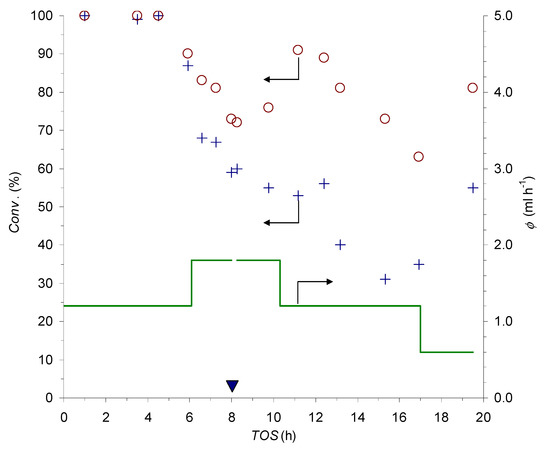
Figure 4.
Set flow-rate ф (continuous line) and conversion values at the outlet of PCFRs R1 (circles) and R2 (crosses) as a function of the time on stream; the filled triangle marks a daily break in the experiment’s course (for the conditions, see Table 3, entries 1 and 3).
In this respect, it is important to note that, while some deactivation is evident for both PCFRs, the devices retained appreciable catalytic activity up to a time on stream (TOS) of almost 20 h. Moreover, due to the large S/C involved in the experiments (around 350 for each device), turnover numbers as high as TON = 304 and TON = 238 could be attained for R1 and R2, respectively.
Given the better performance and apparently lower deactivation rate in the run with R1, all the subsequent experiments made use of mixtures of MeOH and THF. The flow-rate in these experiments was kept fixed at ф = 0.6 mL h−1 and, instead of placing the reactors inside the glove-bag, sodium l-ascorbate (1 mol%) was added to the reactant stream as a more practical measure to prevent catalyst oxidation by adventitious oxygen.
The reuse of R1 and R2 under the said conditions showed that both devices were still active and could promote the CuAAC reaction of benzylazide with propargyl alcohol and 1-hexyne, respectively. Nevertheless, the conversions determined by GC-MS were lower than in the previous runs and ultimately translated into moderate isolated yields (49% and 39%, respectively) of the corresponding triazole products (Table 3, entries 2 and 4).
Further trials with the fresh reactors R3, R4, and R5 aimed to explore the substrate scope (Table 3, entries 5–14). In general, the best results (83%–97% isolated yields) were recorded with polar alkynes, i.e., those provided with an alcohol or amine functional group in proximity of the acetylene unit. On the contrary, under the adopted conditions, 1,7-octadiyne afforded a mixture of mono- and bis-triazole products in a low overall yield (Table 3, entry 10). Even though a more detailed study will be required for clarifying this aspect, these results appear to conform to the conclusion drawn for the batch experiments, about the much reduced reactivity of the purely hydrocarbon substrates under the CuAAC conditions of this work (see Table 1, entries 13–16).
In the case of phenylacetylene, the precipitation of the corresponding triazole product from the MeOH/THF (4:1) reaction medium was an additional issue (Table 3, entry 7). However, when the run was repeated in a THF-enriched mixture, a nearly quantitative yield of the expected triazole product could be easily obtained (Table 3, entry 8).
Irrespective of the exact composition of the solvent mixture, the continuous-flow CuAAC reaction between dipropargylamine (5) and benzylazide gave the bis-triazole product 6 in a high isolated yield (Table 3, entries 11 and 13). However, when the reactor R5 was employed again, in an extended run with the same reactants, 6 and its mono-triazole analogue were only obtained in a moderate overall yield (Table 3, entry 14).
In order to shed some light on these latter findings, selected samples of crude triazole products were evaporated to dryness and assayed by ICP-OES for their copper content. Not unexpectedly, a significantly larger amount of metal was found in the bis-triazole derivative 6 (853 and 257 ppm, Table 3, entries 13 and 14) than in the other (mono-triazole) crudes (138–175 ppm, Table 3, entries 4, 5, and 9). On this basis, it seems reasonable to conclude that the sudden drop in the catalytic activity of R5, noted above, must be related to the possibility for the chelating derivative 6 to effectively compete with the supported TBTA units for Cu(I) complexation. In turn, this resulted in extensive leaching of the active centers in the initial run and, therefore, to a much faster deactivation of the catalytic material than in the CuAAC runs, leading to mono-triazole derivatives. Concerning the latter, it is worth noting that the ≤175 ppm in the dry crude corresponds to <5% of the copper initially loaded in the flow device. Besides comparing favorably with the leaching levels observed in batch with other polystyrene-supported multidentate nitrogen ligands [48], these findings explain how TTON values larger than 400 were accumulated in some of the continuous-flow experiments of this work (e.g., the series of three runs with R4; Table 3, entries 9–11).
4. Materials and Methods
Solvents and reagents were obtained from Sigma-Aldrich (Milano, Italy) and Carlo Erba reagents Srl (Milano, Italy).
All the reactions involving water or air-sensitive compounds were carried out under dry N2, in flame-dried glassware. The solvents for the same reactions were freshly distilled before use from the proper drying agent [49]. For the CuAAC runs, the commercial solvents were degassed by bubbling Ar for 5–10 min. If not noted otherwise, the other commercial compounds were used as received.
Benzylazide, 4-azidomethylstyrene, 1, and 6 were prepared by known methods and showed NMR constants in accordance with the published values [20,50,51].
1H and 13C NMR spectra were recorded as CDCl3 solutions with a Bruker Avance DRX 400 (Bruker BioSpin GmbH, Rheinstetten, Germany) or a Varian Inova 600 spectrometer (Varian Inc., Palo Alto, CA, USA) and are reported in ppm, with the frequency reference made by setting the residual not-deuterated solvent (1H) or deuterated solvent (13C) to the recommended values [52].
The structures of the 1,2,3-triazole products were confirmed by comparing their NMR constants with those reported in the literature [17,51,53,54,55,56,57].
For further experimental details, see the Supporting Information.
4.1. CuAAC in Batch with P7-Supported Catalysts: General Procedure
Catalysis runs in batch were carried out under N2, in Schlenk tubes provided with side glass frits. In the case of P7∙CuCl and reduced P7∙Cu(OTf)2, the supported catalyst was directly suspended in the degassed solution containing the azide (0.5–5 mmol), alkyne (1.0 equiv.), and n-nonane or m-xylene (0.2 equiv., internal standard) in the selected solvent (2.5–25 mL, [azide] = 0.2 M). The reaction progress was determined after 2 and 20 h, by a GC-MS analysis of samples taken from the reaction mixture. Then, the suspension was filtered and the recovered resin beads were washed with the degassed solvent (3 × 10 mL) and dried in vacuo, before being used in the next run. The combined filtrate and washings were evaporated under a reduced pressure to give the crude triazole product.
In the case of P7∙CuSO4, 0.070 g (0.012 mmol Cu, 0.60 mol%) of the supported complex was suspended under N2 in degassed MeOH (10 mL) and treated with 0.021 g (0.11 mmol) of sodium l-ascorbate. The mixture was stirred at r.t. for 15 min, and a degassed solution of the azide (2.0 mmol), alkyne (2.0 mmol), and n-nonane or m-xylene internal standard in distilled THF (0.5 mL) was then added. Reaction monitoring, catalyst recovery, and product isolation were carried out as described above.
4.2. CuAAC in Flow: General Procedure
The working-scheme of the devices used for the CuAAC reactions in flow is shown in Figure S1 (Supporting information).
To effect the reduction of the supported Cu(II) sites, the reactor was flushed with degassed MeOH at ф = 1.2–2.4 mL h−1, followed by a 0.7 wt % solution of sodium l-ascorbate (8 equiv. vs. Cu) in MeOH/H2O (4:1).
In a typical catalysis run, 5.0 mmol of azide, 5.0 mmol of alkyne, 0.010 g (0.050 mmol, 1 mol%) of sodium l-ascorbate, and 0.050 g of n-nonane or m-xylene internal standard were placed in the reagent/solvent reservoir and dissolved with magnetic stirring in the selected solvent mixture (25 mL total volume). After bubbling N2 for 5 min, the feed solution was aspirated into the gas-tight syringe and then infused through the catalytic reactor at ф = 0.6–1.8 mL min−1. The liquid stream at the reactor outlet was collected for product recovery and GC-MS monitoring of the reaction progress. Upon exhaustion of the feed solution, the whole device was repeatedly flushed with degassed MeOH/THF (4:1, at least five column volumes). The washings were combined with the fractions collected in the course of the run and analyzed by GC-MS. After evaporation in vacuo (35 °C, 20 mbar down to 0.1 mbar) to remove the volatiles, the residue was analyzed by 1H NMR and, if required, subjected to flash chromatography for removing the internal standards and unreacted substrates.
The flow device was employed directly in the next run, unless pale-green discoloring of the resin was observed (e.g., after some days being idle). In this case, before re-use, the catalyst bed was subjected to regeneration by flushing with sodium l-ascorbate, as described above.
5. Conclusions
In summary, this paper describes a convenient approach to polystyrene-supported TBTA-Cu(I) catalysts suitable for continuous-flow CuAAC applications. In our opinion, the main merit of the synthetic route disclosed herein is the possibility of its easy and affordable scale-up. This advantage concerns not only the synthesis of the functional monomer 3, but also its covalent immobilization within the insoluble polymer network through robust radical-copolymerization with cheap divinylbenzene.
Irrespective of the technique and conditions adopted for the purpose, either suspension or mold-copolymerization, the resulting highly cross-linked resins (P7 and M7, respectively) have been shown to possess swelling and textural properties that made them fit for applications in batch and in flow. In this regard, it is worth noting that in several circumstances, P7 and M7 were used in liquid media with a high volume fraction of MeOH or H2O, which are two thermodynamically bad solvents for the polystyrene chains. The good results that were nonetheless observed under these conditions demonstrate the suitability of the macromolecular structure (most likely, the high SBET values) of the supported ligands developed in this work.
After charging P7 and M7 with Cu(I) or Cu(II) salts (and, in the case of the latter, reduction with sodium l-ascorbate), the supported complexes showed excellent activity in the copper-catalyzed Huisgen cycloaddition of azides to polar alkyne substrates. Following an initial screening in batch, most of these CuAAC reactions were performed in continuous-flow reactors, either of the monolithic or packed-bed type. In general, good results were obtained with both reactor architectures, with the monolithic ones showing better specific activity, but also some propensity towards clogging.
The CuAAC runs in batch demonstrated the possibility to reach high conversions at room temperature, with calculated space-time yields that, in some cases, exceeded 240 g L−1 h−1. Moreover, at least in the CuAAC processes that led to mono-triazole products, the catalytic flow devices showed a reasonable durability. In some examples, this allowed accumulated turnover numbers larger than 400 with respect to the amount of copper initially loaded onto the functional resin to be easily attained.
In view of the results summarized above, work is currently aimed at increasing the reactivity of the less polar alkyne substrates, further reducing metal leaching, and improving the permeability of the monolithic reactors. Progress in these directions will be reported in due course.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/4/434/s1: Additional experimental details; NMR spectra of 3, 4, 6, and of the 1,2,3-triazole products from the CuAAC runs: typical GC-MS chromatogram for the benchmark CuAAC reaction; Figure S1: Schematic of the continuous-flow reactor used in the CuAAC runs; Figure S2: Appearance and SEM images of the resin materials; Figure S3: FT-IR spectra of P7 and M7; Figure S4: N2 adsorption-desorption isotherms of P7; Figure S5: N2 adsorption-desorption isotherms of M7; Figure S6: N2 adsorption-desorption isotherms of M7∙Cu(OTf)2; Figure S7: EDS spectra and mapping of the Cu content in M7∙Cu(OTf)2.
Author Contributions
Conceptualization, A.M.; validation, G.A., D.S., and A.M.; investigation, A.P., G.A., M.P., A.L., M.C., F.O., M.M., D.S., and A.M.; resources, A.M. and C.E.; writing—original draft preparation, A.M.; writing—review and editing, A.M. and C.E.; visualization, M.M.; supervision, A.M.; project administration, A.M.; funding acquisition, C.E. and A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the University of Pisa, grant number PRA_2018_36, and by MIUR, grant FFABR 2017 and FIRB RBFR10BF5 V “Multifunctional hybrid materials for the development of sustainable catalytic processes”. The APC was partially funded by the University of Pisa.
Acknowledgments
A. Manariti (Dip. di Chimica e Chimica Industriale, Univeristà di Pisa) and N. Scotti (SCITEC-CNR) are gratefully acknowledged for the recording of the FT-IR spectra and BET analyses, respectively.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Gil, M.V.; Arevalo, M.J.; Lopez, O. Click chemistry—What‘s in a name? Triazole synthesis and beyond. Synthesis 2007, 2007, 1589–1620. [Google Scholar] [CrossRef]
- Wang, C.; Ikhlef, D.; Kahlal, S.; Saillard, J.-Y.; Astruc, D. Metal-catalyzed azide-alkyne “click” reactions: Mechanistic overview and recent trends. Coord. Chem. Rev. 2016, 316, 1–20. [Google Scholar] [CrossRef]
- Tabacaru, A.; Furdui, B.; Ghinea, I.O.; Carac, G.; Dinica, R.M. Recent advances in click chemistry reactions mediated by transition metal based systems. Inorg. Chim. Acta 2017, 455, 329–349. [Google Scholar] [CrossRef]
- Huo, J.; Hu, H.; Zhang, M.; Hu, X.; Chen, M.; Chen, D.; Liu, J.; Xiao, G.; Wang, Y.; Wen, Z. A mini review of the synthesis of poly-1,2,3-triazole-based functional materials. RSC Adv. 2017, 7, 2281–2287. [Google Scholar] [CrossRef]
- Pickens, C.J.; Johnson, S.N.; Pressnall, M.M.; Leon, M.A.; Berkland, C.J. Practical considerations, challenges, and limitations of bioconjugation via azide-alkyne cycloaddition. Bioconjugate Chem. 2018, 29, 686–701. [Google Scholar] [CrossRef]
- Haldon, E.; Nicasio, M.C.; Perez, P.J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as Copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Rodionov, V.O.; Presolski, S.I.; Gardinier, S.; Lim, Y.-H.; Finn, M.G. Benzimidazole and related ligands for Cu-catalyzed azide−alkyne cycloaddition. J. Am. Chem. Soc. 2007, 129, 12696–12704. [Google Scholar] [CrossRef]
- Wang, C.; Wang, D.; Yu, S.; Cornilleau, T.; Ruiz, J.; Salmon, L.; Astruc, D. Design and applications of an efficient amphiphilic “click” Cu(I) catalyst in water. ACS Catal. 2016, 6, 5424–5431. [Google Scholar] [CrossRef]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M.G. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed]
- Dervaux, B.; Du Prez, F.E. Heterogeneous azide-alkyne click chemistry: Towards metal-free end products. Chem. Sci. 2012, 3, 959–966. [Google Scholar] [CrossRef]
- Monguchi, Y.; Sawama, Y.; Sajiki, H. Synthesis of triazole, indole, and five or six-membered saturated heterocyclic compounds. Heterocycles 2015, 91, 239–264. [Google Scholar]
- Chassaing, S.; Beneteau, V.; Pale, P. When CuAAC ’click chemistry’ goes heterogeneous. Catal. Sci. Technol. 2016, 6, 923–957. [Google Scholar] [CrossRef]
- Mandoli, A. Recent advances in recoverable systems for the copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC). Molecules 2016, 21, 1174. [Google Scholar] [CrossRef]
- Chan, T.R.; Fokin, V.V. Polymer-supported copper(I) catalysts for the experimentally simplified azide–alkyne cycloaddition. QSAR Comb. Sci. 2007, 26, 1274–1279. [Google Scholar] [CrossRef]
- Sigma-Aldrich Product Cat. 696773. Available online: https://www.sigmaaldrich.com/catalog/product/aldrich/696773 (accessed on 28 February 2020).
- De Bo, G.; Kuschel, S.; Leigh, D.A.; Lewandowski, B.; Papmeyer, M.; Ward, J.W. Efficient assembly of threaded molecular machines for sequence-specific synthesis. J. Am. Chem. Soc. 2014, 136, 5811–5814. [Google Scholar] [CrossRef]
- Lammens, M.; Skey, J.; Wallyn, S.; O’Reilly, R.; Du Prez, F. Polymeric ligands as homogeneous, reusable catalyst systems for copper assisted click chemistry. Chem. Commun. 2010, 46, 8719–8721. [Google Scholar] [CrossRef]
- Wallyn, S.; Lammens, M.; O’Reilly, R.K.; Du Prez, F. Highly active, thermo-responsive polymeric catalytic system for reuse in aqueous and organic CuAAC reactions. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 2878–2885. [Google Scholar] [CrossRef]
- Movahedi, A.; Moth-Poulsen, K.; Ekloef, J.; Nyden, M.; Kann, N. One-pot synthesis of TBTA-functionalized coordinating polymers. React. Funct. Polym. 2014, 82, 1–8. [Google Scholar] [CrossRef]
- Fernandes, A.E.; Ye, Q.; Collard, L.; Le Duff, C.; d’Haese, C.; Deumer, G.; Haufroid, V.; Nysten, B.; Riant, O.; Jonas, A.M. Effects of thickness and grafting density on the activity of polymer-brush-immobilized tris(triazolyl) Copper(I) catalysts. ChemCatChem 2015, 7, 856–864. [Google Scholar] [CrossRef]
- Yamada Yoichi, M.A.; Ohno, A.; Sato, T.; Uozumi, Y. Instantaneous click chemistry by a copper-containing polymeric-membrane-installed microflow catalytic reactor. Chem. Eur. J. 2015, 21, 17269–17273. [Google Scholar] [CrossRef] [PubMed]
- Mandity, I.M.; Oetvoes, S.B.; Szolosi, G.; Fueloep, F. Harnessing the versatility of continuous-flow processes: Selective and efficient reactions. Chem. Rec. 2016, 16, 1018–1033. [Google Scholar] [CrossRef]
- Pan, S.; Yan, S.; Osako, T.; Uozumi, Y. Batch and continuous-flow Huisgen 1,3-dipolar cycloadditions with an amphiphilic resin-supported triazine-based polyethyleneamine dendrimer copper catalyst. ACS Sustain. Chem. Eng. 2017, 5, 10722–10734. [Google Scholar] [CrossRef]
- Manzano, J.S.; Weinstein, Z.B.; Sadow, A.D.; Slowing, I.I. Direct 3D printing of catalytically active structures. ACS Catal. 2017, 7, 7567–7577. [Google Scholar] [CrossRef]
- Hatit, M.Z.C.; Reichenbach, L.F.; Tobin, J.M.; Vilela, F.; Burley, G.A.; Watson, A.J.B. A flow platform for degradation-free CuAAC bioconjugation. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Wen, J.; Wu, K.; Yang, D.; Tian, J.; Huang, Z.; Filatov, A.S.; Lei, A.; Lin, X.-M. Low-pressure flow chemistry of CuAAC click reaction catalyzed by nanoporous AuCu membrane. ACS Appl. Mater. Interfaces 2018, 10, 25930–25935. [Google Scholar] [CrossRef]
- Mandoli, A. Catalyst recycling in continuous flow reactors. In Catalyst Immobilization. Methods and Applications; Benaglia, M., Puglisi, A., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp. 257–306. [Google Scholar]
- Santos de Sa, D.; Bustamante, R.d.A.; Rocha, C.E.R.; Diniz da Silva, V.; Rodrigues, E.J.d.R.; Muller, C.D.B.; Ghavami, K.; Massi, A.; Pandoli, O.G. Fabrication of lignocellulose-based microreactors: Copper-functionalized bamboo for continuous-flow CuAAC click reactions. ACS Sustain. Chem. Eng. 2019, 7, 3267–3273. [Google Scholar] [CrossRef]
- Zhang, C.; Feng, X.; Wang, B.; Mao, Z.; Xu, H.; Zhong, Y.; Zhang, L.; Chen, X.; Sui, X. Nanocellulose sponges as efficient continuous flow reactors. Carbohydr. Polym. 2019, 224, 115184. [Google Scholar] [CrossRef]
- Orlandi, S.; Mandoli, A.; Pini, D.; Salvadori, P. An insoluble polymer-bound bis-oxazoline copper(II) complex: A highly efficient heterogeneous catalyst for the enantioselective Mukaiyama aldol reaction. Angew. Chem. Int. Ed. 2001, 40, 2519–2521. [Google Scholar] [CrossRef]
- Mandoli, A.; Orlandi, S.; Pini, D.; Salvadori, P. Insoluble polystyrene-bound bis(oxazoline): Batch and continuous-flow heterogeneous enantioselective glyoxylate-ene reaction. Tetrahedron Asymmetry 2004, 15, 3233–3244. [Google Scholar] [CrossRef]
- Mandoli, A.; Lessi, M.; Pini, D.; Evangelisti, C.; Salvadori, P. Remarkable efficiency improvement in the preparation of insoluble polymer-bound (IPB) enantioselective catalytic systems by the use of silicone chemistry. Adv. Synth. Catal. 2008, 350, 375–379. [Google Scholar] [CrossRef]
- Mandoli, A.; Garzelli, R.; Orlandi, S.; Pini, D.; Lessi, M.; Salvadori, P. Some factors affecting the catalytic efficiency in the enantioselective cyclopropanation of olefins by the use of insoluble polystyrene-bound bisoxazoline-copper(I) complex. Catal. Today 2009, 140, 51–57. [Google Scholar] [CrossRef]
- Jumde, R.P.; Evangelisti, C.; Mandoli, A.; Scotti, N.; Psaro, R. Aminopropyl-silica-supported Cu nanoparticles: An efficient catalyst for continuous-flow Huisgen azide-alkyne cycloaddition (CuAAC). J. Catal. 2015, 324, 25–31. [Google Scholar] [CrossRef]
- Balerna, A.; Evangelisti, C.; Tiozzo, C. XAFS structural characterization of cu vapour derived catalysts supported on poly-4-vinylpyridine and carbon. X-ray Spectrom. 2017, 46, 82–87. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhu, L. Structural determinants of alkyne reactivity in copper-catalyzed azide-alkyne cycloadditions. Molecules 2016, 21, 1697. [Google Scholar] [CrossRef]
- Svec, F.; Frechet, J.M.J. Kinetic control of pore formation in macroporous polymers. Formation of “molded” porous materials with high flow characteristics for separations or catalysis. Chem. Mater. 1995, 7, 707–715. [Google Scholar] [CrossRef]
- Viklund, C.; Svec, F.; Fréchet, J.M.J.; Irgum, K. Monolithic, “molded”, porous materials with high flow characteristics for separations, catalysis, or solid-phase chemistry: Control of porous properties during polymerization. Chem. Mater. 1996, 8, 744–750. [Google Scholar] [CrossRef]
- Peters, E.C.; Svec, F.; Fréchet, J.M.J. Rigid macroporous polymer monoliths. Adv. Mater. (Weinh. Ger.) 1999, 11, 1169–1181. [Google Scholar] [CrossRef]
- Altava, B.; Burguete, M.I.; Fraile, J.M.; García, J.I.; Luis, S.V.; Mayoral, J.A.; Vicent, M.J. How important is the inert matrix of supported enantiomeric catalysts? Reversal of topicity with two polystyrene backbones. Angew. Chem. Int. Ed. 2000, 39, 1503–1506. [Google Scholar] [CrossRef]
- Sherrington, D.C. Preparation, structure and morphology of polymer supports. Chem. Commun. 1998, 2275–2286. [Google Scholar] [CrossRef]
- Hu, H.; Ohno, A.; Sato, T.; Mase, T.; Uozumi, Y.; Yamada, Y.M.A. Self-assembled polymeric pyridine copper catalysts for Huisgen cycloaddition with alkynes and acetylene gas: Application in synthesis of tazobactam. Org. Process Rev. Dev. 2019, 23, 493–498. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Huang, X.; Zhang, Q. Determination, correlation, and application of sodium l-ascorbate solubility in nine pure solvents and two binary solvents at temperatures from 278.15 to 323.15 k. J. Chem. Eng. Data 2018, 63, 233–245. [Google Scholar] [CrossRef]
- El Kadib, A.; Chimenton, R.; Sachse, A.; Fajula, F.; Galarneau, A.; Coq, B. Functionalized inorganic monolithic microreactors for high productivity in fine chemicals catalytic synthesis. Angew. Chem. Int. Ed. 2009, 48, 4969–4972. [Google Scholar] [CrossRef]
- Megia-Fernandez, A.; Ortega-Muñoz, M.; Lopez-Jaramillo, J.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. Non-magnetic and magnetic supported Copper(I) chelating adsorbents as efficient heterogeneous catalysts and copper scavengers for click chemistry. Adv. Synth. Catal. 2010, 352, 3306–3320. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Campbell-Verduyn, L.; Elsinga, P.H.; Mirfeizi, L.; Dierckx, R.A.; Feringa, B.L. Copper-free ‘click’: 1,3-dipolar cycloaddition of azides and arynes. Org. Biomol. Chem. 2008, 6, 3461–3463. [Google Scholar] [CrossRef]
- Presolski, S.I.; Hong, V.; Cho, S.-H.; Finn, M.G. Tailored ligand acceleration of the Cu-catalyzed azide−alkyne cycloaddition reaction: Practical and mechanistic implications. J. Am. Chem. Soc. 2010, 132, 14570–14576. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Díez-González, S.; Correa, A.; Cavallo, L.; Nolan, S.P. (nhc)Copper(I)-catalyzed [3+2] cycloaddition of azides and mono- or disubstituted alkynes. Chem. Eur. J. 2006, 12, 7558–7564. [Google Scholar] [CrossRef]
- Sarkar, A.; Mukherjee, T.; Kapoor, S. PVP-stabilized copper nanoparticles: A reusable catalyst for “click” reaction between terminal alkynes and azides in nonaqueous solvents. J. Phys. Chem. C 2008, 112, 3334–3340. [Google Scholar] [CrossRef]
- Sharghi, H.; Khalifeh, R.; Doroodmand, M.M. Copper nanoparticles on charcoal for multicomponent catalytic synthesis of 1,2,3-triazole derivatives from benzyl halides or alkyl halides, terminal alkynes and sodium azide in water as a “green” solvent. Adv. Synth. Catal. 2009, 351, 207–218. [Google Scholar] [CrossRef]
- Borgati, T.F.; Alves, R.B.; Teixeira, R.R.; de Freitas, R.P.; Perdigao, T.G.; da Silva, S.F.; dos Santos, A.A.; de Jesus, O.; Bastidas, A. Synthesis and phytotoxic activity of 1,2,3-triazole derivatives. J. Braz. Chem. Soc. 2013, 24, 953–961. [Google Scholar] [CrossRef]
- Chahdoura, F.; Pradel, C.; Gómez, M. Copper(I) oxide nanoparticles in glycerol: A convenient catalyst for cross-coupling and azide–alkyne cycloaddition processes. ChemCatChem 2014, 6, 2929–2936. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).