Efficient Waste to Energy Conversion Based on Co-CeO2 Catalyzed Water-Gas Shift Reaction



 and
and 
Abstract
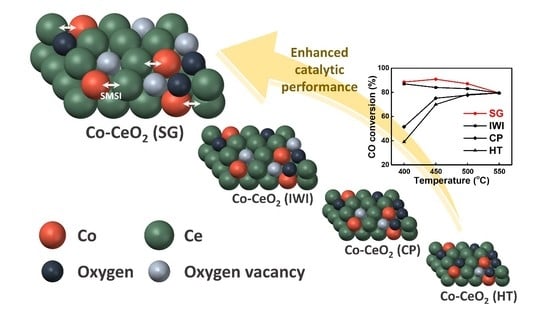
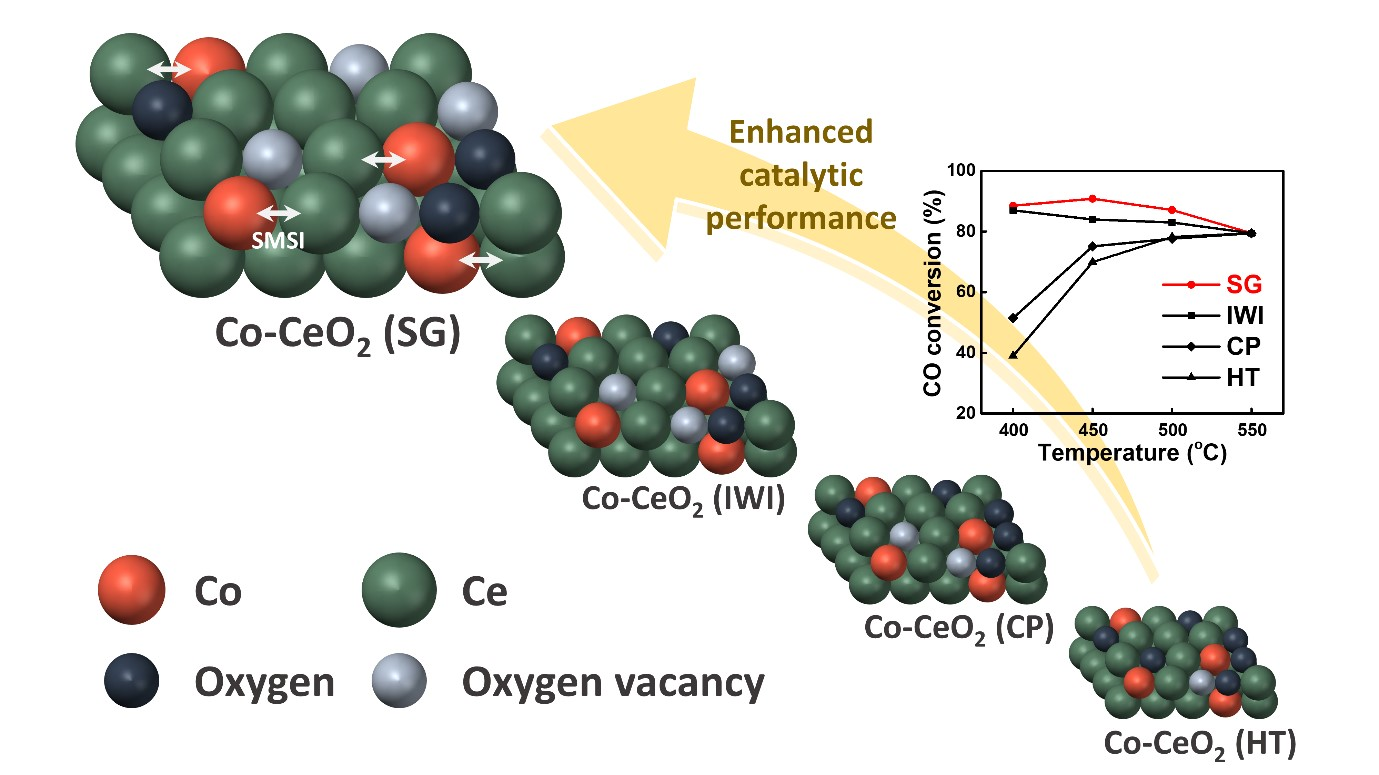
1. Introduction
2. Results and Discussion
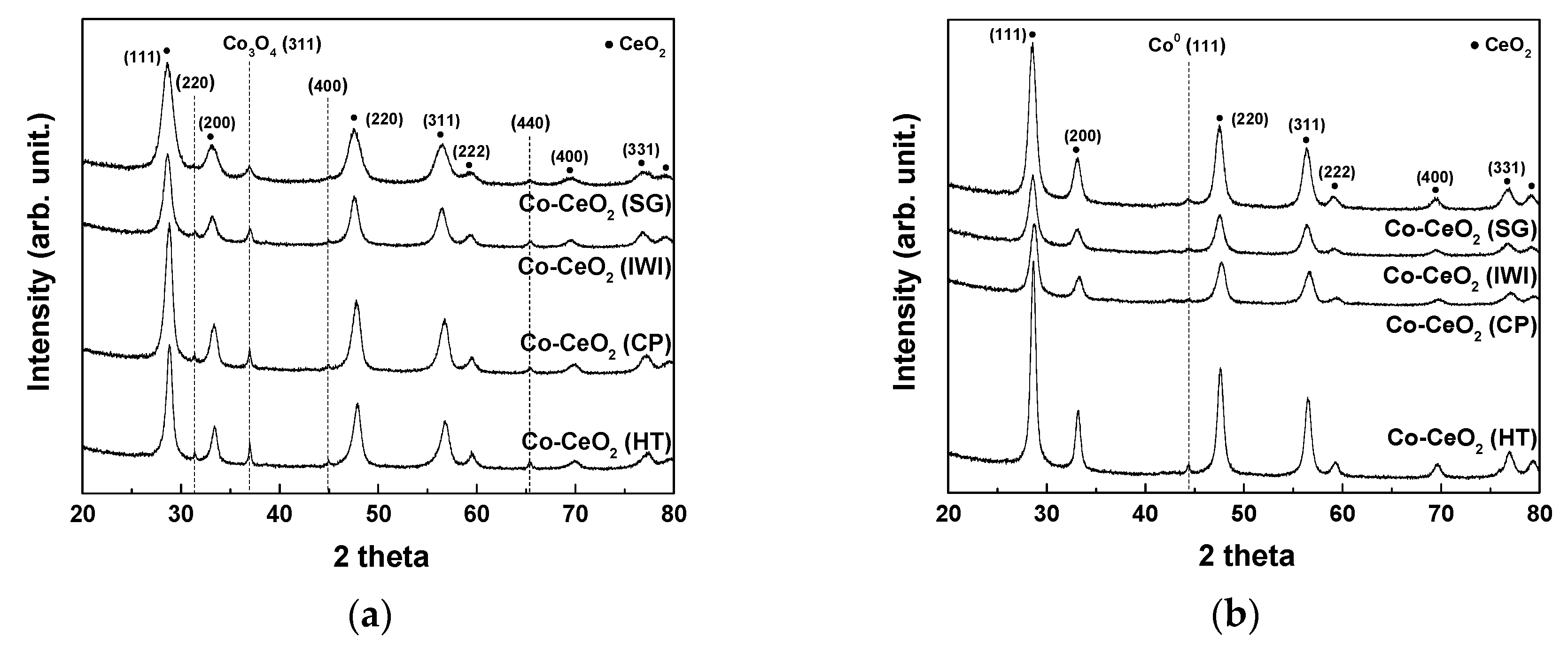
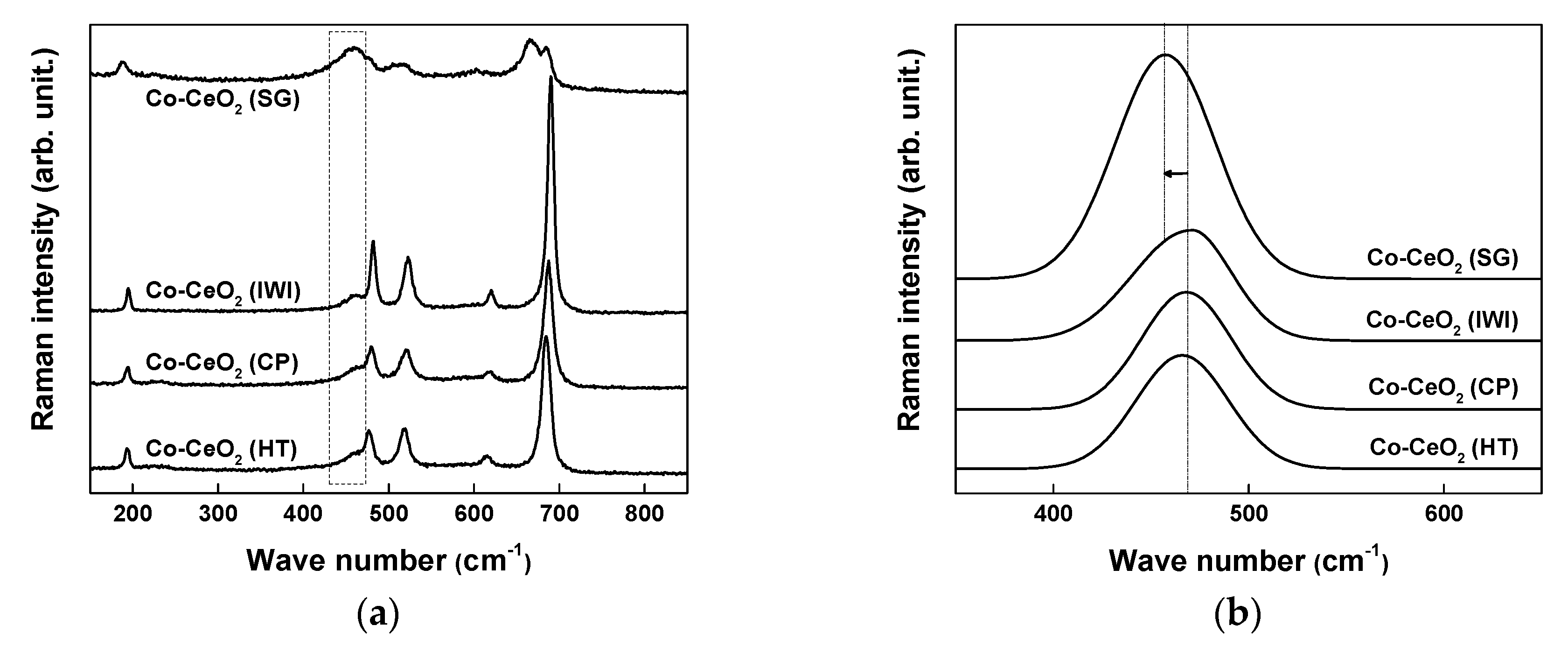
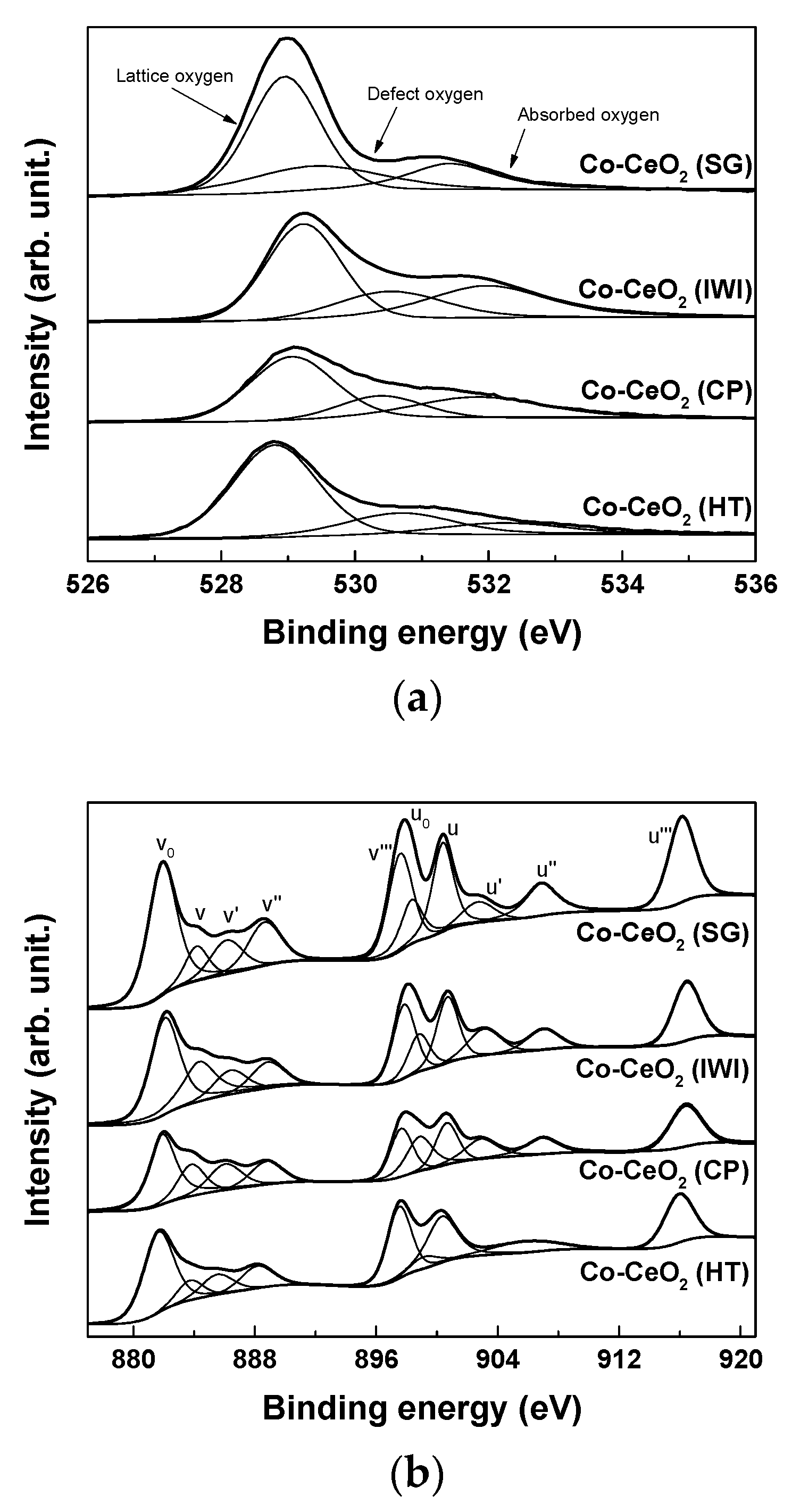
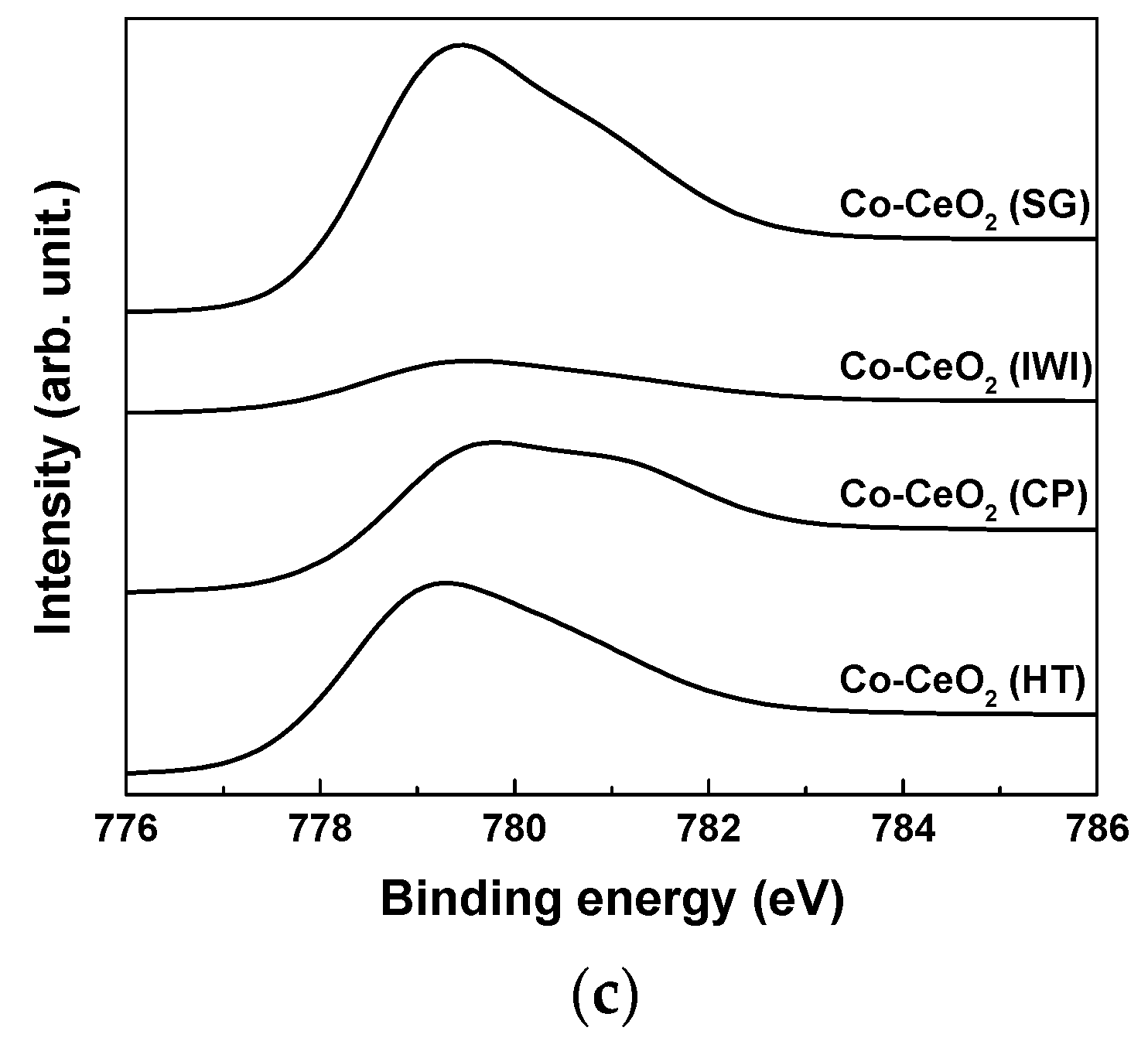
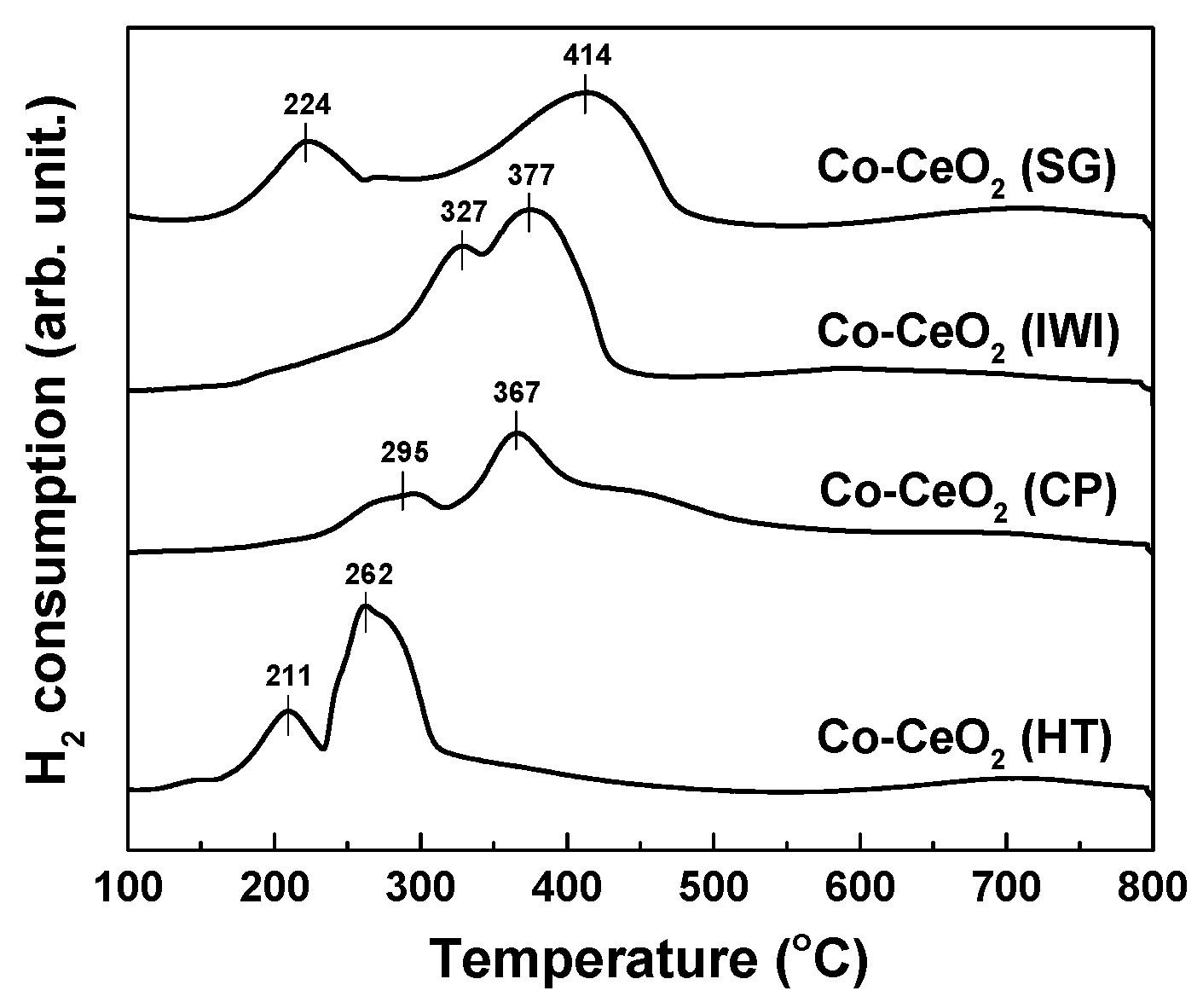
2.1. Catalyst Characterization
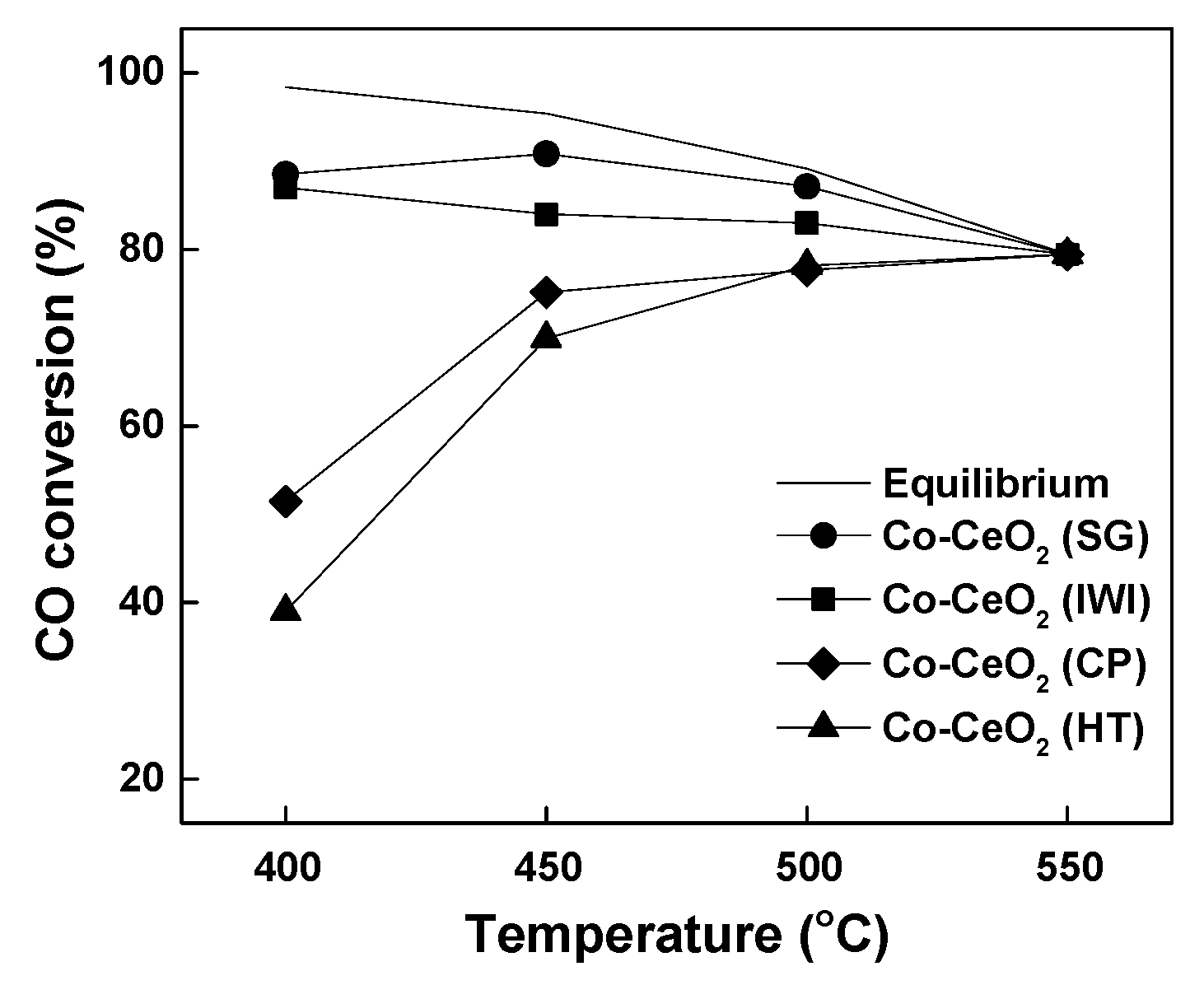
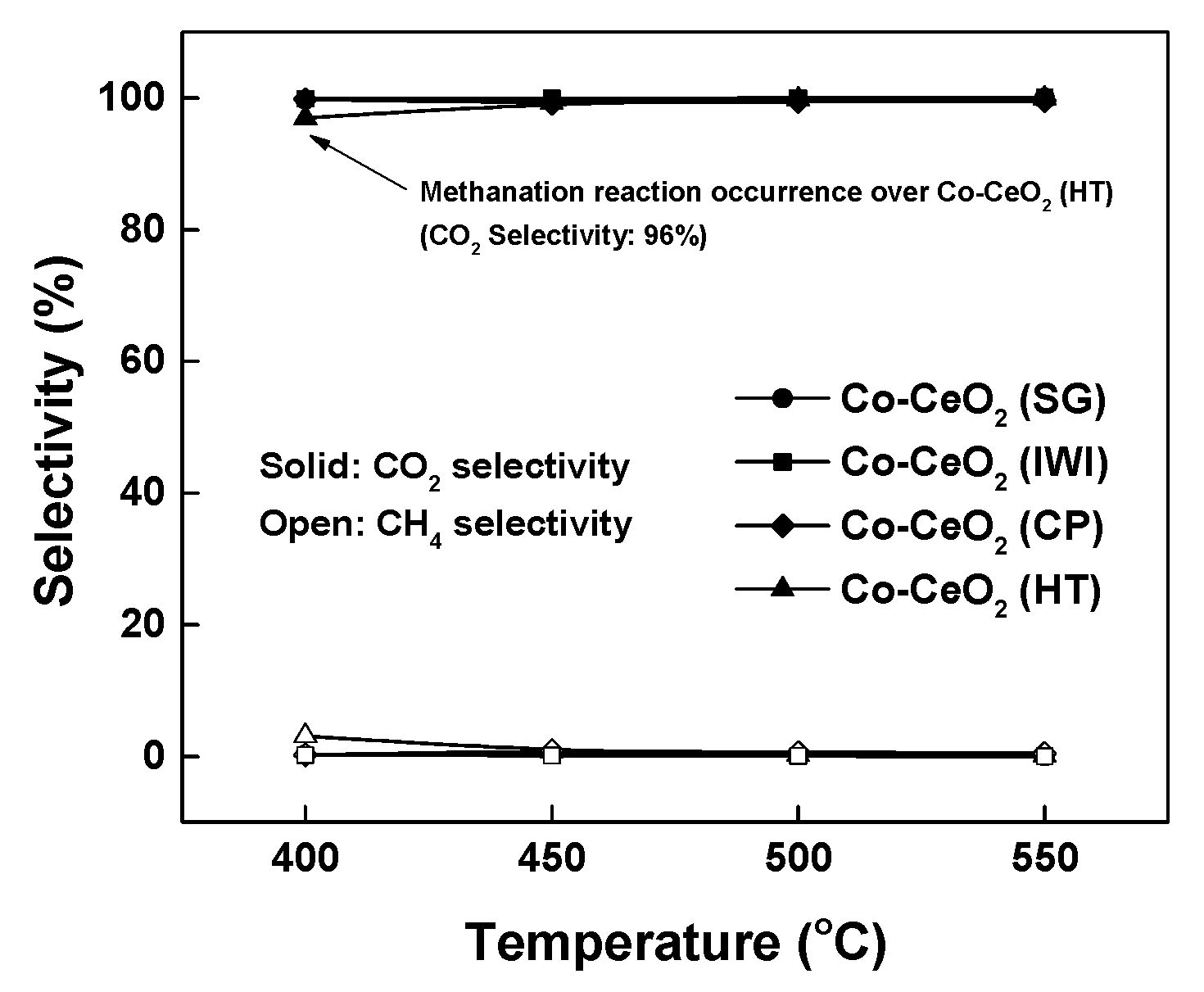
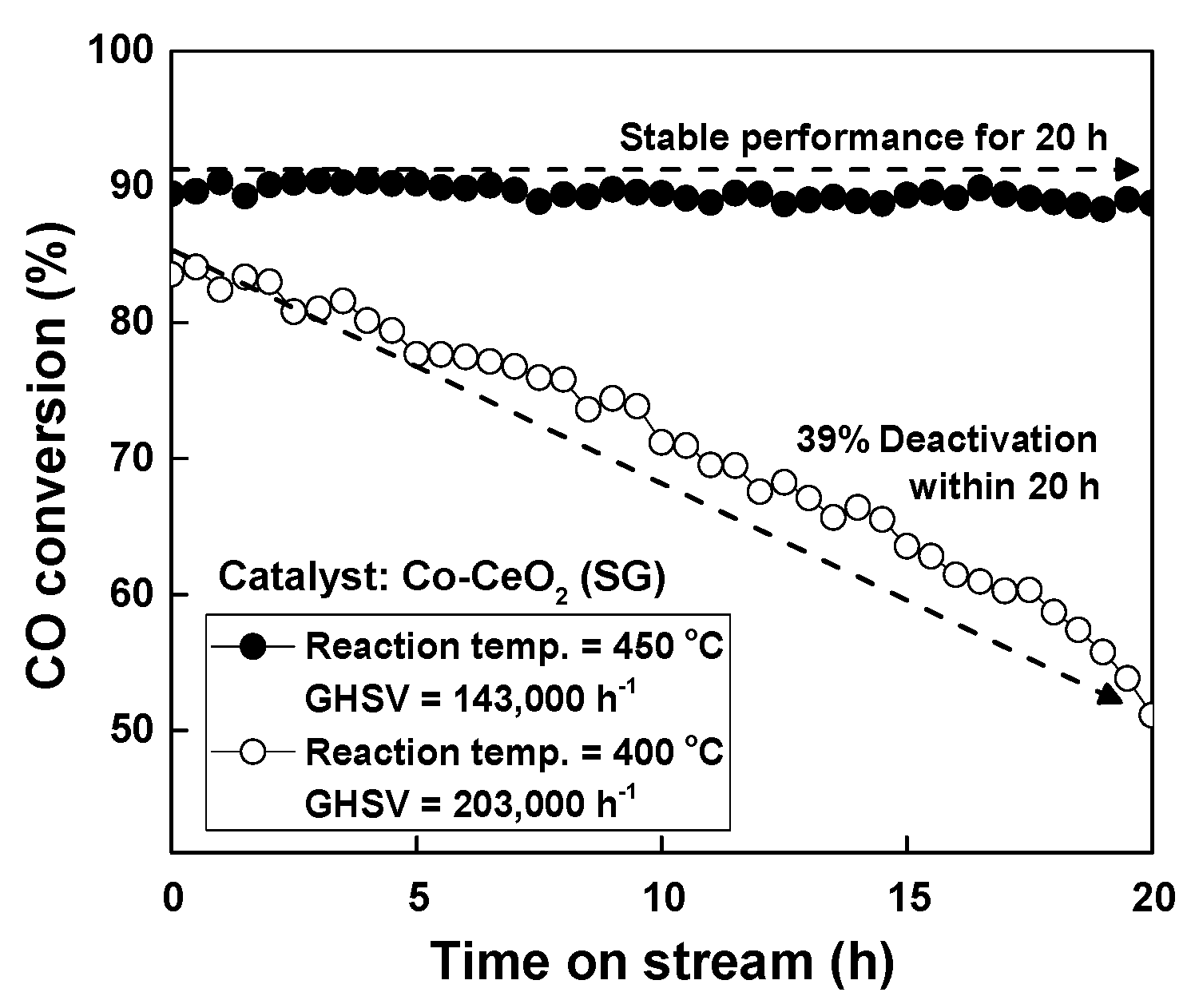
2.2. Reaction Results
3. Materials and Methods
3.1. Preparation of Catalysts
3.2. Catalyst Characterization
3.3. Catalyst Performance Test
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kaza, S.; Yao, L.; Bhada-Tata, P.; Woreden, F.V. What a Waste 2.0: A Global Snapshot of Solid Waste Management to 2050, 1st ed.; World Bank: Washington, DC, USA, 2018; pp. 1–292. [Google Scholar]
- Wilson, D.C.; Rodic, L.; Modak, P.; Soos, R.; Carpintero, A.; Velis, K.; Iyer, M.; Simonett, O. Global Waste Management Outlook; United Nations Environment Programme: Nairobi, Kenya, 2015; pp. 1–332. [Google Scholar]
- Arena, U. Process and technological aspects of municipal solid waste gasification. A review. Waste Manag. 2012, 32, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.G.; Silva, J.N.; Oliveira, J.L.; Machado, C.S. Sustainable energy: A review of gasification technologies. Renew. Sust. Energ. Rev. 2012, 16, 4753–4762. [Google Scholar] [CrossRef]
- Jang, W.-J.; Shim, J.-O.; Jeon, K.-W.; Na, H.-S.; Kim, H.-M.; Lee, Y.-L.; Roh, H.-S.; Jeong, D.-W. Design and scale-up of a Cr-free Fe-Al-Cu catalyst for hydrogen production from waste-derived synthesis gas. Appl. Catal. B Environ. 2019, 249, 72–81. [Google Scholar] [CrossRef]
- Fan, M.-S.; Abdullah, A.Z.; Bhatia, S. Catalytic Technology for Carbon Dioxide Reforming of Methane to Synthesis Gas. ChemCatChem. 2009, 1, 192–208. [Google Scholar] [CrossRef]
- Koo, K.Y.; Lee, S.-H.; Jung, U.H.; Roh, H.-S.; Yoon, W.L. Syngas production via combined steam and carbon dioxide reforming of methane over Ni–Ce/MgAl2O4 catalysts with enhanced coke resistance. Fuel Process. Technol. 2014, 119, 151–157. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.-W.; Lee, Y.-L.; Jang, W.-J.; Shim, J.-O.; Jeon, K.-W.; Rode, C.V.; Roh, H.-S. Chromium free high temperature water–gas shift catalyst for the production of hydrogen from waste derived synthesis gas. Appl. Catal. A Gen. 2016, 522, 21–31. [Google Scholar] [CrossRef]
- Jang, W.-J.; Shim, J.-O.; Kim, H.-M.; Yoo, S.-Y.; Roh, H.-S. A review on dry reforming of methane in aspect of catalytic properties. Catal. Today. 2019, 324, 15–26. [Google Scholar] [CrossRef]
- Jeon, K.-W.; Na, H.-S.; Lee, Y.-L.; Ahn, S.-Y.; Kim, K.-J.; Shim, J.-O.; Jang, W.-J.; Jeong, D.-W.; Nah, I.W.; Roh, H.-S. Catalytic deoxygenation of oleic acid over a Ni-CeZrO2 catalyst. Fuel 2019, 258, 116179–116186. [Google Scholar] [CrossRef]
- Ventura-Espinosa, D.; Sabater, S.; Carretero-Cerdán, A.; Baya, M.; Mata, J.A. High Production of Hydrogen on Demand from Silanes Catalyzed by Iridium Complexes as a Versatile Hydrogen Storage System. ACS Catal. 2018, 8, 2558–2566. [Google Scholar] [CrossRef]
- Ismagilov, Z.R.; Matus, E.V.; Ismagilov, I.Z.; Sukhova, O.B.; Yashnik, S.A.; Ushakov, V.A.; Kerzhentsev, M.A. Hydrogen production through hydrocarbon fuel reforming processes over Ni based catalysts. Catal. Today. 2019, 323, 166–182. [Google Scholar] [CrossRef]
- Kurtz, J.; Sprik, S.; Bradley, T.H. Review of transportation hydrogen infrastructure performance and reliability. Int. J. Hydrogen Energ. 2019, 44, 12010–12023. [Google Scholar] [CrossRef]
- Shim, J.-O.; Na, H.-S.; Jha, A.; Jang, W.-J.; Jeong, D.-W.; Nah, I.W.; Jeon, B.-H.; Roh, H.-S. Effect of preparation method on the oxygen vacancy concentration of CeO2-promoted Cu/γ-Al2O3 catalysts for HTS reactions. Chem. Eng. J. 2016, 306, 908–915. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.-W.; Shim, J.-O.; Jang, W.-J.; Lee, Y.-L.; Rode, C.V.; Roh, H.-S. Hydrogen production by the water-gas shift reaction using CuNi/Fe2O3 catalyst. Catal. Sci. Technol. 2015, 5, 2752–2760. [Google Scholar] [CrossRef]
- Shim, J.-O.; Na, H.-S.; Ahn, S.-Y.; Jeon, K.-W.; Jang, W.-J.; Jeon, B.-H.; Roh, H.-S. An important parameter for synthesis of Al2O3 supported Cu-Zn catalysts in low-temperature water-gas shift reaction under practical reaction condition. Int. J. Hydrogen Energ. 2019, 44, 14853–14860. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Jha, A.; Jang, W.-J.; Shim, J.-O.; Rode, C.V.; Jeon, B.-H.; Bae, J.W.; Roh, H.-S. Effect of alkali and alkaline earth metal on Co/CeO2 catalyst for the water-gas shift reaction of waste derived synthesis gas. Appl. Catal. A Gen. 2018, 551, 63–70. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.-W.; Jang, W.-J.; Lee, Y.-L.; Roh, H.-S. Hydrogen production from water–gas shift reaction over Ni–Cu–CeO2 oxide catalyst: The effect of preparation methods. Int. J. Hydrogen Energ. 2015, 40, 9209–9216. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Jang, W.-J.; Shim, J.-O.; Han, W.-B.; Jeon, K.-W.; Seo, Y.-C.; Roh, H.-S.; Gu, J.H.; Lim, Y.T. A comparison study on high-temperature water–gas shift reaction over Fe/Al/Cu and Fe/Al/Ni catalysts using simulated waste-derived synthesis gas. J. Mater. Cycles Waste. 2014, 16, 650–656. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Jang, W.-J.; Jha, A.; Han, W.-B.; Jeon, K.-W.; Kim, S.-H.; Roh, H.-S. The Effect of Metal on Catalytic Performance over MFe2O4 Catalysts for High Temperature Water-Gas Shift Reaction. J. Nanoelectron. Optoe. 2015, 10, 530–534. [Google Scholar] [CrossRef]
- Lee, D.-W.; Lee, M.S.; Lee, J.Y.; Kim, S.; Eom, H.-J.; Moon, D.J.; Lee, K.-Y. The review of Cr-free Fe-based catalysts for high-temperature water-gas shift reactions. Catal. Today. 2013, 210, 2–9. [Google Scholar] [CrossRef]
- Jha, A.; Jeong, D.-W.; Lee, Y.-L.; Nah, I.W.; Roh, H.-S. Enhancing the catalytic performance of cobalt oxide by doping on ceria in the high temperature water–gas shift reaction. RSC Adv. 2015, 5, 103023–103029. [Google Scholar] [CrossRef]
- Jha, A.; Lee, Y.-L.; Jang, W.-J.; Shim, J.-O.; Jeon, K.-W.; Na, H.-S.; Kim, H.-M.; Roh, H.-S.; Jeong, D.-W.; Jeon, S.G.; et al. Effect of the redox properties of support oxide over cobalt-based catalysts in high temperature water-gas shift reaction. Mol. Catal. 2017, 433, 145–152. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Jha, A.; Jang, W.-J.; Shim, J.-O.; Jeon, K.-W.; Na, H.-S.; Kim, H.-M.; Lee, D.-W.; Yoo, S.-Y.; Jeon, B.-H.; et al. Optimization of Cobalt Loading in Co-CeO2 Catalyst for the High Temperature Water–Gas Shift Reaction. Top. Catal. 2017, 60, 721–726. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Kim, K.-J.; Jang, W.-J.; Shim, J.-O.; Jeon, K.-W.; Na, H.-S.; Kim, H.-M.; Bae, J.W.; Nam, S.C.; Jeon, B.-H.; et al. Increase in stability of BaCo/CeO2 catalyst by optimizing the loading amount of Ba promoter for high-temperature water-gas shift reaction using waste-derived synthesis gas. Renew. Energ. 2020, 145, 2715–2722. [Google Scholar] [CrossRef]
- Fazio, B.; Spadaro, L.; Trunfio, G.; Negro, J.; Arena, F. Raman scattering of MnOx–CeOx composite catalysts: Structural aspects and laser-heating effects. J. Raman Spectrosc. 2011, 42, 1583–1588. [Google Scholar] [CrossRef]
- Arena, F.; Trunfio, G.; Negro, J.; Spadaro, L. Synthesis of highly dispersed MnCeOx catalysts via a novel ‘‘redox-precipitation’’ route. Mater. Res. Bull. 2008, 43, 539–545. [Google Scholar] [CrossRef]
- Arena, F.; Chio, R.D.; Filiciottoa, L.; Trunfio, G.; Espro, C.; Palella, A.; Patti, A.; Spadaro, L. Probing the functionality of nanostructured MnCeOx catalysts in the carbon monoxide oxidation Part II. Reaction mechanism and kinetic modelling. Appl. Catal. B Environ. 2017, 218, 803–809. [Google Scholar] [CrossRef]
- Arena, F.; Chio, R.D.; Espro, C.; Palella, A.; Spadaro, L. A definitive assessment of the CO oxidation pattern of a nanocomposite MnCeOx catalyst. React. Chem. Eng. 2018, 3, 293–300. [Google Scholar] [CrossRef]
- Arena, F.; Chio, R.D.; Espro, C.; Fazio, B.; Palella, A.; Spadaro, L. A New Class of MnCeOx Materials for the Catalytic Gas Exhausts Emission Control: A Study of the CO Model Compound Oxidation. Top. Catal. 2019, 62, 259–265. [Google Scholar] [CrossRef]
- Arena, F.; Famulari, P.; Interdonato, N.; Bonura, G.; Frusteri, F.; Spadaro, L. Physico-chemical properties and reactivity of Au/CeO2 catalysts in total and selective oxidation of CO. Catal. Today. 2006, 116, 384–390. [Google Scholar] [CrossRef]
- Liotta, L.F.; Ousmane, M.; Carlo, G.D.; Pantaleo, G.; Deganello, G.; Marcì, G.; Retailleau, L.; Giroir-Fendler, A. Total oxidation of propene at low temperature over Co3O4–CeO2 mixed oxides: Role of surface oxygen vacancies and bulk oxygen mobility in the catalytic activity. Appl. Catal. A Gen. 2008, 347, 81–88. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Delgado, J.J.; Ivanov, I.; Idakiev, V.; Tabakova, T.; Centeno, M.A.; Odriozola, J.A. Viability of Au/CeO2–ZnO/Al2O3 Catalysts for Pure Hydrogen Production by the Water–Gas Shift Reaction. ChemCatChem. 2014, 6, 1401–1409. [Google Scholar] [CrossRef]
- Reina, T.R.; Moreno, A.A.; Ivanova, S.; Odriozola, J.A.; Centeno, M.A. Influence of Vanadium or Cobalt Oxides on the CO Oxidation Behavior of Au/MOx/CeO2–Al2O3 Systems. ChemCatChem. 2012, 4, 512–520. [Google Scholar] [CrossRef]
- Wang, J.A.; Morales, A.; Bokhimi, X.; Novaro, O.; López, T.; Gómez, R. Cationic and Anionic Vacancies in the Crystalline Phases of Sol−Gel Magnesia−Alumina Catalysts. Chem. Mater. 1999, 11, 308–313. [Google Scholar] [CrossRef]
- Kakihana, M. Invited review “sol-gel” preparation of high temperature superconducting oxides. J. Sol-Gel Sci. Techn. 1996, 6, 7–55. [Google Scholar] [CrossRef]
- Shojaie-Bahaabad, M.; Taheri-Nassaj, E. Economical synthesis of nano alumina powder using an aqueous sol–gel method. Mater. Lett. 2008, 62, 3364–3366. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Matralis, H. Influence of the preparation method on the performance of CuO–CeO2 catalysts for the selective oxidation of CO. Appl. Catal. B Environ. 2005, 56, 87–93. [Google Scholar] [CrossRef]
- Qiao, D.; Lu, G.; Liu, X.; Guo, Y.; Wang, Y.; Guo, Y. Preparation of Ce1−xFexO2 solid solution and its catalytic performance for oxidation of CH4 and CO. J. Mater. Sci. 2011, 46, 3500–3506. [Google Scholar] [CrossRef]
- Megarajan, S.K.; Rayalu, S.; Teraoka, Y.; Labhsetwar, N. High NO oxidation catalytic activity on non-noble metal based cobalt-ceria catalyst for diesel soot oxidation. J. Mol. Catal. A Chem. 2014, 385, 112–118. [Google Scholar] [CrossRef]
- Na, H.-S.; Ahn, S.-Y.; Shim, J.-O.; Jeon, K.-W.; Kim, H.-M.; Lee, Y.-L.; Jang, W.-J.; Jeon, B.-H.; Roh, H.-S. Effect of precipitation on physico-chemical and catalytic properties of Cu-Zn-Al catalyst for water-gas shift reaction. Korean, J. Chem. Eng. 2019, 36, 1243–1248. [Google Scholar] [CrossRef]
- Shim, J.-O.; Jeon, K.-W.; Jang, W.-J.; Na, H.-S.; Cho, J.-W.; Kim, H.-M.; Lee, Y.-L.; Jeong, D.-W.; Roh, H.-S.; Ko, C.H. Facile production of biofuel via solvent-free deoxygenation of oleic acid using a CoMo catalyst. Appl. Catal. B Environ. 2018, 239, 644–653. [Google Scholar] [CrossRef]
- Shim, J.-O.; Jang, W.-J.; Jeon, K.-W.; Lee, D.-W.; Na, H.-S.; Kim, H.-M.; Lee, Y.-L.; Yoo, S.-Y.; Jeon, B.-H.; Roh, H.-S.; et al. Petroleum like biodiesel production by catalytic decarboxylation of oleic acid over Pd/Ce-ZrO2 under solvent-free condition. Appl. Catal. A Gen. 2018, 563, 163–169. [Google Scholar] [CrossRef]
- Banerjee, A.M.; Pai, M.R.; Tewari, R.; Raje, N.; Tripathi, A.K.; Bharadwaj, S.R.; Das, D. A comprehensive study on Pt/Al2O3 granular catalyst used for sulfuric acid decomposition step in sulfur–iodine thermochemical cycle: Changes in catalyst structure, morphology and metal-support interaction. Appl. Catal. B Environ. 2015, 162, 327–337. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.; Huang, J.; Lee, J.; Kim, D.H.; Frenkel, A.I.; Kim, T. Effects of Molecular and Electronic Structures in CoOx/CeO2 Catalysts on NO Reduction by CO. J. Phys. Chem. C. 2019, 123, 7166–7177. [Google Scholar] [CrossRef]
- Gómez, L.E.; Boix, A.V.; Miró, E.E. Co/ZrO2, Co/CeO2 and MnCoCe structured catalysts for COPrOx. Catal. Today. 2013, 216, 246–253. [Google Scholar] [CrossRef]
- Gómez, L.E.; Múnera, J.F.; Sollier, B.M.; Miró, E.E.; Boix, A.V. Raman in situ characterization of the species present in Co/CeO2 and Co/ZrO2 catalysts during the COPrOx reaction. Int. J. Hydrogen Energ. 2016, 41, 4993–5002. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Gamarra, D.; Fernández-García, M.; Wang, X.Q.; Hanson, J.C.; Rodriguez, J.A. Comparative study on redox properties of nanosized CeO2 and CuO/CeO2 under CO/O2. J. Catal. 2006, 240, 1–7. [Google Scholar] [CrossRef]
- Jang, W.-J.; Kim, H.-M.; Shim, J.-O.; Yoo, S.-Y.; Jeon, K.-W.; Na, H.-S.; Lee, Y.-L.; Jeong, D.-W.; Bae, J.W.; Nah, I.W.; et al. Key properties of Ni-MgO-CeO2, Ni-MgO-ZrO2, and Ni-MgO-Ce(1−x)Zr(x)O2 catalysts for the reforming of methane with carbon dioxide. Green Chem. 2018, 20, 1621–1633. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Idriss, H.; Diagne, C.; Hindermann, J.P.; Kiennemann, A.; Barteau, M.A. Reactions of Acetaldehyde on CeO2 and CeO2-Supported Catalysts. J. Catal. 1995, 155, 219–237. [Google Scholar] [CrossRef]
- Kumar, P.A.; Tanwar, M.D.; Russo, N.; Pirone, R.; Fino, D. Synthesis and catalytic properties of CeO2 and Co/CeO2 nanofibres for diesel soot combustion. Catal. Today. 2012, 184, 279–287. [Google Scholar] [CrossRef]
- Lin, S.S.-Y.; Kim, D.H.; Engelhard, M.H.; Ha, S.Y. Water-induced formation of cobalt oxides over supported cobalt/ceria–zirconia catalysts under ethanol-steam conditions. J. Catal. 2010, 273, 229–235. [Google Scholar] [CrossRef]
- Aramendía, M.A.; Colmenares, J.C.; Marinas, A.; Marinas, J.M.; Moreno, J.M.; Navío, J.A.; Urbano, F.J. Effect of the redox treatment of Pt/TiO2 system on its photocatalytic behaviour in the gas phase selective photooxidation of propan-2-ol. Catal. Today. 2007, 128, 235–244. [Google Scholar] [CrossRef]
- Weerachawanasak, P.; Mekasuwandumrong, O.; Arai, M.; Fujita, S.-I.; Praserthdam, P.; Panpranot, J. Effect of strong metal–support interaction on the catalytic performance of Pd/TiO2 in the liquid-phase semihydrogenation of phenylacetylene. J. Catal. 2009, 262, 199–205. [Google Scholar] [CrossRef]
- Kim, W.-J.; Moon, S.H. Modified Pd catalysts for the selective hydrogenation of acetylene. Catal. Today. 2012, 185, 2–16. [Google Scholar] [CrossRef]
- Fichtl, M.B.; Schumann, J.; Kasatkin, I.; Jacobsen, N.; Behrens, M.; Schlögl, R.; Muhler, M.; Hinrichsen, O. Counting of Oxygen Defects versus Metal Surface Sites in Methanol Synthesis Catalysts by Different Probe Molecules. Angew. Chem. Int. Ed. 2014, 53, 7043–7047. [Google Scholar] [CrossRef]
- Wang, N.; Qian, W.; Chu, W.; Wei, F. Crystal-plane effect of nanoscale CeO2 on the catalytic performance of Ni/CeO2 catalysts for methane dry reforming. Catal. Sci. Technol. 2016, 6, 3594–3605. [Google Scholar] [CrossRef]
- Feng, X.; Guo, J.; Wen, X.; Xu, M.; Chu, Y.; Yuan, S. Enhancing performance of Co/CeO2 catalyst by Sr doping for catalytic combustion of toluene. Appl. Surf. Sci. 2018, 445, 145–153. [Google Scholar] [CrossRef]
- Zhan, Y.; Liu, Y.; Peng, X.; Zhao, W.; Zhang, Y.; Wang, X.; Au, C.; Jiang, L. Molecular-level understanding of reaction path optimization as a function of shape concerning the metal–support interaction effect of Co/CeO2 on water-gas shift catalysis. Catal. Sci. Technol. 2019, 9, 4928–4937. [Google Scholar] [CrossRef]
- Gamboa-Rosales, N.K.; Ayastuy, J.L.; Iglesias-González, A.; González-Marcos, M.P.; Gutiérrez-Ortiz, M.A. Oxygen-enhanced WGS over ceria-supported Au–Co3O4 bimetallic catalysts. Chem. Eng. J. 2012, 207–208, 49–56. [Google Scholar] [CrossRef]
- Tepamatr, P.; Laosiripojana, N.; Sesuk, T.; Charojrochkul, S. Effect of samarium and praseodymium addition on water gas shift performance of Co/CeO2 catalysts. J. Rare. Earth. 2019, in press. [Google Scholar] [CrossRef]
- Jianqiang, W.; Meiqing, S.; Jun, W.; Jidong, G.; Jie, M.; Shuangxi, L. Effect of cobalt doping on ceria-zirconia mixed oxide: Structural characteristics, oxygen storage/release capacity and three-way catalytic performance. J. Rare. Earth. 2012, 30, 878–883. [Google Scholar] [CrossRef]
- Li, C.; Li, Z.; Park, S.B.; Hong, G.H.; Park, J.S.; Oh, H.Y.; Kim, J.M. Ordered Mesoporous Cu–Co–CeO2 Catalyst for Water-Gas Shift Reaction at High Temperature. J. Nanosci. Nanotechno. 2017, 17, 8149–8152. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Surface Area (m2/g) a | Dispersion (%) b | O22−/O2− c | Ce3+ (%) c | OSC (10-5 gmol/gcat) d | ||
|---|---|---|---|---|---|---|---|
| Fresh | Used | Fresh | Used | ||||
| Co–CeO2 (SG) | 30 | 24 | 1.61 | 0.42 (74% ↓) | 0.50 | 44.6 | 3.9 |
| Co–CeO2 (IWI) | 82 | 65 | 1.91 | 0.17 (91% ↓) | 0.43 | 43.8 | 2.8 |
| Co–CeO2 (CP) | 82 | 73 | 1.09 | 0.15 (86% ↓) | 0.37 | 40.7 | 2.6 |
| Co–CeO2 (HT) | 69 | 48 | 2.14 | 0.68 (68% ↓) | 0.29 | 36.9 | 2.5 |
| Catalysts (Preparation Method) | Reaction Condition | Conversion | Reference | |||
|---|---|---|---|---|---|---|
| GHSV or Flow Rate | Feed Gas Ratio | Press (bar) | Reaction Temp. (°C) | CO Conv. [Equil. CO Conv.] (%) | ||
| 15%Co–CeO2 (Sol-gel method) | 143,000 h−1 | 17.02% CO, 9.55% CO2, 1.03% CH4, 13.14% H2, 55.20% H2O, 4.06 %N2 | 1 | 450 | ~90 [~95] | This work |
| 10%Co/CeO2-spindles (Wet impregnation method) | 6000 h−1 | 7.5% CO, 3% CO2, 27.5% H2, 50% H2O, 12% N2 | 1 | 300 | ~91 [~95] | [60] |
| 1%Au–10%Co3O4/CeO2 (Deposition precipitation method) | 12,000 h−1 | 4% CO, 3% CO2, 37.9 H2, 9.4% H2O, 45.7% He | 1 | 350 | ~30 [~60] | [61] |
| 1%Co/Ce5%SmO (Impregnation method | 100 mL min−1 | 5% CO, 10% H2O, 85% N2 | 1 | 450 | ~86 [~88] | [62] |
| Pd/Co0.1Ce0.6Zr0.3Ox (Impregnation method) | 50,000 h−1 | 2% CO, 5% H2O, 93% N2 | 1 | 400 | ~95 | [63] |
| Cu0.1Co0.1Ce0.8O2 (nano-replication method) | 64 mL min−1 | 3% CO balanced with He and H2O | 1 | 350 | ~99 | [64] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-J.; Lee, Y.-L.; Na, H.-S.; Ahn, S.-Y.; Shim, J.-O.; Jeon, B.-H.; Roh, H.-S. Efficient Waste to Energy Conversion Based on Co-CeO2 Catalyzed Water-Gas Shift Reaction. Catalysts 2020, 10, 420. https://doi.org/10.3390/catal10040420
Kim K-J, Lee Y-L, Na H-S, Ahn S-Y, Shim J-O, Jeon B-H, Roh H-S. Efficient Waste to Energy Conversion Based on Co-CeO2 Catalyzed Water-Gas Shift Reaction. Catalysts. 2020; 10(4):420. https://doi.org/10.3390/catal10040420
Chicago/Turabian StyleKim, Kyoung-Jin, Yeol-Lim Lee, Hyun-Suk Na, Seon-Yong Ahn, Jae-Oh Shim, Byong-Hun Jeon, and Hyun-Seog Roh. 2020. "Efficient Waste to Energy Conversion Based on Co-CeO2 Catalyzed Water-Gas Shift Reaction" Catalysts 10, no. 4: 420. https://doi.org/10.3390/catal10040420
APA StyleKim, K.-J., Lee, Y.-L., Na, H.-S., Ahn, S.-Y., Shim, J.-O., Jeon, B.-H., & Roh, H.-S. (2020). Efficient Waste to Energy Conversion Based on Co-CeO2 Catalyzed Water-Gas Shift Reaction. Catalysts, 10(4), 420. https://doi.org/10.3390/catal10040420