Nitric Acid Functionalization of Petroleum Coke to Access Inherent Sulfur



Abstract
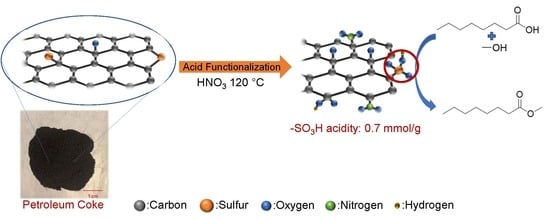
1. Introduction
2. Results and Discussion
2.1. Chemical Properties of Petcoke Treated with Various Acids
2.2. Physical Properties of Petcoke Treated with Various Acids
2.3. Mechanisms of Surface Group Formation
2.4. Esterification Reactions
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Sample Preparation
3.3. Characterization of Material
3.4. Esterification Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manasrah, A.D.; Nassar, N.N.; Ortega, L.C. Conversion of petroleum coke into valuable products using oxy-cracking technique. Fuel 2018, 215, 865–878. [Google Scholar] [CrossRef]
- Hill, J.M. Sustainable and/or waste sources for catalysts: Porous carbon development and gasification. Catal. Today 2017, 285, 204–210. [Google Scholar] [CrossRef]
- Shan, J.; Huang, J.J.; Li, J.Z.; Li, G.; Zhao, J.T.; Fang, Y.T. Insight into transformation of sulfur species during KOH activation of high sulfur petroleum coke. Fuel 2018, 215, 258–265. [Google Scholar] [CrossRef]
- Tripathi, N.; Singh, R.S.; Hills, C.D. Microbial removal of sulphur from petroleum coke (petcoke). Fuel 2019, 235, 1501–1505. [Google Scholar] [CrossRef]
- Zhao, P.J.; Ma, C.; Wang, J.T.; Qiao, W.M.; Ling, L.C. Almost total desulfurization of high-sulfur petroleum coke by Na2CO3 -promoted calcination combined with ultrasonic-assisted chemical oxidation. Xinxing Tan Cailiao/New Carbon Mater. 2018, 33, 587–594. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, W.; Wei, K.; Wu, J.; Li, S.; Zhang, C.; Yu, Z.; Xie, K.; Yu, J. Detailed vacuum-assisted desulfurization of high-sulfur petroleum coke. Sep. Purif. Technol. 2017, 175, 115–121. [Google Scholar] [CrossRef]
- Agarwal, P.; Sharma, D.K. Studies on the desulfurization of petroleum coke by organorefining and other chemical and biochemical techniques under milder ambient pressure conditions. Pet. Sci. Technol. 2011, 29, 1482–1493. [Google Scholar] [CrossRef]
- Wright, P.A. Acid Catalysts. In Encyclopedia of Materials: Science and Technology; Elsevier: Amsterdam, The Netherlands, 2001; pp. 1–6. [Google Scholar]
- Zeng, D.; Zhang, Q.; Chen, S.; Liu, S.; Wang, G. Synthesis porous carbon-based solid acid from rice husk for esterification of fatty acids. Microporous Mesoporous Mater. 2016, 219, 54–58. [Google Scholar] [CrossRef]
- Ngaosuwan, K.; Goodwin, J.G.; Prasertdham, P. A green sulfonated carbon-based catalyst derived from coffee residue for esterification. Renew. Energy 2016, 86, 262–269. [Google Scholar] [CrossRef]
- Kastner, J.R.; Miller, J.; Geller, D.P.; Locklin, J.; Keith, L.H.; Johnson, T. Catalytic esterification of fatty acids using solid acid catalysts generated from biochar and activated carbon. Catal. Today 2012, 190, 122–132. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, H.; Ao, Z.; Zhang, S. Hydrothermal carbon enriched with sulfonic and carboxyl groups as an efficient solid acid catalyst for butanolysis of furfuryl alcohol. Catal. Commun. 2019, 123, 109–113. [Google Scholar] [CrossRef]
- Trejda, M.; Nurwita, A.; Kryszak, D. Synthesis of solid acid catalysts for esterification with the assistance of elevated pressure. Microporous Mesoporous Mater. 2019, 278, 115–120. [Google Scholar] [CrossRef]
- Russo, P.A.; Antunes, M.M.; Neves, P.; Wiper, P.V.; Fazio, E.; Neri, F.; Barreca, F.; Mafra, L.; Pillinger, M.; Pinna, N.; et al. Solid acids with SO3H groups and tunable surface properties: Versatile catalysts for biomass conversion. J. Mater. Chem. A 2014, 2, 11813–11824. [Google Scholar] [CrossRef]
- Zeng, D.; Liu, S.; Gong, W.; Wang, G.; Qiu, J.; Tian, Y. Acid properties of solid acid from petroleum coke by chemical activation and sulfonation. Communications 2013, 40, 5–8. [Google Scholar] [CrossRef]
- Wu, M.; Wang, Y.; Wang, D.; Tan, M.; Li, P.; Wu, W.; Tsubaki, N. SO3H-modified petroleum coke derived porous carbon as an efficient solid acid catalyst for esterification of oleic acid. J. Porous Mater. 2016, 23, 263–271. [Google Scholar] [CrossRef]
- Landwehr, J.; Steldinger, H.; Etzold, B.J.M. Introducing sulphur surface groups in microporous carbons: A mechanistic study on carbide derived carbons. Catal. Today 2018, 301, 191–195. [Google Scholar] [CrossRef]
- Wu, J.; Montes, V.; Virla, L.D.; Hill, J.M. Impacts of amount of chemical agent and addition of steam for activation of petroleum coke with KOH or NaOH. Fuel Process. Technol. 2018, 181, 53–60. [Google Scholar] [CrossRef]
- Xiao, Y.; Hill, J.M. Chemosphere Solid acid catalysts produced by sulfonation of petroleum coke: Dominant role of aromatic hydrogen. Chemosphere 2020, 248, 125981. [Google Scholar] [CrossRef]
- Wepasnick, K.A.; Smith, B.A.; Schrote, K.E.; Wilson, H.K.; Diegelmann, S.R.; Fairbrother, D.H. Surface and structural characterization of multi-walled carbon nanotubes following different oxidative treatments. Carbon 2011, 49, 24–36. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Ferro-García, M.A.; Joly, J.P.; Bautista-Toledo, I.; Carrasco-Marín, F.; Rivera-Utrilla, J. Activated Carbon Surface Modifications by Nitrcc Acid, Hydrogen Peroxide, and Ammonium Peroxydisulfate Treatmenss. Langmuir 1995, 11, 4386–4392. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Carrasco-Marín, F.; Parejo-Pérez, C.; López Ramón, M.V. Dehydration of methanol to dimethyl ether catalyzed by oxidized activated carbons with varying surface acidic character. Carbon 2001, 39, 869–875. [Google Scholar] [CrossRef]
- Saleh, T.A. The influence of treatment temperature on the acidity of MWCNT oxidized by HNO3 or a mixture of HNO3/H2SO4. Appl. Surf. Sci. 2011, 257, 7746–7751. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Lin, W.H.; Chang, Y.C. The influence of treatment duration on multi-walled carbon nanotubes functionalized by H2SO4/HNO3 oxidation. Appl. Surf. Sci. 2011, 257, 2401–2410. [Google Scholar] [CrossRef]
- Toebes, M.L.; Van Heeswijk, J.M.P.; Bitter, J.H.; Jos van Dillen, A.; De Jong, K.P. The influence of oxidation on the texture and the number of oxygen-containing surface groups of carbon nanofibers. Carbon 2004, 42, 307–315. [Google Scholar] [CrossRef]
- Tammer, M.G. Sokrates: Infrared and Raman characteristic group frequencies: Tables and charts. Colloid Polym. Sci. 2004, 283, 235. [Google Scholar] [CrossRef]
- Yao, H.; You, Z.; Li, L.; Goh, S.W.; Lee, C.H.; Yap, Y.K.; Shi, X. Rheological properties and chemical analysis of nanoclay and carbon microfiber modified asphalt with Fourier transform infrared spectroscopy. Constr. Build. Mater. 2013, 38, 327–337. [Google Scholar] [CrossRef]
- Zawadzki, J. IR spectroscopy investigations of acidic character of carbonaceous films oxidized with HNO3 solution. Carbon 1981, 19, 19–25. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Schuttlefield, J.; Jensen, J.H.; Grassian, V.H. FTIR spectroscopy combined with quantum chemical calculations to investigate adsorbed nitrate on aluminium oxide surfaces in the presence and absence of co-adsorbed water. Phys. Chem. Chem. Phys. 2007, 9, 4970–4980. [Google Scholar] [CrossRef]
- Figueiredo, J.; Pereira, M.F.; Freitas, M.M.; Órfão, J.J. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Stein, A.; Wang, Z.; Fierke, M.A. Functionalization of porous carbon materials with designed pore architecture. Adv. Mater. 2009, 21, 265–293. [Google Scholar] [CrossRef]
- Collins, J.; Ngo, T.; Qu, D.; Foster, M. Spectroscopic investigations of sequential nitric acid treatments on granulated activated carbon: Effects of surface oxygen groups on π density. Carbon 2013, 57, 174–183. [Google Scholar] [CrossRef]
- Phillips, C.R.; Chao, K.S. Desulphurization of Athabasca petroleum coke by (a) chemical oxidation and (b) solvent extraction. Fuel 1977, 56, 70–72. [Google Scholar] [CrossRef]
- Sun, D.; Yang, J.; Yan, X. Hierarchically porous and nitrogen, sulfur-codoped graphene-like microspheres as a high capacity anode for lithium ion batteries. Chem. Commun. 2015, 51, 2134–2137. [Google Scholar] [CrossRef] [PubMed]
- Thushari, I.; Babel, S. Sustainable utilization of waste palm oil and sulfonated carbon catalyst derived from coconut meal residue for biodiesel production. Bioresour. Technol. 2018, 248, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.A. University of T. Lange’s Handbook of Chemistry, 15th ed.; Esposito, R., Ed.; McGRAW-HILL, INC: New York, NY, USA, 1999. [Google Scholar]
- Yang, J.; Yue, L.; Lin, B.; Wang, L.; Zhao, Y.; Lin, Y.; Chang, K.; DaCosta, H.; Hu, X. CO2 Adsorption of Nitrogen-Doped Carbons Prepared from Nitric Acid Preoxidized Petroleum Coke. Energy Fuels 2017, 31, 11060–11068. [Google Scholar] [CrossRef]
- Gęsikiewicz-Puchalska, A.; Zgrzebnicki, M.; Michalkiewicz, B.; Narkiewicz, U.; Morawski, A.W.; Wrobel, R.J. Improvement of CO2 uptake of activated carbons by treatment with mineral acids. Chem. Eng. J. 2017, 309, 159–171. [Google Scholar] [CrossRef]
- Thommes, M. Physical adsorption characterization of nanoporous materials. Chemie-Ingenieur-Technik 2010, 82, 1059–1073. [Google Scholar] [CrossRef]
- Ternero-Hidalgo, J.J.; Rosas, J.M.; Palomo, J.; Valero-Romero, M.J.; Rodríguez-Mirasol, J.; Cordero, T. Functionalization of activated carbons by HNO3treatment: Influence of phosphorus surface groups. Carbon 2016, 101, 409–419. [Google Scholar] [CrossRef]
- Song, X.; Liu, H.; Cheng, L.; Qu, Y. Surface modification of coconut-based activated carbon by liquid-phase oxidation and its effects on lead ion adsorption. Desalination 2010, 255, 78–83. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Carrasco-Marín, F.; Maldonado-Hódar, F.J.; Rivera-Utrilla, J. Effects of non-oxidant and oxidant acid treatments on the surface properties of an activated carbon with very low ash content. Carbon 1998, 36, 145–151. [Google Scholar] [CrossRef]
- Johra, F.T.; Lee, J.W.; Jung, W.G. Facile and safe graphene preparation on solution based platform. J. Ind. Eng. Chem. 2014, 20, 2883–2887. [Google Scholar] [CrossRef]
- Paton-Carrero, A.; Valverde, J.L.; Garcia-Alvarez, E.; Lavin-Lopez, M.P.; Romero, A. Influence of the oxidizing agent in the synthesis of graphite oxide. J. Mater. Sci. 2019, 55, 2333–2342. [Google Scholar] [CrossRef]
- Cao, J.; He, P.; Mohammed, M.A.; Zhao, X.; Young, R.J.; Derby, B.; Kinloch, I.A.; Dryfe, R.A.W. Two-Step Electrochemical Intercalation and Oxidation of Graphite for the Mass Production of Graphene Oxide. J. Am. Chem. Soc. 2017, 139, 17446–17456. [Google Scholar] [CrossRef]
- Chingombe, P.; Saha, B.; Wakeman, R.J. Surface modification and characterisation of a coal-based activated carbon. Carbon 2005, 43, 3132–3143. [Google Scholar] [CrossRef]
- Vinke, P.; van der Eijk, M.; Verbree, M.; Voskamp, A.F.; van Bekkum, H. Modification of the surfaces of a gas-activated cabron and a chemically activated carbon with nitric acid. Carbon 1994, 32, 675–686. [Google Scholar] [CrossRef]
- D’Alessandro, N.; Tonucci, L.; Bonetti, M.; Di Deo, M.; Bressan, M.; Morvillo, A. Oxidation of dibenzothiophene by hydrogen peroxide or monopersulfate and metal-sulfophthalocyanine catalysts: An easy access to biphenylsultone or 2-(2′-hydroxybiphenyl)sulfonate under mild conditions. New J. Chem. 2003, 27, 989–993. [Google Scholar] [CrossRef]
- Ouellette, R.J.; Rawn, J.D.; Towson, U. Organic Chemistry: Structure, Mechanism, and Synthesis; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Hara, M.; Yoshida, T.; Takagaki, A.; Takata, T.; Kondo, J.N.; Hayashi, S.; Domen, K. A carbon material as a strong protonic acid. Angew. Chem. Int. Ed. 2004, 43, 2955–2958. [Google Scholar] [CrossRef]
- Ogino, I.; Suzuki, Y.; Mukai, S.R. Esterification of levulinic acid with ethanol catalyzed by sulfonated carbon catalysts: Promotional effects of additional functional groups. Catal. Today 2018, 314, 62–69. [Google Scholar] [CrossRef]
- Melero, J.A.; Morales, G.; Iglesias, J.; Paniagua, M.; Hernández, B.; Penedo, S. Efficient conversion of levulinic acid into alkyl levulinates catalyzed by sulfonic mesostructured silicas. Appl. Catal. A Gen. 2013, 466, 116–122. [Google Scholar] [CrossRef]
- Patil, C.R.; Niphadkar, P.S.; Bokade, V.V.; Joshi, P.N. Esterification of levulinic acid to ethyl levulinate over bimodal micro-mesoporous H/BEA zeolite derivatives. Catal. Commun. 2014, 43, 188–191. [Google Scholar] [CrossRef]
- Yamaguchi, D.; Hayashi, S.; Suganuma, S.; Kato, H.; Kitano, M.; Nakajima, K.; Hara, M. Adsorption-Enhanced Hydrolysis of β-1,4-Glucan on Graphene-Based Amorphous Carbon Bearing SO 3 H, COOH, and OH Groups. Langmuir 2009, 25, 5068–5075. [Google Scholar]
- do Fraga, A.C.; Quitete, C.P.B.; Ximenes, V.L.; Sousa-Aguiar, E.F.; Fonseca, I.M.; Rego, A.M.B. Biomass derived solid acids as effective hydrolysis catalysts. J. Mol. Catal. A Chem. 2016, 422, 248–257. [Google Scholar] [CrossRef]
- Zhang, S.; Tsang, D.C.W.; Yu, I.K.M.; Chen, S.S.; Xiong, X.; Cao, L.; Kwon, E.E.; Song, H.; Poon, C.S.; Ok, Y.S. Sulfonated biochar as acid catalyst for sugar hydrolysis and dehydration. Catal. Today 2018, 314, 52–61. [Google Scholar]
- Boudart, M. Turnover Rates in Heterogeneous Catalysis. Chem. Rev. 1995, 95, 661–666. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Elemental Analysis 1 (at%) | Acidity (mmol/g) | Aromatic Hydrogen 4 (mmol/g) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C | O | S | N | O/C | S/C | Strong Acid 2 | Total 3 | ||
| Petcoke | 72.4 | 20.5 | 2.2 | 1.1 | 0.28 | 0.03 | nd 5 | 0.34 | 1.5 |
| P-S-24 | 84 | 11.7 | 2.5 | 1.8 | 0.14 | 0.03 | 0.25 | 1.49 | 0.6 |
| P-N-24 | 67.6 | 26.6 | 1.4 | 4.4 | 0.39 | 0.02 | 0.70 | 5.25 | 0.9 |
| P-N/S-24 | 68.3 | 25.0 | 1.8 | 4.9 | 0.37 | 0.03 | 0.73 | 5.46 | 0.6 |
| Sample | N2 Adsorption 1 | CO2 Adsorption 1 | Ash Content 2 (wt%) | ||
|---|---|---|---|---|---|
| SA (m2/g) | V (cm3/g) | SA (m2/g) | V (cm3/g) | ||
| Petcoke | 1.5 | 0.005 | 84 | 0.021 | 7.7 |
| P-S-24 | 4.6 | 0.013 | 174 | 0.045 | 2.1 |
| P-N-24 | 4.1 | 0.015 | 151 | 0.035 | 2.1 |
| P-N/S-24 | 2.3 | 0.009 | 125 | 0.028 | 3.4 |
| No. | Reactants | Catalyst (Concentration of Catalytic Acid Sites) | Reaction Conditions | TOF 1 (h–1) | Ref. |
|---|---|---|---|---|---|
| 1 | AcA 2 + EtOH 3 | Sulfonated naphthalene derived carbon (4.9 mmol/g) | 70 °C, AcA/EtOH = 1/10 (molar ratio) | 78 | [50] |
| 2 | LA 4 + EtOH | Sulfonated activated carbon (0.5 mmol/g) | 60 °C, LA/EtOH = 1/3 (molar ratio) | 40 | [51] |
| 3 | LA + EtOH | Sulfonated carbide-derived carbon (0.8 mmol/g) | 60 °C, LA/EtOH = 1/5 (molar ratio) | 2 | [17] |
| 4 | CA 5+ MeOH 6 | Sulfonated activated carbon from coffee residue (0.45–0.72 mmol/g) | 60 °C, CA/MeOH = 1/3 (molar ratio) | 25–47 | [10] |
| 5 | CA+ MeOH | Amberlyst-15 (4.3 mmol/g) | 60 °C, CA/MeOH = 1/3 (molar ratio) | 4 | [10] |
| 6 | LA + EtOH | Amberlyst-15 (4.8 mmol g–1) | 75 °C, LA/EtOH = 1/5 (molar ratio) | 5 | [52] |
| 7 | LA + EtOH | Micro-mesoporous Beta zeolite (0.69 mmol/g) | 70 °C, LA/EtOH = 1/6 (molar ratio) | 6 | [53] |
| 8 | OA 7+ MeOH | Sulfonated high sulfur petroleum coke (0.7 mmol/g) | 60 °C, OA/MeOH = 1/20 (molar ratio) | 31 | This work |
| 9 | OA+ MeOH | Nitric acid oxidized high sulfur petroleum coke (0.7 mmol/g) | 60 °C, OA/MeOH = 1/20 (molar ratio) | 7 | This work |
| 10 | OA+ MeOH | Nitric acid and sulfuric acid mixture oxidized high sulfur petroleum coke (0.73 mmol/g) | 60 °C, OA/MeOH = 1/20 (molar ratio) | 11 | This work |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Schafranski, A.S.; Hazlett, M.J.; Xiao, Y.; Hill, J.M. Nitric Acid Functionalization of Petroleum Coke to Access Inherent Sulfur. Catalysts 2020, 10, 259. https://doi.org/10.3390/catal10020259
Huang Q, Schafranski AS, Hazlett MJ, Xiao Y, Hill JM. Nitric Acid Functionalization of Petroleum Coke to Access Inherent Sulfur. Catalysts. 2020; 10(2):259. https://doi.org/10.3390/catal10020259
Chicago/Turabian StyleHuang, Qing, Annelisa S. Schafranski, Melanie J. Hazlett, Ye Xiao, and Josephine M. Hill. 2020. "Nitric Acid Functionalization of Petroleum Coke to Access Inherent Sulfur" Catalysts 10, no. 2: 259. https://doi.org/10.3390/catal10020259
APA StyleHuang, Q., Schafranski, A. S., Hazlett, M. J., Xiao, Y., & Hill, J. M. (2020). Nitric Acid Functionalization of Petroleum Coke to Access Inherent Sulfur. Catalysts, 10(2), 259. https://doi.org/10.3390/catal10020259