Preparation of Carbon-Based Solid Acid Catalysts Using Rice Straw Biomass and Their Application in Hydration of α-Pinene

 ,
,
Abstract
:1. Introduction
2. Results
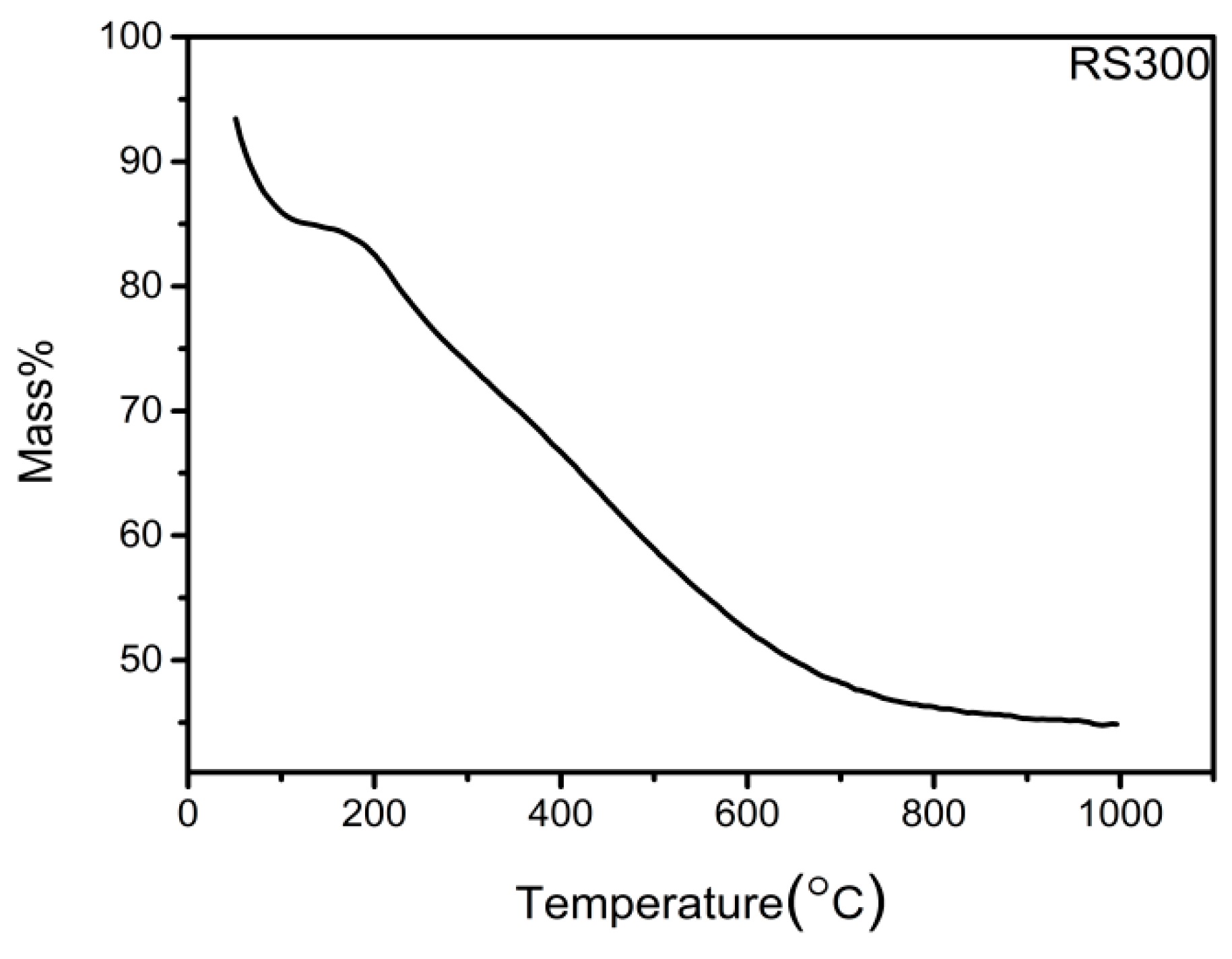
2.1. Thermal Stability
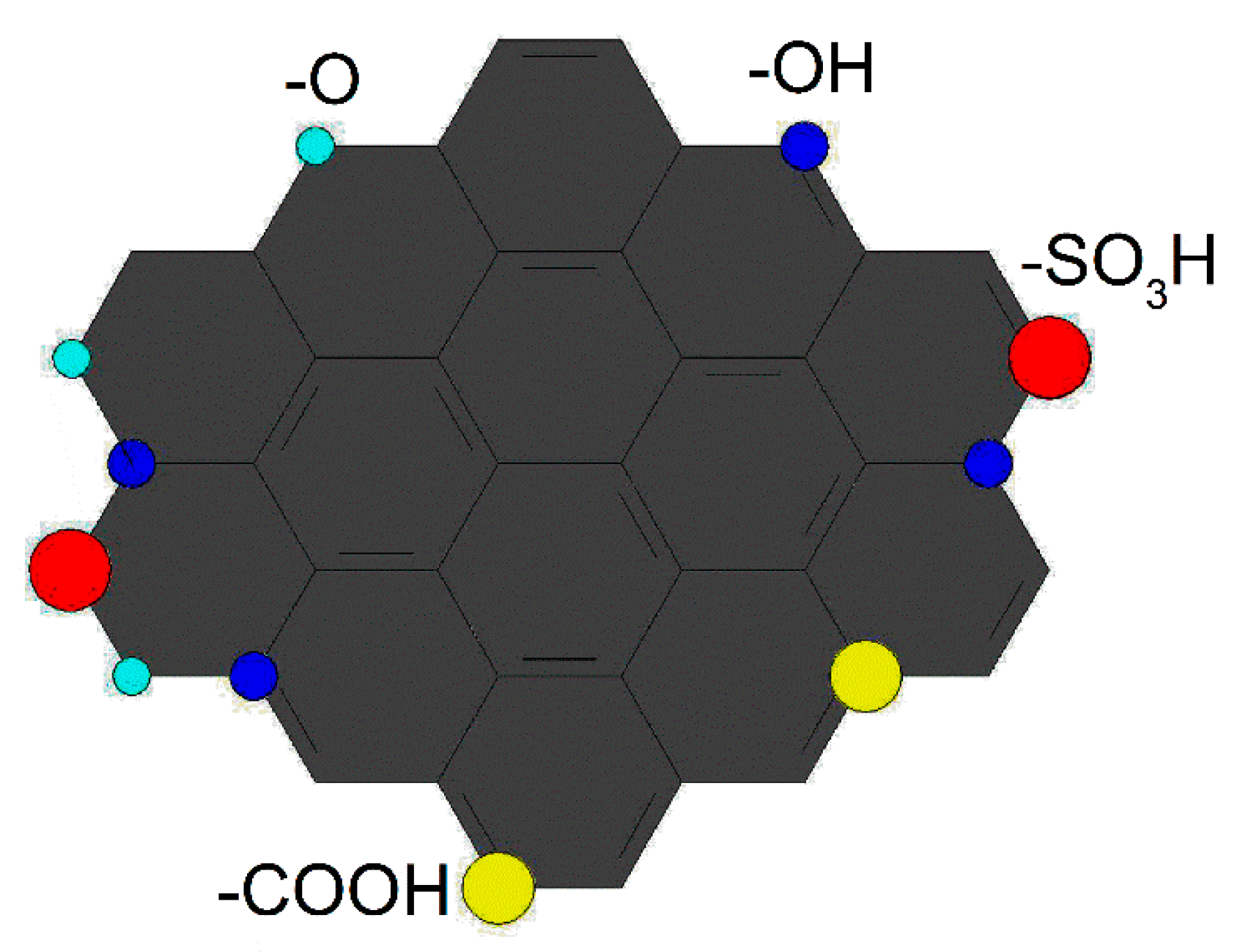
2.2. Characterization of Catalysts
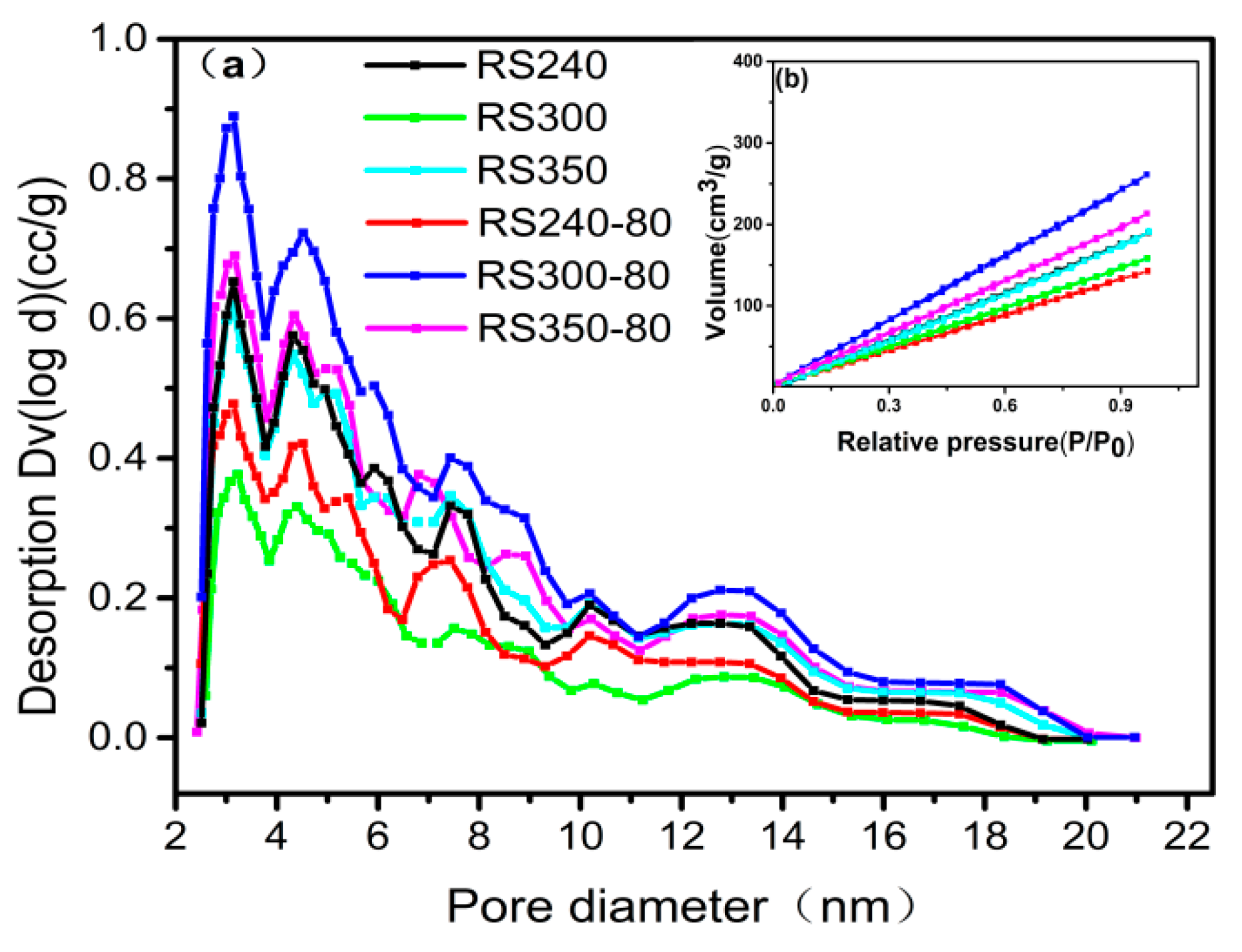
2.3. N2-physisorption
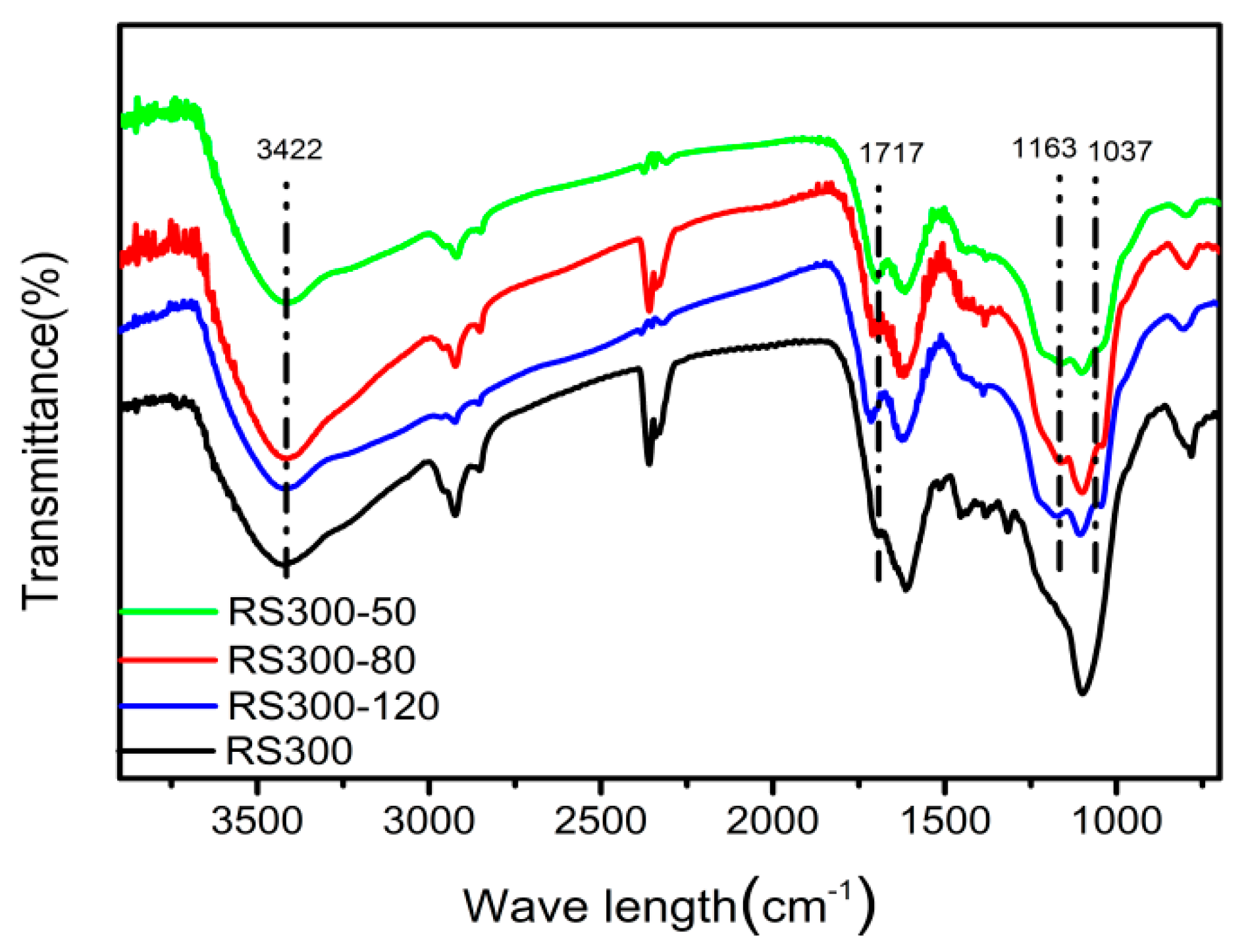
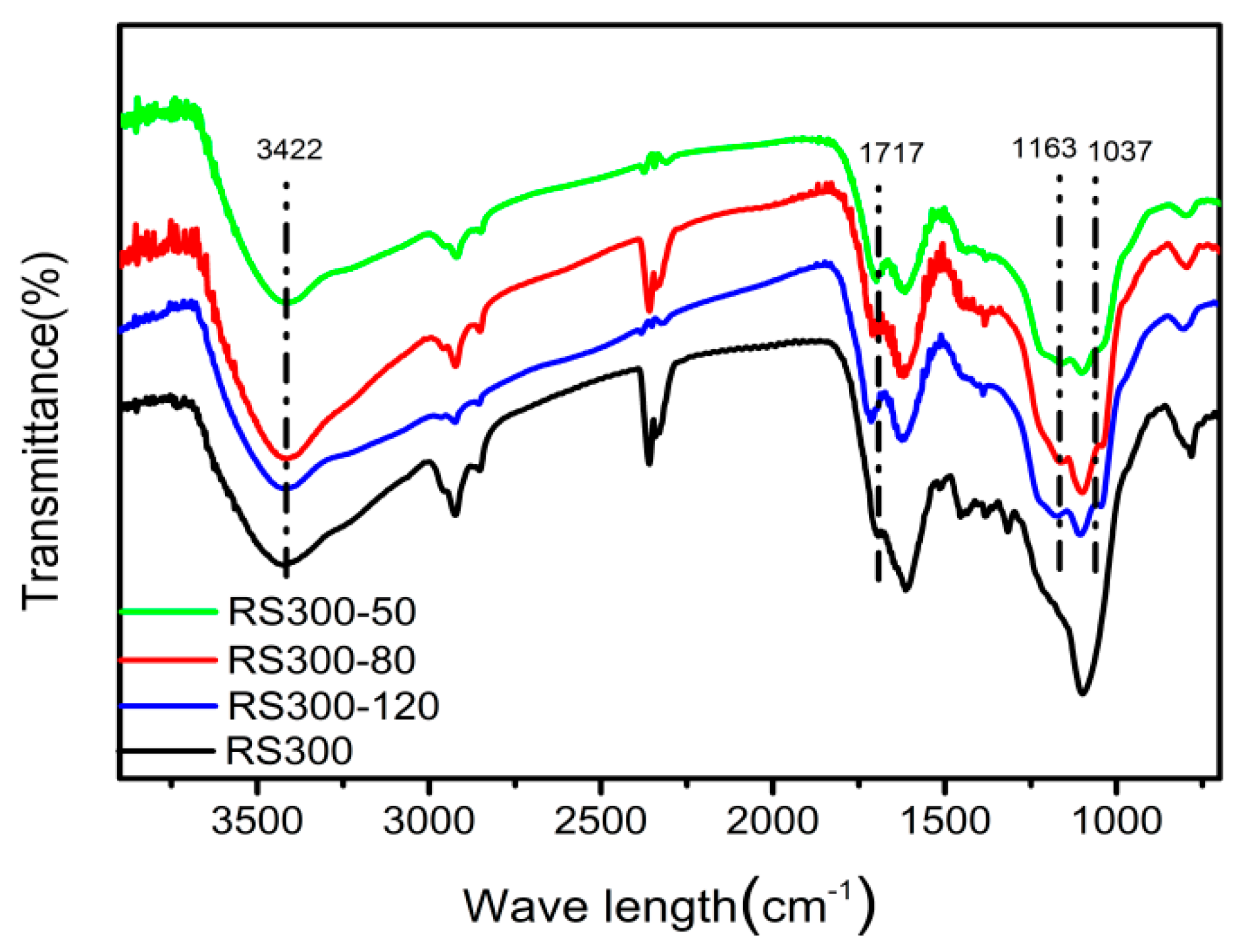
2.4. FT-IR Analysis
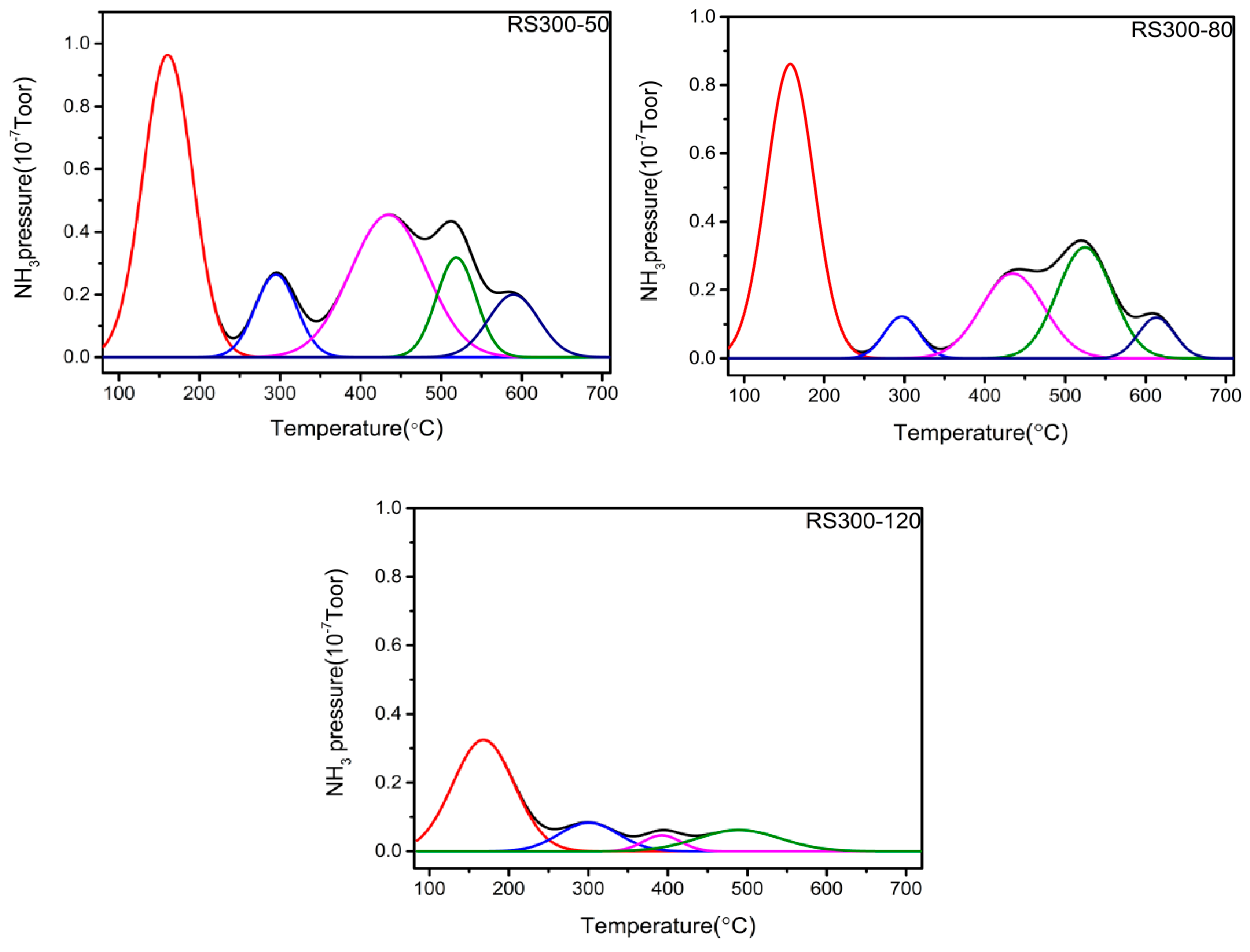
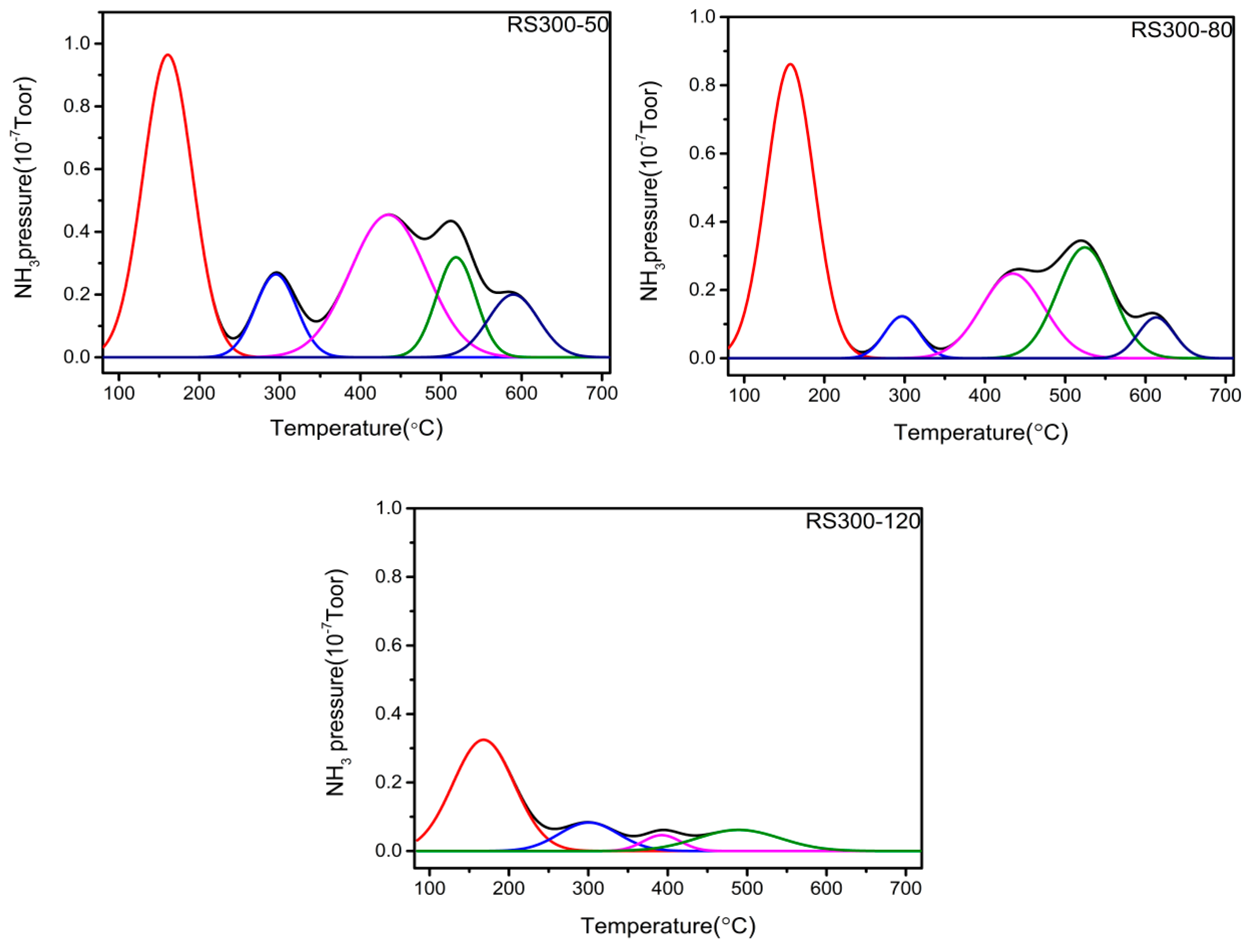
2.5. TPD Analysis
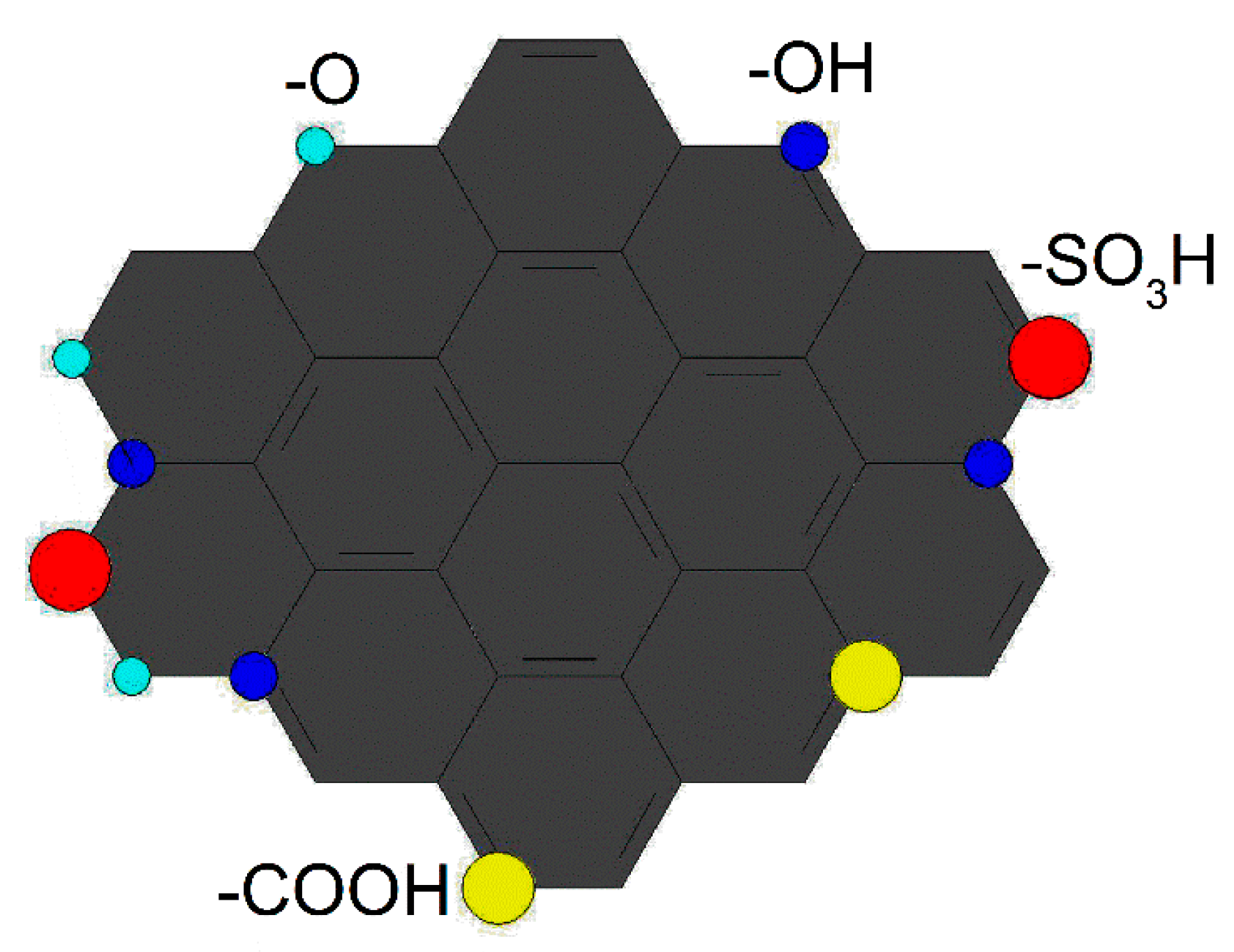
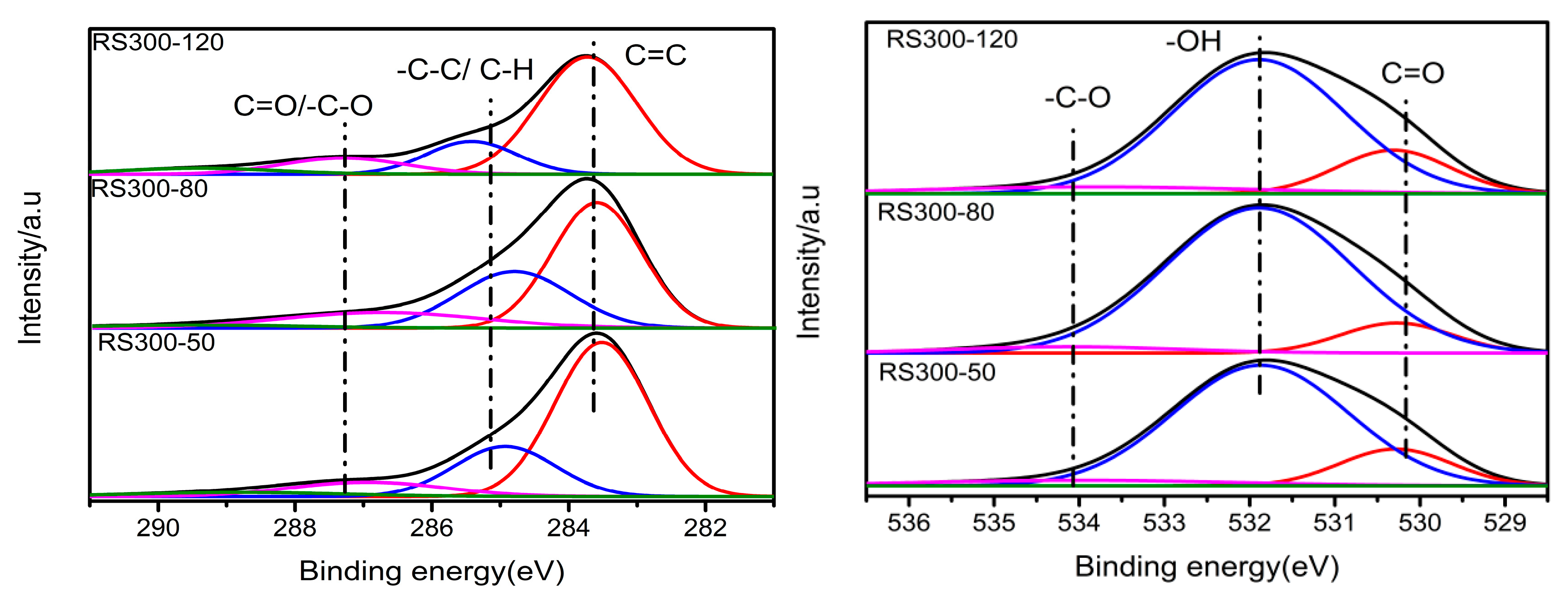
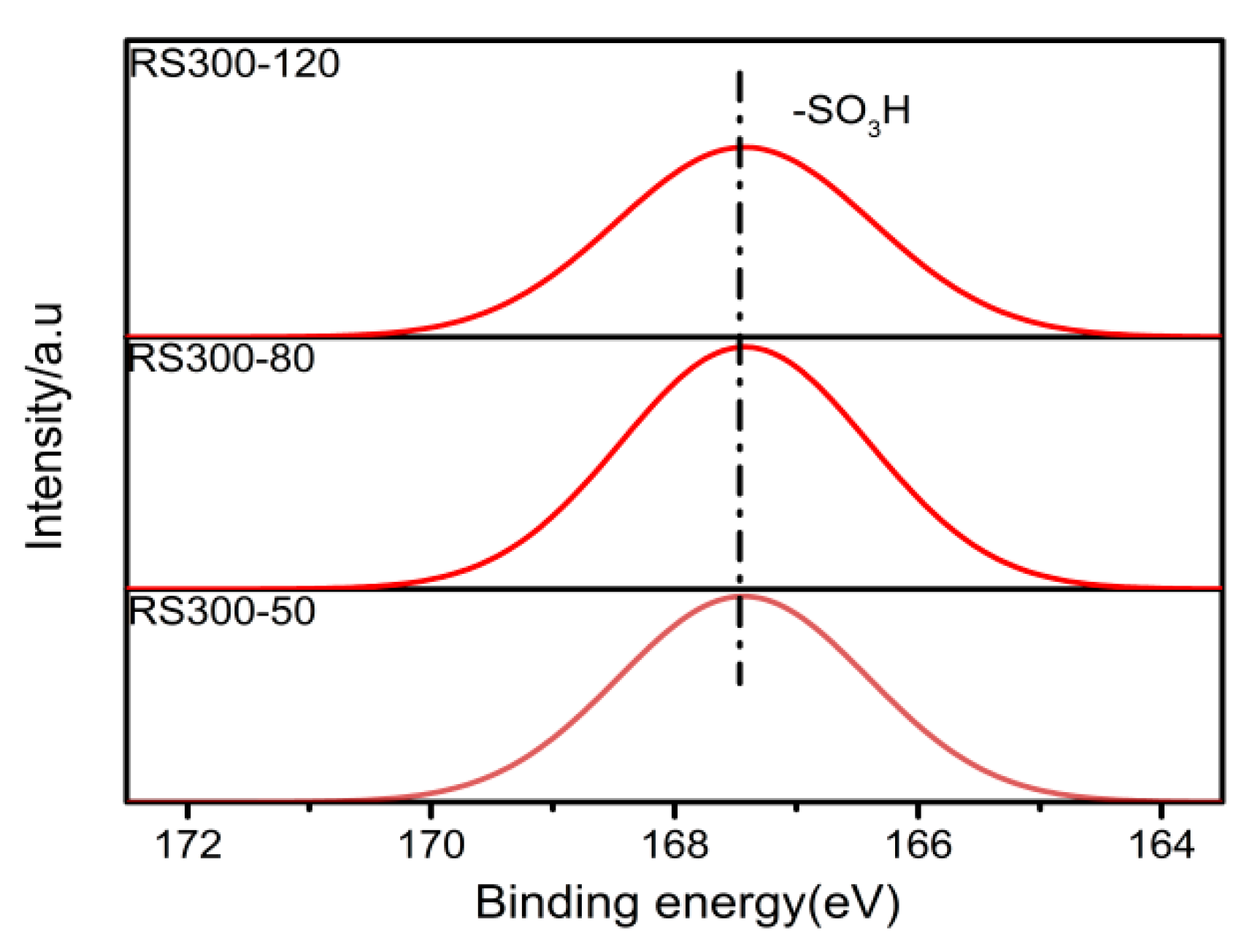
2.6. XPS Analysis
3. Materials and Methods
3.1. Material
3.2. Preparation of Catalyst
3.3. Characterization of the Catalysts
3.4. Investigatment of Catalytic Activity
3.5. Products Analysis
4. Activity Tests of Catalysts
4.1. Effect of Carbonization Temperature
4.2. Effect of Sulfonation Temperature
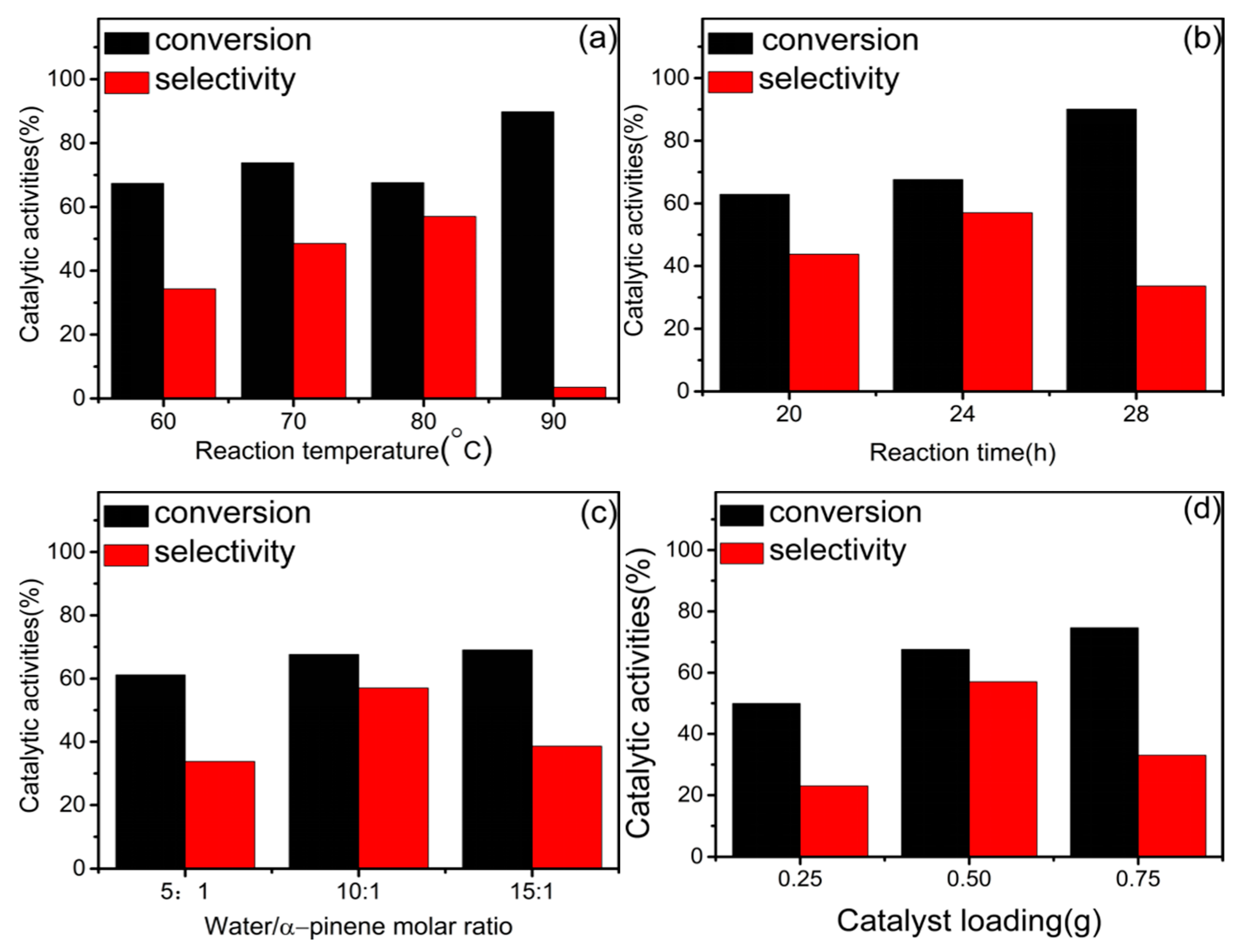
4.3. Effect of Reaction Temperature
4.4. Effect of Reaction Time
4.5. Effect of Water/α-Pinene Molar Ratio
4.6. Effect of Catalyst Loading
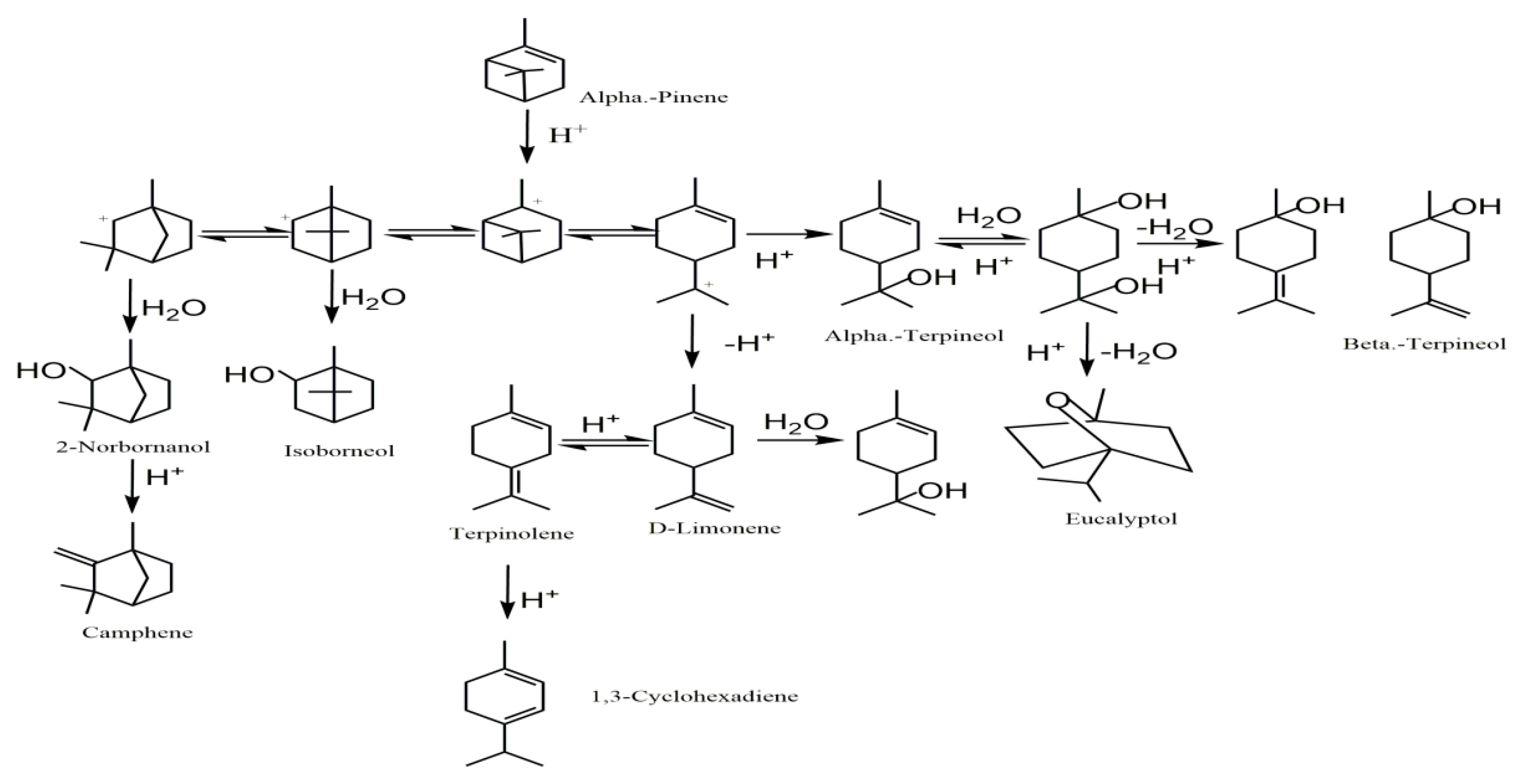
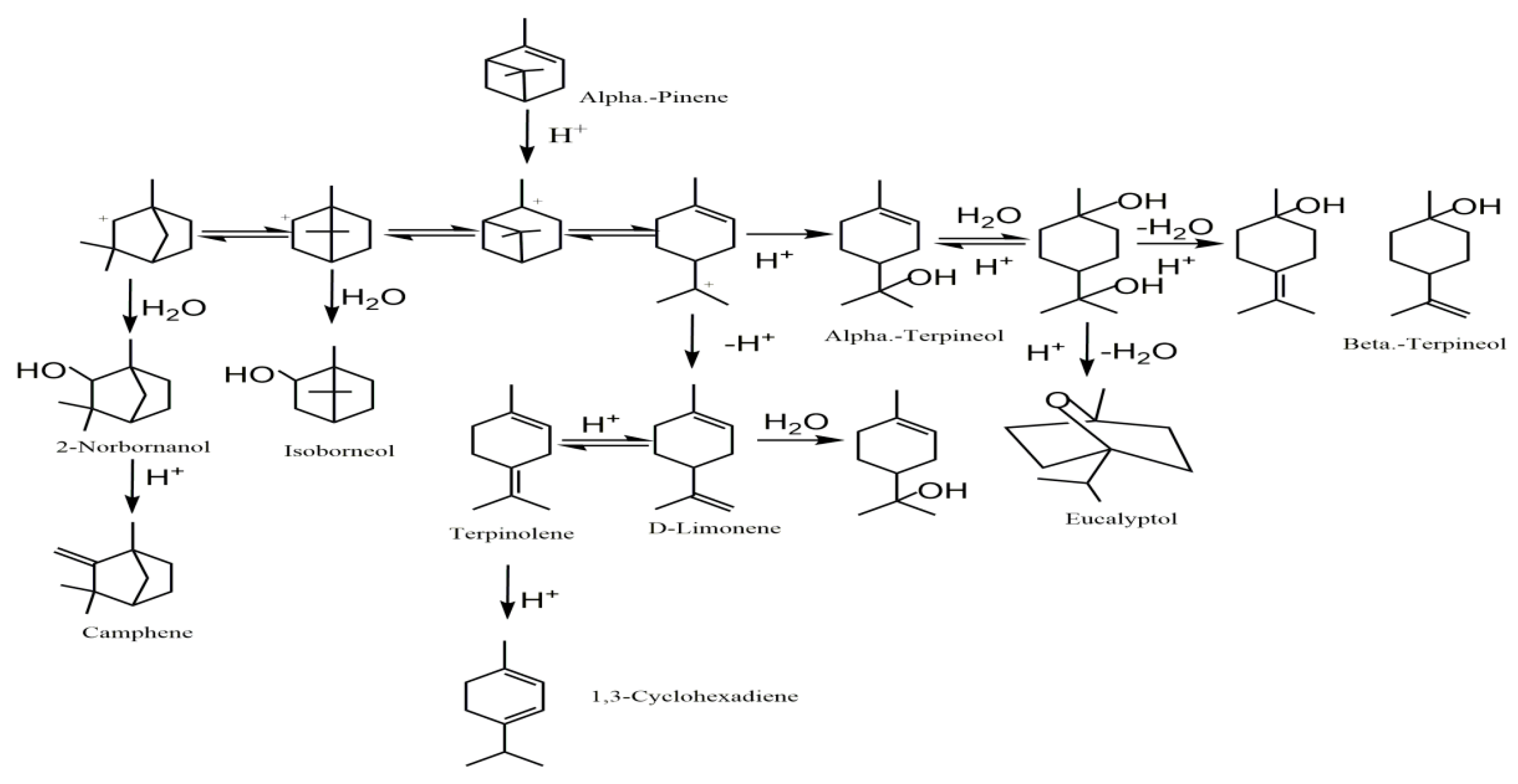
5. Postulated Reaction Mechanism
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wright, G. The value of turpentine in gall-stone operations. Br. Med. J. 1908, 2503, 1808. [Google Scholar]
- Pakdel, H.; Sarron, S.; Roy, C. α-Terpineol from Hydration of Crude Sulfate Turpentine Oil. J. Agric. Food Chem. 2001, 49, 4337–4341. [Google Scholar] [CrossRef]
- Román-Aguirre, M.; De la Torre-Saenz, L.; Flores, W.A.; Robau-Sanchez, A.; Elguezabal, A.A. Synthesis of terpineol from α-pinene by homogeneous acid catalysis. Catal. Today 2005, 108, 310–314. [Google Scholar] [CrossRef]
- Yang, G.; Liu, Y.; Zhou, Z.; Zhang, Z. Kinetic study of the direct hydration of turpentine. Chem. Eng. J. 2011, 168, 351–358. [Google Scholar] [CrossRef]
- Ávila, M.C.; Comelli, N.A.; Rodríguez-Castellón, E.; Jiménez-López, A.; Carrizo Flores, R.; Ponzi, E.N.; Ponzi, M.I. Study of solid acid catalysis for the hydration of α-pinene. J. Mol. Catal. A Chem. 2010, 322, 106–112. [Google Scholar] [CrossRef]
- Vital, J.; Ramos, A.M.; Silva, I.F.; Valente, H.; Castanheiro, J.E. Hydration of α-pinene over zeolites and activated carbons dispersed in polymeric membranes. Catal. Today 2000, 56, 167–172. [Google Scholar] [CrossRef]
- Vital, J.; Ramos, A.M.; Silva, I.F.; Castanheiro, J.E. The effect of α-terpineol on the hydration of α-pinene over zeolites dispersed in polymeric membranes. Catal. Today 2001, 67, 217–223. [Google Scholar] [CrossRef]
- Thushari, I.; Babel, S. Preparation of solid acid catalysts from waste biomass and their application for microwave-assisted biodiesel production from waste palm oil. Waste Manag. Res. 2018, 36, 719–728. [Google Scholar] [CrossRef]
- Xie, J.; Han, Q.; Feng, B.; Liu, Z. Preparation of amphiphilic mesoporous carbon-based solid acid from kraft lignin activated by phosphoric acid and its catalytic performance for hydration of α-pinene. BioResources 2019, 14, 4284–4303. [Google Scholar]
- Jin, S.; Chen, H. Near-infrared analysis of the chemical composition of rice straw. Ind. Crops Prod. 2007, 26, 207–211. [Google Scholar] [CrossRef]
- Mardhiah, H.H.; Ong, H.C.; Masjuki, H.H.; Lim, S.; Pang, Y.L. Investigation of carbon-based solid acid catalyst from Jatropha curcas biomass in biodiesel production. Energy Convers. Manag. 2017, 144, 10–17. [Google Scholar] [CrossRef]
- Nakajima, K.; Hara, M. Amorphous Carbon with SO3H Groups as a Solid Brønsted Acid Catalyst. ACS Catal. 2012, 2, 1296–1304. [Google Scholar] [CrossRef]
- Shu, Q.; Gao, J.; Nawaz, Z.; Liao, Y.; Wang, D.; Wang, J. Synthesis of biodiesel from waste vegetable oil with large amounts of free fatty acids using a carbon-based solid acid catalyst. Appl. Energy 2010, 87, 2589–2596. [Google Scholar] [CrossRef]
- Inagaki, M. Pores in carbon materials-importance of their control. New Carbon Mater. 2009, 24, 193–232. [Google Scholar] [CrossRef]
- Tao, M.L.; Guan, H.Y.; Wang, X.H.; Liu, Y.C.; Louh, R.F. Fabrication of sulfonated carbon catalyst from biomass waste and its use for glycerol esterification. Fuel Process. Technol. 2015, 138, 355–360. [Google Scholar] [CrossRef]
- Fukuhara, K.; Nakajima, K.; Kitano, M.; Kato, H.; Hayashi, S. Structure and Catalysis of Cellulose-Derived Amorphous Carbon Bearing SO3 H Groups. ChemSusChem 2011, 4, 778–784. [Google Scholar] [CrossRef]
- Chen, G.; Fang, B. Preparation of solid acid catalyst from glucose-starch mixture for biodiesel production. Bioresour. Technol. 2011, 102, 2635–2640. [Google Scholar] [CrossRef]
- Ding, S.; Li, Z.; Li, F.; Wang, Z.; Li, J.; Zhao, T.; Lin, H.; Chen, C. Catalytic hydrogenation of stearic acid over reduced NiMo catalysts: Structure–activity relationship and effect of the hydrogen-donor. Appl. Catal. A Gen. 2018, 566, 146–154. [Google Scholar] [CrossRef]
- Rao, B.V.S.K.; Chandra Mouli, K.; Rambabu, N.; Dalai, A.K.; Prasad, R.B.N. Carbon-based solid acid catalyst from de-oiled canola meal for biodiesel production. Catal. Commun. 2011, 14, 20–26. [Google Scholar] [CrossRef]
- Nakajima, K.; Hara, M.; Hayashi, S. Environmentally benign production of chemicals and energy using a carbon-based strong solid acid. J. Am. Ceram. Soc. 2007, 90, 3725–3734. [Google Scholar] [CrossRef]
- Ishimaru, K.; Hata, T.; Bronsveld, P.; Meier, D.; Imamura, Y. Spectroscopic analysis of carbonization behavior of wood, cellulose and lignin. J. Mater. Sci. 2007, 42, 122–129. [Google Scholar] [CrossRef]
- Toda, M.; Takagaki, A.; Okamura, M.; Kondo, J.N.; Hayashi, S.; Domen, K.; Hara, M. Biodiesel made with sugar catalyst. Nature 2005, 438, 178. [Google Scholar] [CrossRef] [PubMed]
- Laohapornchaiphan, J.; Smith, C.B.; Smith, S.M. One-step Preparation of Carbon-based Solid Acid Catalyst from Water Hyacinth Leaves for Esterification of Oleic Acid and Dehydration of Xylose. Chem.-Asian J. 2017, 12, 3178–3186. [Google Scholar] [CrossRef]
- Kitano, M.; Arai, K.; Kodama, A.; Kousaka, T.; Nakajima, K.; Hayashi, S.; Hara, M. Preparation of a Sulfonated Porous Carbon Catalyst with High Specific Surface Area. Catal. Lett. 2009, 242–249. [Google Scholar] [CrossRef]
- Prakoso, T.; Hanley, J.; Soebianta, M.N.; Soerawidjaja, T.H.; Indarto, A. Synthesis of Terpineol from α-Pinene Using Low-Price Acid Catalyst. Catal. Lett. 2018, 148, 725–731. [Google Scholar] [CrossRef]
- Wijayati, N.; Hidayah, N.; Mursiti, S.; Kusumastuti, E. Catalytic activity of P2O5-natural zeolite on hydration reaction of turpentine into α-terpineol. IOP Conf. Ser. Mater. Sci. Eng. 2019, 509, 012093. [Google Scholar] [CrossRef]
- Lopez, D.E.; Goodwin, J.G., Jr.; Bruce, D.A.; Lotero, E. Transesterification of triacetin with methanol on solid acid and base catalysts. Appl. Catal. A Gen. 2005, 295, 97–105. [Google Scholar] [CrossRef]
- Li, Y.; Shen, S.; Wang, C.; Peng, X.; Yuan, S. The effect of difference in chemical composition between cellulose and lignin on carbon based solid acids applied for cellulose hydrolysis. Cellulose 2018, 25, 1851–1863. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | SBET (m2/g) | VTotal (cm3/g) | Dpore (nm) |
|---|---|---|---|
| RS240 | 306.8 | 2.94 | 3.82 |
| RS300 | 260.5 | 2.44 | 3.74 |
| RS350 | 312.1 | 2.95 | 3.76 |
| RS240-80 | 222.1 | 2.22 | 3.98 |
| RS300-80 | 420.9 | 4.05 | 3.84 |
| RS350-80 | 337.7 | 3.30 | 3.90 |
| Catalyst | Total Acidity/mmol/g | Percentage of Acid Sites (% of Total Acid) | ||
|---|---|---|---|---|
| Weak Acid (50–190 °C) | Medium Acid (190–400 °C) | Strong Acid (> 400 °C) | ||
| RS300-50 | 2.07 | 36.52 | 36.45 | 27.03 |
| RS300-80 | 2.87 | 48.25 | 23.13 | 28.62 |
| RS300-120 | 1.67 | 67.37 | 13.15 | 19.48 |
| Catalyst | C (wt %) | O (wt %) | S (wt %) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C=C | –C–C/C–H | –C=O | C=O | –OH | –O–C | –SO3H | |||
| RS300-50 | 28.13 | 8.53 | 7.22 | 8.27 | 40.16 | 4.09 | 3.01 | ||
| RS300-80 | 27.31 | 12.33 | 8.74 | 5.13 | 41.11 | 2.20 | 3.16 | ||
| RS300-120 | 30.41 | 10.87 | 6.78 | 7.43 | 37.89 | 3.60 | 2.59 |
| Catalyst | Conversion (%) | Selectivity (%) |
|---|---|---|
| RS240-80 | 57.23 | 35.27 |
| RS300-80 | 67.60 | 57.07 |
| RS350-80 | 84.16 | 13.20 |
| RS300-50 | 55.33 | 36.85 |
| RS300-120 | 74.05 | 34.02 |
| NO. | Product | Molecular Formula | Relative Content (%) | Similarity (%) | |
|---|---|---|---|---|---|
| (a) | (b) | ||||
| 1 | α-Pinene | C10H16 | 7.23 | 17.33 | 97 |
| 2 | Camphene | C10H16 | 9.80 | 3.69 | 97 |
| 3 | 1,3-Cyclohexadiene | C10H16 | 7.23 | 0.53 | 96 |
| 4 | D-Limonene | C10H16 | 15.95 | 8.30 | 90 |
| 5 | Eucalyptol | C10H18O | 1.69 | 1.63 | 93 |
| 6 | 1,4-Cyclohexadiene | C10H16 | 3.98 | 3.97 | 97 |
| 7 | Terpinolene | C10H16 | 14.10 | 9.19 | 96 |
| 8 | 2-Norbornanol | C10H18O | 8.14 | 6.97 | 96 |
| 9 | β-Terpineol | C10H18O | 2.37 | 2.20 | 93 |
| 10 | Isoborneol | C10H18O | 13.34 | 1.55 | 97 |
| 11 | α-Terpineol | C10H18O | 16.17 | 44.64 | 86 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Z.; Xiong, D.; Duan, P.; Ding, S.; Li, Y.; Li, L.; Niu, P.; Chen, X. Preparation of Carbon-Based Solid Acid Catalysts Using Rice Straw Biomass and Their Application in Hydration of α-Pinene. Catalysts 2020, 10, 213. https://doi.org/10.3390/catal10020213
Wei Z, Xiong D, Duan P, Ding S, Li Y, Li L, Niu P, Chen X. Preparation of Carbon-Based Solid Acid Catalysts Using Rice Straw Biomass and Their Application in Hydration of α-Pinene. Catalysts. 2020; 10(2):213. https://doi.org/10.3390/catal10020213
Chicago/Turabian StyleWei, Zhaozhou, Deyuan Xiong, Pengzhi Duan, Shilei Ding, Yuanlin Li, Lisi Li, Peirong Niu, and Xusong Chen. 2020. "Preparation of Carbon-Based Solid Acid Catalysts Using Rice Straw Biomass and Their Application in Hydration of α-Pinene" Catalysts 10, no. 2: 213. https://doi.org/10.3390/catal10020213
APA StyleWei, Z., Xiong, D., Duan, P., Ding, S., Li, Y., Li, L., Niu, P., & Chen, X. (2020). Preparation of Carbon-Based Solid Acid Catalysts Using Rice Straw Biomass and Their Application in Hydration of α-Pinene. Catalysts, 10(2), 213. https://doi.org/10.3390/catal10020213