Rh-induced Support Transformation and Rh Incorporation in Titanate Structures and Their Influence on Catalytic Activity






Abstract
1. Introduction
1.1. General Surway
1.2. Literature Review of Phase Transformation of Heat-Treated Pristine (H-Titanate) and Rh-Decorated Titanates
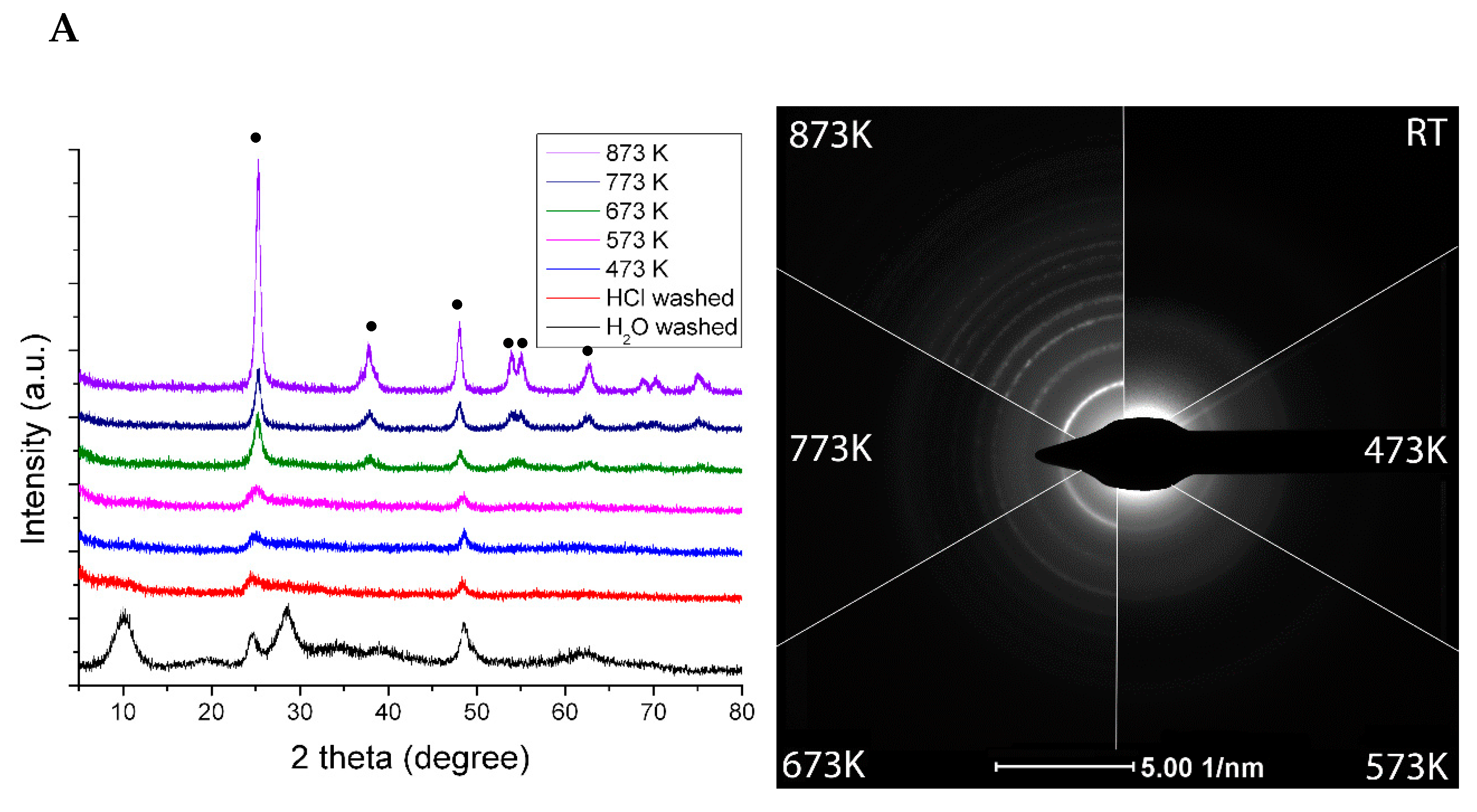
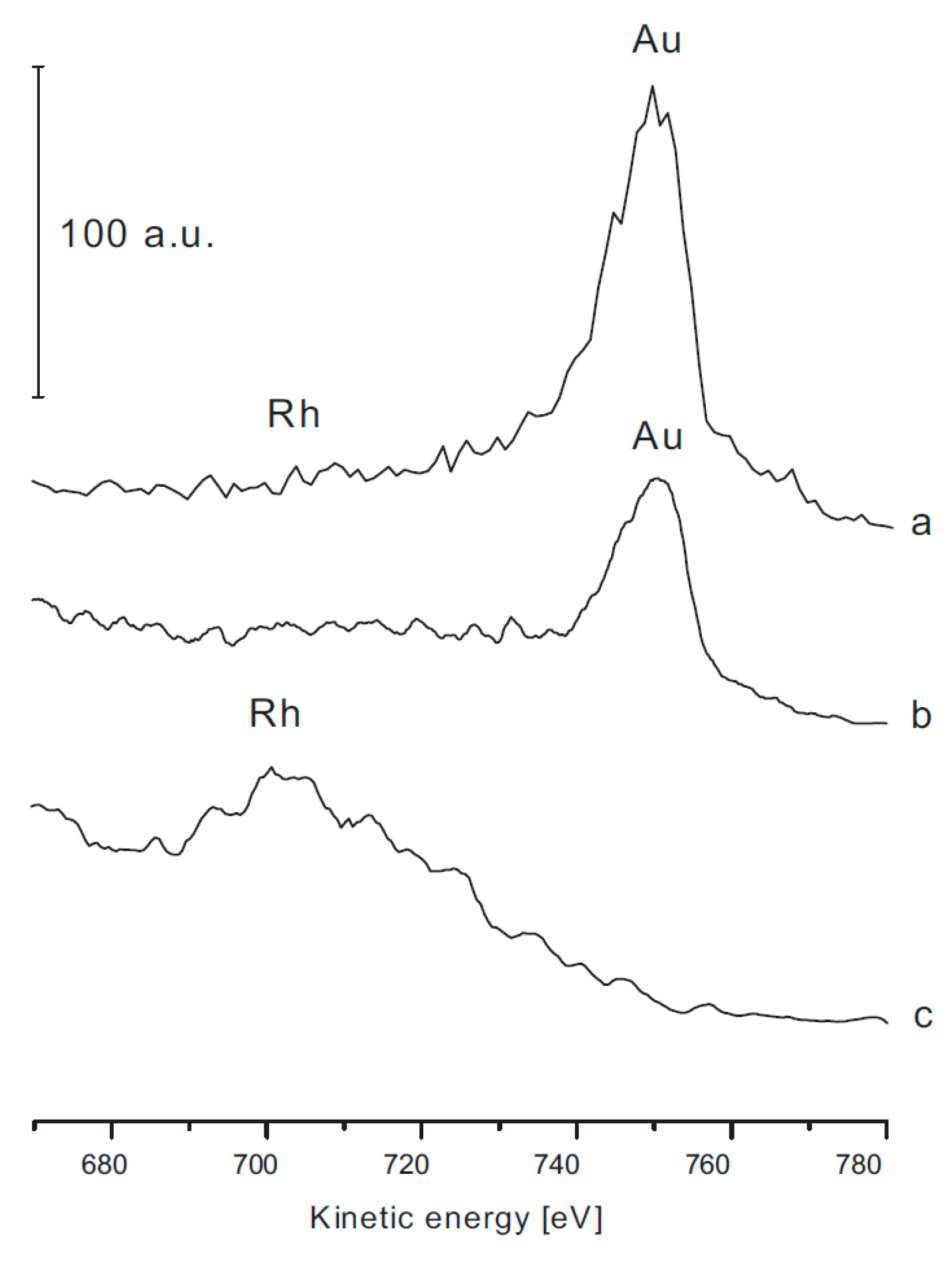
1.3. Summary Results on the Morphology and Chemical State of Rh Nanoparticles on Titanates
2. Materials and Methods
3. Effect of Titania Structure and Form of the Rh Metal on Heterogeneous Catalytic Reactions
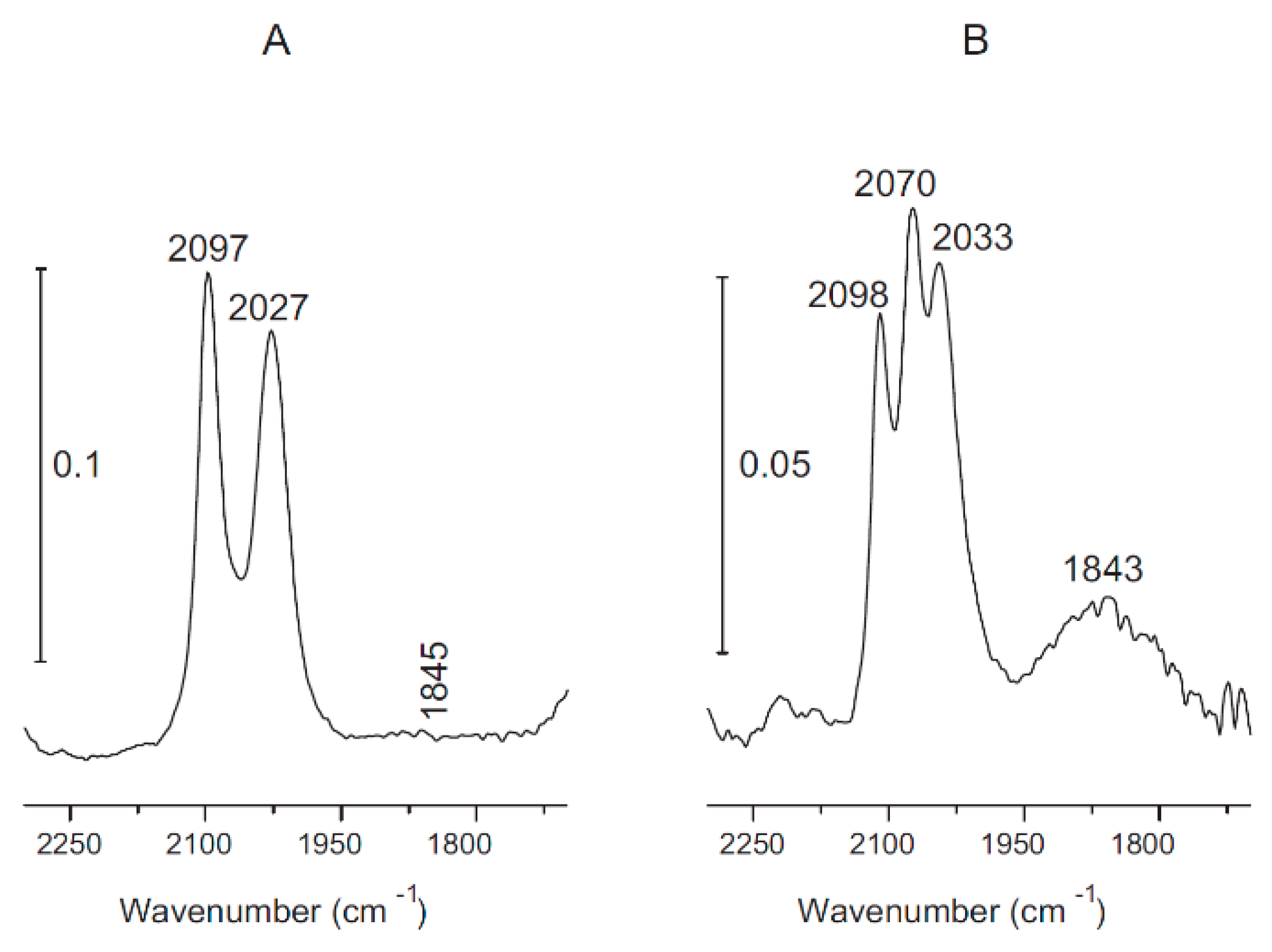
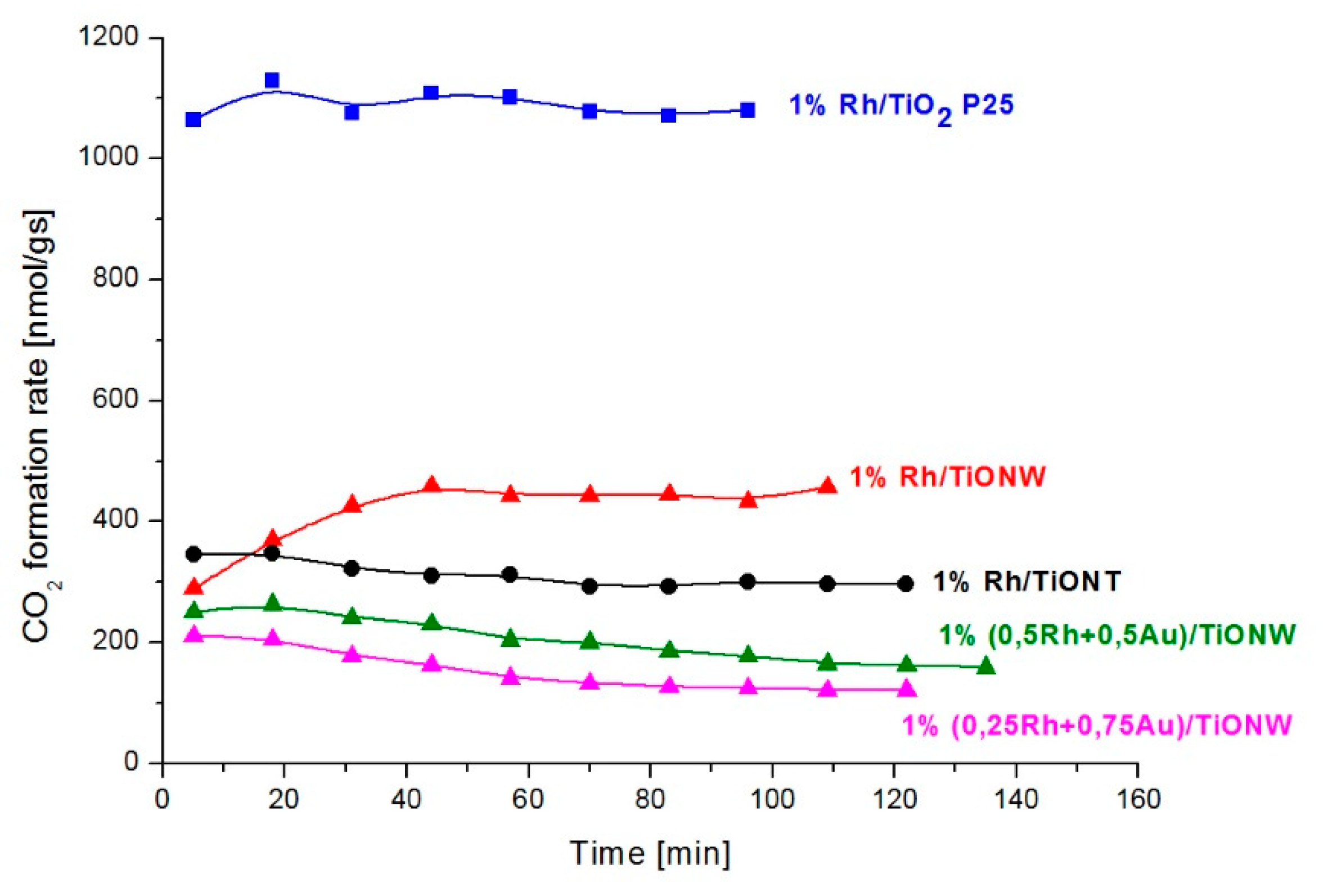
3.1. CO2 Hydrogenation on Titania and Titanate Supported Rh
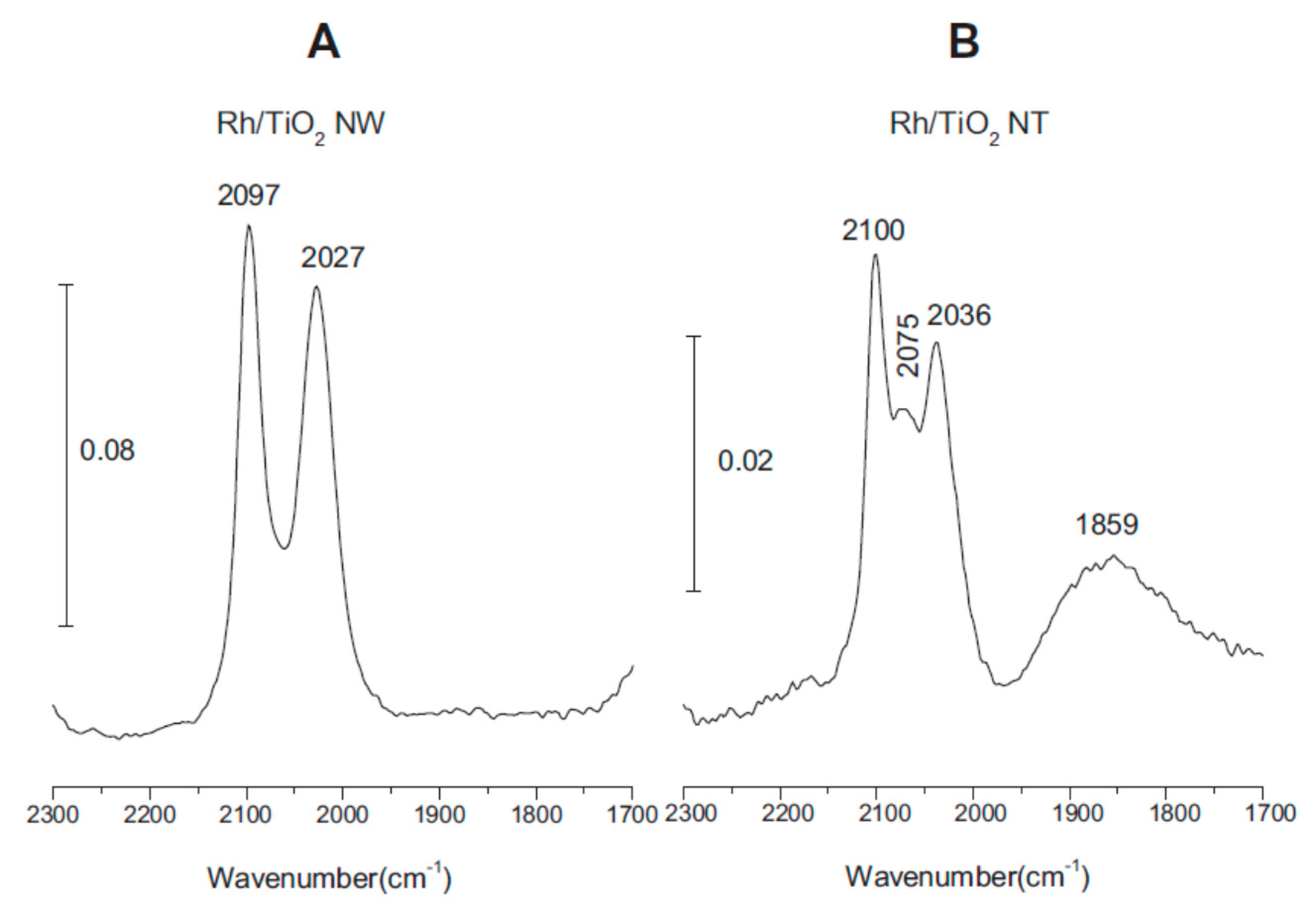
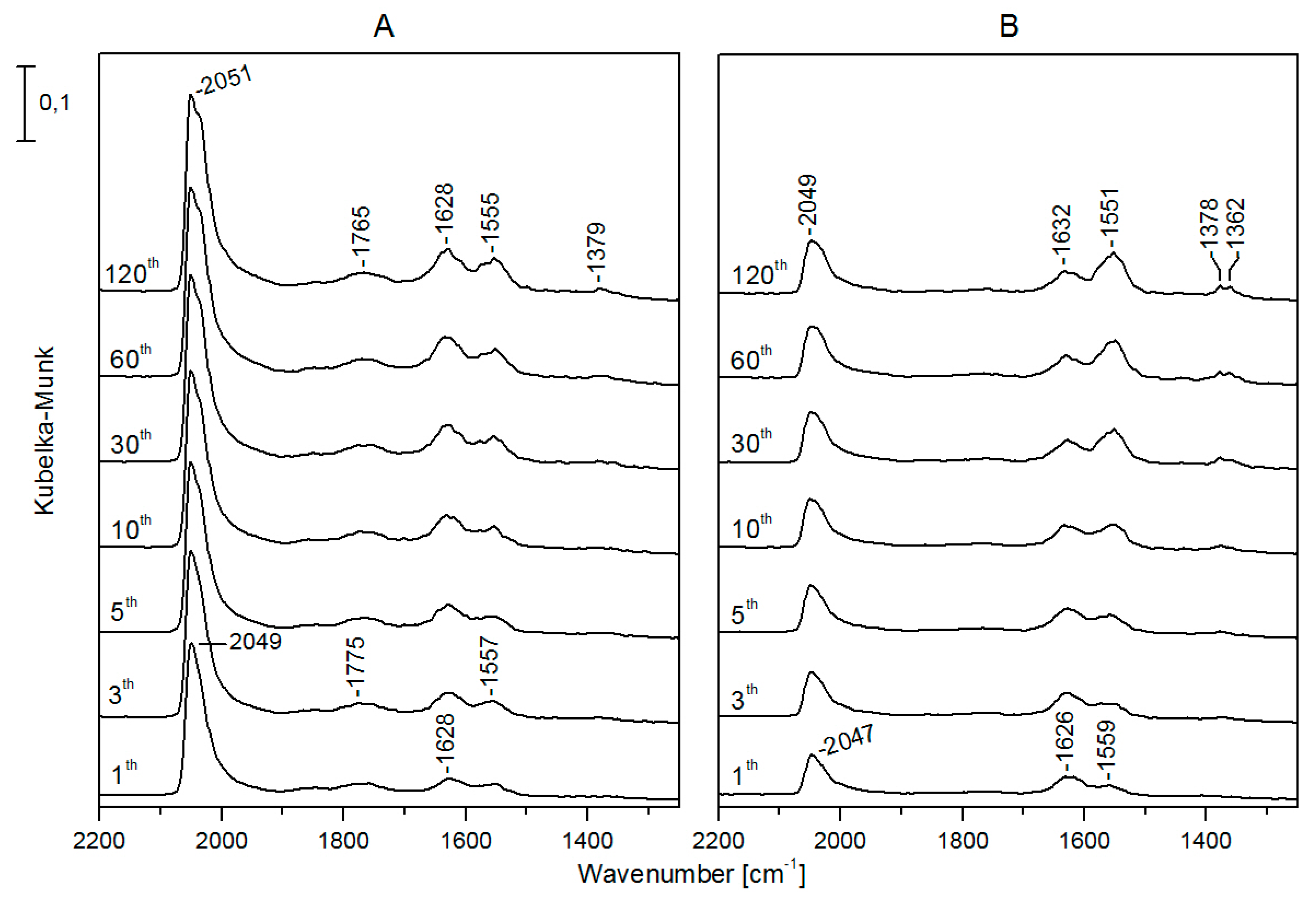
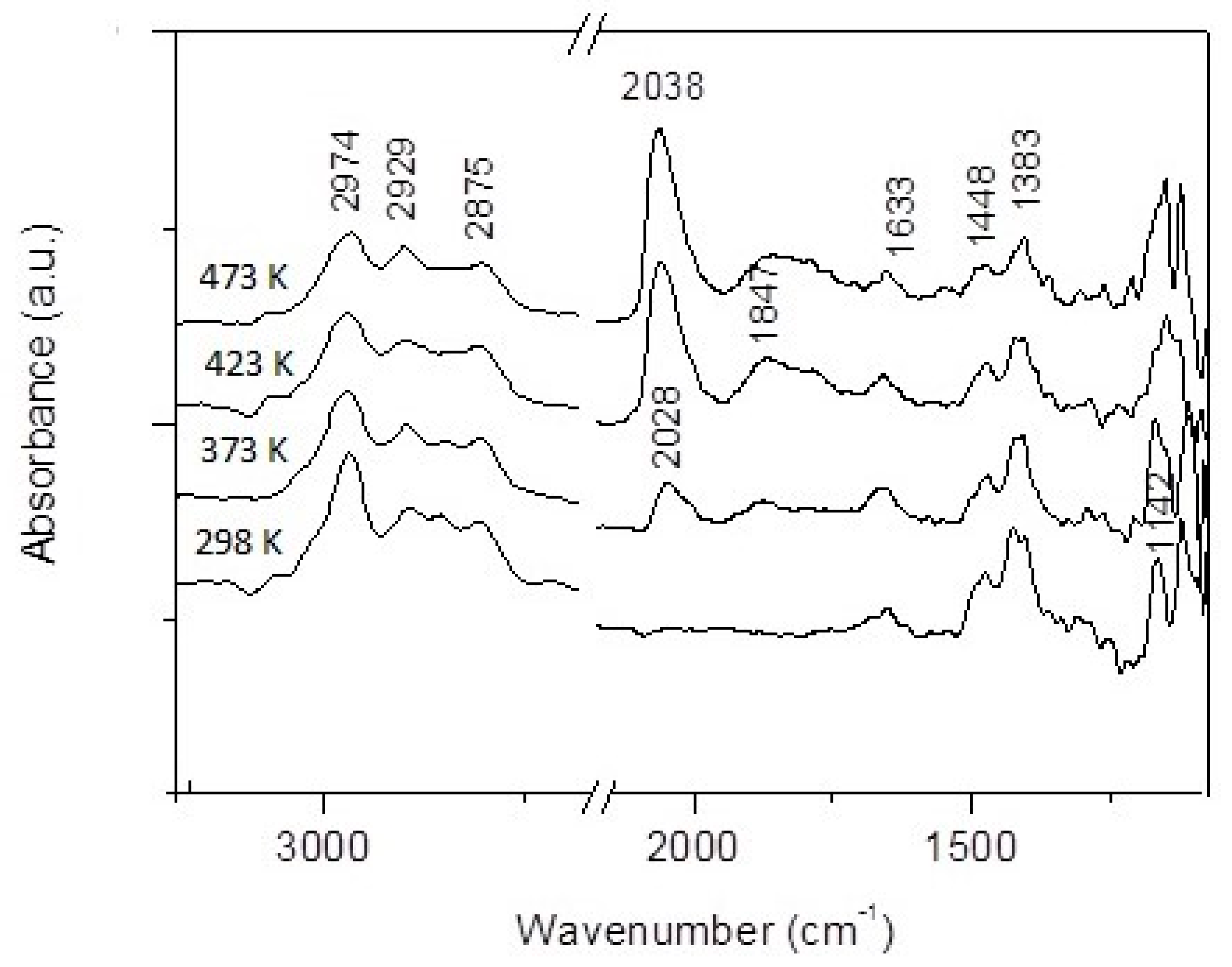
3.2. CO + H2O Reaction on Rh/TiONW, Rh/TiONT and Rh/TiO2
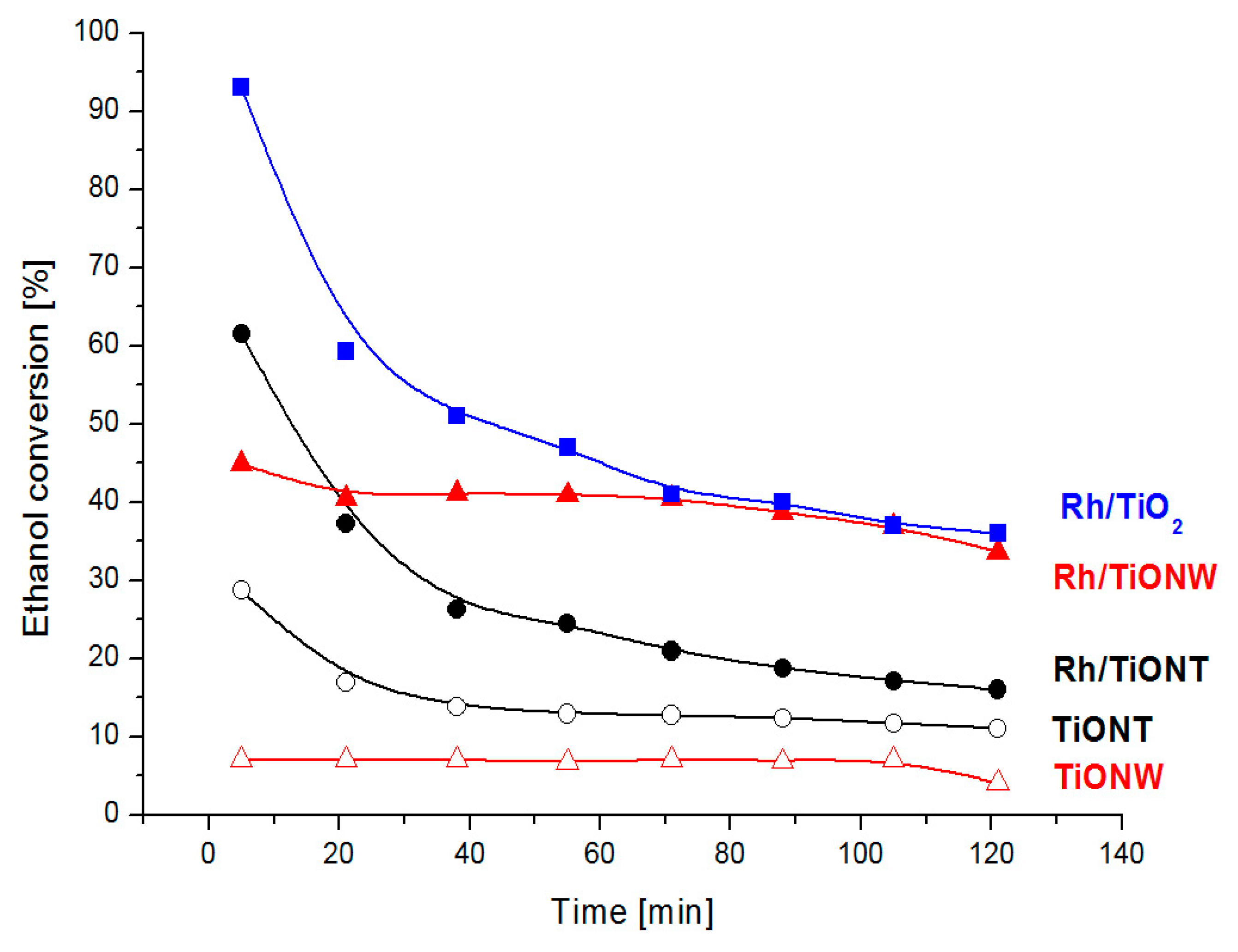
3.3. CH5OH Decomposition
4. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ertl, G.; Knözinger, H.; Weitkamp, J. Handbook of Heterogeneous Catalysis; VCH: Weinheim, German, 1997; p. 2478. [Google Scholar]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable conversion of carbon dioxide: An integrated review of catalysis and life cycle assessment. Chem. Rev. 2017, 118, 434–504. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G.O.; Pérez-Ramírez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef]
- Studt, F.; Sharafutdinov, I.; Abild-Pedersen, F.; Elkjær, C.F.; Hummelshøj, J.S.; Dahl, S.; Chorkendorff, I.; Nørskov, J.K. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat. Chem. 2014, 6, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Leonzio, G. State of art and perspectives about the production of methanol, dimethyl ether and syngas by carbon dioxide hydrogenation. J. CO2 Util. 2018, 27, 326–354. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, W.; Li, Y.; Chen, J.; Yu, B.; Wang, J.; Zhang, L.; Zhang, J. Energy related CO2 conversion and utilization: Advanced materials/nanomaterials, reaction mechanisms and technologies. Nano Energy 2017, 40, 512–539. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Freund, H.J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef]
- Kiss, J.; Révész, K.; Solymosi, F. Photoelectron Spectroscopic Studies of the Adsorption of CO2 on Potassium-Promoted Rh(111). Surf. Sci. 1988, 207, 36–54. [Google Scholar] [CrossRef][Green Version]
- László, B.; Baán, K.; Varga, E.; Oszkó, A.; Erdőhelyi, A.; Kónya, Z.; Kiss, J. Photo-induced reactions in the CO2-methane system on titanate nanotubes modified with Au and Rh nanoparticles. Appl. Catal. B Environ. 2016, 199, 473–484. [Google Scholar] [CrossRef]
- Kiss, J.; Kukovecz, Á.; Kónya, Z. Beyond Nanoparticles; The Role of Sub-nanosized Metal Species in Heterogeneous Catalysis. Catal. Lett. 2019, 149, 1441–1454. [Google Scholar] [CrossRef]
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A Review. Renew. Sustain. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Ratnasamy, C.; Wagner, J.P. Water Gas Shift Catalysis. Catal.Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Mattos, L.V.; Jacobs, G.; Davis, B.H.; Noronha, F.B. Production of hydrogen from ethanol: Review of reaction mechanism and catalytic deactivation. Chem. Rev. 2012, 112, 4094–4123. [Google Scholar] [CrossRef]
- Contreras, J.L.; Salmones, J.; Colin-Luna, J.A.; Nuno, L.; Quintana, B.; Cordova, I.; Zeifert, B.H.; Tapia, C.; Fuentes, G. A. Cataysts for H2 production using the ethanol steam reforming (a review). Int. J. Hydrog. Energy 2014, 39, 18835–18853. [Google Scholar] [CrossRef]
- Punase, K.D.; Rao, N.; Vija, P. A review on mechanistic kinetic models of ethanol steam reforming for hydrogen production using a fixed bed reactor. Chem. Pap. 2019, 73, 1027–1042. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdőhelyi, A.; Bánsági, T. Methanation of CO2 on supported rhodium catalyst. J. Catal. 1981, 68, 371–382. [Google Scholar] [CrossRef]
- Henderson, M.A.; Worley, S.D. An infrared study of the hydrogenation of carbon dioxide on supported rhodium catalysts. J. Phys. Chem. 1985, 89, 1417–1423. [Google Scholar] [CrossRef]
- Trovarelli, A.; Mustazza, C.; Dolcetti, G.; Kaspar, F.; Grazioni, M. Carbon-dioxide hydrogenation on Rhodium supported on transition-metal oxides-effect of reduction temperature on product distribution. Appl. Catal. 1990, 65, 129–142. [Google Scholar] [CrossRef]
- Solymosi, F.; Tombácz, I.; Koszta, J. Effect of variation of electric properties of TiO2 support on hydrogenation of CO and CO2 over Rh catalysts. J. Catal. 1985, 95, 578–586. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Kladi, A.; Verykios, X.E. Effects of Carrier Doping on Kinetic Parameters of CO2 Hydrogenation on Supported Rhodium Catalysts. J. Catal. 1994, 148, 737–747. [Google Scholar] [CrossRef]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Henderson, M.A. A surface science perspective on photocatalysis. Surf. Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Wang, L.; Sasaki, T. Titanium Oxide Nanosheets: Graphene Analogues with Versatile Functionalities. Chem. Rev. 2014, 114, 9455–9486. [Google Scholar] [CrossRef]
- Peña, M.A.; Fierro, J.L.G. Chemical Structures and Performance of Perovskite Oxides. Chem. Rev. 2001, 101, 1981–2018. [Google Scholar] [CrossRef]
- Roy, P.; Berger, S.; Schmuki, P. TiO2 nanotubes: Synthesis and applications. Angew. Chem. Int. Ed. Engl. 2011, 50, 2904–2939. [Google Scholar] [CrossRef]
- Macák, J.M.; Tsuchiya, H.; Schmiuki, P. High-aspect-ratio TiO2 nanotubes by anodization of titanium. Angew. Chem. Int. Ed. Engl. 2005, 44, 2100–2102. [Google Scholar] [CrossRef]
- Bavykin, D.V.; Lapkin, A.A.; Plucinski, P.K.; Friderich, J.M.; Walsh, F.C. Reversible Storage of Molecular Hydrogen by Sorption into Multilayered TiO2 Nanotubes. J. Phys. Chem. B 2005, 109, 19422–19427. [Google Scholar] [CrossRef]
- Huang, R.W.J.M.; Chung, F.; Kelder, E.M. Impedance Simulation of a Li-Ion Battery with Porous Electrodes and Spherical Li+. Intercalation Part. J. Electrochem. Soc. 2006, 153, A1459–A1465. [Google Scholar] [CrossRef][Green Version]
- Kukovecz, Á.; Pótári, G.; Oszkó, A.; Kónya, Z.; Erdőhelyi, A.; Kiss, J. Probing the interaction of Au, Rh and bimetallic Au-Rh clusters with the TiO2 nanowire and nanotube support. Surf. Sci. 2011, 605, 1048–1055. [Google Scholar] [CrossRef]
- Madarász, D.; Pótári, G.; Sápi, A.; László, B.; Csudai, C.; Oszkó, A.; Kukovecz, Á.; Erdőhelyi, A.; Kónya, Z.; Kiss, J. Metal loading determines the stabilization pathway for Co2+ in the titanate nanowires: Ion exchange vs. cluster formation. J. Phys. Chem. Chem. Phys. 2013, 15, 15917–15925. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kasuga, T.; Hiramatsu, M.; Hoson, A.; Sekino, T.; Niihara, K. Formation of Titanium Oxide Nanotube. Langmuir 1998, 14, 3160–3163. [Google Scholar] [CrossRef]
- Horváth, E.; Kukovecz, Á.; Kónya, Z.; Kiricsi, I. Hydrothermal Conversion of Self-Assembled Titanate Nanotubes into Nanowires in a Revolving Autoclave. Chem. Mater. 2007, 19, 927–931. [Google Scholar] [CrossRef]
- Kukovecz, Á.; Hodos, M.; Horváth, E.; Radnóczi, G.; Kónya, Z.; Kiricsi, I. Oriented Crystal Growth Model Explains the Formation of Titania Nanotubes. J. Phys. Chem. B 2005, 109, 17781–17783. [Google Scholar] [CrossRef] [PubMed]
- Torrente-Murciano, L.; Lapkin, A.A.; Chadwick, D. Synthesis of high aspect ratio titanate nanotubes. J. Mater. Chem. 2010, 20, 6484–6489. [Google Scholar] [CrossRef]
- Sun, X.; Li, Y. Synthesis and Characterization of Ion-Exchangeable Titanate Nanotubes. Chem. Eur. J. 2003, 9, 2229–2238. [Google Scholar] [CrossRef]
- Cesano, F.; Bertarione, S.; Uddin, M.J.; Agostini, G.; Scarano, D.; Zeccina, A. Designing TiO2 Based Nanostructures by Control of Surface Morphology of Pure and Silver Loaded Titanate Nanotubes. J. Phys. Chem. C 2010, 114, 169–178. [Google Scholar] [CrossRef]
- Pusztai, P.; Puskás, R.; Varga, E.; Erdőhelyi, A.; Kukovecz, Á.; Kónya, Z.; Kiss, J. Influence of gold additives on the stability and phase transformation of titanate nanostructures. Phys. Chem. Chem. Phys. 2014, 16, 26786–26797. [Google Scholar] [CrossRef]
- Bavykin, D.V.; Walsh, F.C. Titanate and Titania Nanotubes: Synthesis, Properties and Application; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Kukovecz, Á.; Kordás, K.; Kiss, J.; Kónya, Z. Atomic scale characterization and surface chemistry of metal modified titanate nanotubes and nanowires. Surf. Sci. Rep. 2016, 71, 473–546. [Google Scholar] [CrossRef]
- Bavykin, D.V.; Friedrich, J.M.; Walsh, F.C. Protonated Titanates and TiO2 Nanostructured Materials: Synthesis, Properties, and Applications. Adv. Mater. 2006, 18, 2807–2824. [Google Scholar] [CrossRef]
- Kiss, J.; Pusztai, P.; Óvári, L.; Baán, K.; Merza, G.; Erdőhelyi, A.; Kukovecz, Á.; Kónya, Z. Decoration of Titanate Nanowires and Nanotubes by Gold Nanoparticles: XPS, HRTEM and XRD Characterizatione. J. Surf. Sci. Nanotechnol. 2014, 12, 252–258. [Google Scholar] [CrossRef][Green Version]
- Akita, T.; Okumura, M.; Tanaka, K.; Ohkuma, K.; Kohyama, M.; Koyanagi, T.; Data, M.; Tsubota, S.; Haruta, M. Transmission electron microscopy observation of the structure of TiO2 nanotube and Au/TiO2 nanotube catalyst. Surf. Interf. Anal. 2005, 37, 265–269. [Google Scholar] [CrossRef]
- Malwadkar, S.S.; Gholap, R.S.; Awate, S.V.; Korake, P.V.; Chaskar, M.G.; Gupta, N.M. Physico-chemical, photo-catalytic and O2-adsorption properties of TiO2 nanotubes coated with gold nanoparticles. Photochem. Photobiol. A Chem. 2009, 203, 24–31. [Google Scholar] [CrossRef]
- Turki, A.; Kochkar, H.; Guillard, C.; Berhault, G.; Ghorbel, A. Effect of Na content and thermal treatment of titanate nanotubes on the photocatalytic degradation of formic acid. Appl. Catal. B Environ. 2013, 138–139, 401–415. [Google Scholar] [CrossRef]
- Idakiev, V.; Yuan, Z.Y.; Tabakova, T.; Su, B.L. Titanium oxide nanotubes as supports of nano-sized gold catalysts for low temperature water-gas shift reaction. Appl. Catal. A Gen. 2005, 281, 149–155. [Google Scholar] [CrossRef]
- Méndez-Cruz, M.; Ramírez-Solís, J.; Zanella, R. CO oxidation on gold nanoparticles supported over titanium oxide nanotubes. Catal. Today 2011, 166, 172–179. [Google Scholar] [CrossRef]
- Zhu, B.; Guo, Q.; Huang, X.; Wang, S.; Zhang, S.; Wu, S.; Huang, W. Characterization and catalytic performance of TiO2 nanotubes-supported gold and copper particles. J. Mol. Catal. A Chem. 2006, 249, 211–217. [Google Scholar] [CrossRef]
- László, B.; Baán, K.; Ferencz, Z.; Galbács, G.; Oszkó, A.; Kiss, J.; Kónya, Z.; Erdőhelyi, A. Gold size effect in the thermal-induced reaction of CO2 and H2 on titania and titanate nanotube-supported gold catalysts. J. Nanosci. Nanotechnol. 2019, 19, 470–477. [Google Scholar] [CrossRef]
- László, B.; Baán, K.; Oszkó, A.; Erdőhelyi, A.; Kiss, J.; Kónya, Z. Hydrogen evolution in the photocatalytic reaction between methane and water in the presence of CO2 on titanate and titania supported Rh an Au catalysts. Top. Catal. 2018, 61, 875–888. [Google Scholar] [CrossRef]
- Bavykin, D.V.; Lapkin, A.A.; Plucinski, P.K.; Torrente-Murciano, L.; Friedrich, J.M.; Walsh, F.C. Deposition of Pt, Pd, Ru, and Au on the Surface of Titanate Nanotubes. Top. Catal. 2006, 39, 151–160. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Kiatkittipong, K.; Scott, J.; Amal, R. Hydrothermal synthesized titanate nanostructures: Impact of heat treatment of particle characteristics and photocatalytic properties. ACS Appl. Mater. Inteface 2011, 3, 3988–3996. [Google Scholar] [CrossRef] [PubMed]
- Pirila, M.; Lennkeri, R.; Goldman, W.M.; Kordas, K.; Huuhtanen, M.; Keiski, L. Photocatalytic Degradation of Butanol in Aqueous Solution by TiO2 Nanofibers. Top. Catal. 2013, 56, 630–636. [Google Scholar] [CrossRef]
- Wu, M.-C.; Sapi, A.; Avila, A.; Szabó, M.; Hiltunen, J.; Tóth, G.; Kukovecz, Á.; Kónya, Z.; Keiski, R.; Su, W.-F.; Jantunen, H.; et al. Enhanced Photocatalytic Activity of TiO2 Nanofiber and Their Flexible Composite Films: Decomposition of Organic Dyes and Efficent H2 Generationfrom Ethanol-Water Mixtures. Nano Res. 2011, 4, 360–369. [Google Scholar] [CrossRef]
- Wu, M.-C.; Hiltunen, J.; Avila, A.; Larson, W.; Liao, H.-C.; Huuthanen, M.; Tóth, G.; Schhukarev, A.; Laufer, N.; Kukovecz, Á.; Kónya, Z.; et al. Nitrogen-Doped Anatase Nanofibers Decorated with Noble Metal Nanoparticles for Photocatalytic Production of Hydrogen. ACS Nano 2010, 5, 5025–5030. [Google Scholar] [CrossRef]
- Wu, M.-C.; Tóth, G.; Sápi, A.; Leoni, A.R.; Kónya, Z.; Kukovecz, Á.; Su, W.-F.; Kordás, K. Synthesis and photocatalytic performance of titanium dioxide nanofibers and fabrication of flexible composite films fromnanofibers. J. Nanosci. Nanotech. 2012, 12, 1421–1424. [Google Scholar] [CrossRef]
- Kordás, K.; Mohl, M.; Kónya, Z.; Kukovecz, Á. Layered Titanate Nanostructures: Perspectives and for Industrial Exploitation. Transl. Mater. Res. 2015, 2, 015003. [Google Scholar] [CrossRef]
- Hoang, S.; Lu, X.; Tang, W.; Wang, S.; Du, S.; Nam, C.-Y.; Ding, Y.; Vinluan, R.D.; Zheng, J.; Gao, P.-X. High Performance Diesel Oxidation Catalysts Using Ultra-Low Pt Loading on Titania Nanowire Array Integrated Cordierite Honeycombs. Catal. Today 2019, 320, 2–10. [Google Scholar] [CrossRef]
- Lu, X.; Tang, W.; Du, S.; Wen, L.; Weng, J.; Ding, Y.; Willis, W.S.; Suib, S.L.; Gao, P.X. Ion-Exchange Loading Promoted Stability of Platinum Catalysts Supported on Layered Protonated Titania-Deived Nanoarrays. ACS Appl. Mater. Interfaces 2019, 11, 21515–21525. [Google Scholar] [CrossRef]
- Kolen’ko, Y.V.; Kovnir, K.A.; Gavrilov, A.I.; Garshev, A.V.; Frantti, J.; Lebedev, O.I.; Churagulov, B.R.; Van Tendeloo, G.; Yoshimura, M. Hydrothermal Synthesis and Characterization of Nanorods of Various Titanates and Titanium Dioxide. J. Phys. Chem. B 2006, 110, 4030–4038. [Google Scholar] [CrossRef]
- Ma, R.; Fukuda, K.; Sasaki, T.; Osada, M.; Bando, Y. Structural Features of Titanate Nanotubes/Nanobelts Revealed by Raman, X-ray Absorption Fine Structure and Electron Diffraction Characterizations. J. Phys. Chem. B 2005, 109, 6210–6214. [Google Scholar] [CrossRef] [PubMed]
- Ohsaka, T.; Izumi, F.; Fujiki, Y. Raman spectrum of anatase, TiO2. J. Raman Spectrosc. 1978, 7, 321–324. [Google Scholar] [CrossRef]
- Du, Y.L.; Deng, Y.; Zhang, M.S. Variable-temperature Raman scattering study on anatase titanium dioxide nanocrystals. J. Phys. Chem. Solids 2006, 67, 2405–2408. [Google Scholar] [CrossRef]
- Scepanovic, M.J.; Grujic-Brojcin, M.; Dohcevic-Mitrovic, Z.D.; Popovic, Z.V. Characterization of Anatase TiO2 Nanopowder by Variable-Temperature Raman Spectroscopy. Sci. Sinter. 2009, 41, 67–73. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Z.Y.; Wu, J.N.; Lu, Q. Preparation and electrochemical capacitance behaviour of TiO2-B nanotubes for hybrid supercapacitor. Mater. Lett. 2012, 71, 120–122. [Google Scholar] [CrossRef]
- Pótári, G.; Madarász, D.; Nagy, L.; László, B.; Sápi, A.; Oszkó, A.; Kukovecz, A.; Erdőhelyi, A.; Kónya, Z.; Kiss, J. Rh-induced Support Transformation Phenomena in Titanate Nanowire and Nanotube Catalysts. Langmuir 2013, 29, 3061–3072. [Google Scholar] [CrossRef]
- Poirier, G.E.; Hance, B.K.; White, J.M. Scanning tunneling microscopic and Auger electron spectroscopic characterization of a model catalyst: Rhodium on titania(001). J. Phys. Chem. 1993, 97, 5965–5972. [Google Scholar] [CrossRef]
- Berkó, A.; Ménesi, G.; Solymosi, F. STM study of rhodium deposition on the TiO2(110)-(1x2) surface. Surf. Sci. 1997, 372, 202–210. [Google Scholar] [CrossRef]
- Óvári, L.; Kiss, J. Growth of Rh nanoclusters on TiO2(110): XPS and LEIS studies. Appl. Surf. Sci. 2006, 252, 8624–8629. [Google Scholar] [CrossRef]
- Evans, J.; Hayden, B.E.; Newton, M.A. A comparison of the chemistry of RhI(acac)(CO)2 and RhI(CO)2Cl adsorbed on TiO2[110]: Development of particulate Rh and oxidative disruption by CO. Surf. Sci. 2000, 462, 169–180. [Google Scholar] [CrossRef]
- Khosravian, H.; Liang, Z.; Uhl, A.; Trenary, M.; Meyer, R. Controlled Synthesis of Rh Nanoparticles on TiO2(110) via Rh(CO)2(acac). J. Phys. Chem. C 2012, 116, 11987–11993. [Google Scholar] [CrossRef]
- Berkó, A.; Ménesi, G.; Solymosi, F. Scanning tunneling microscopy study of the CO-induced structural changes of Rh crystallites supported by TiO2(110). J. Phys. Chem. 1996, 100, 17732–17734. [Google Scholar] [CrossRef]
- Solymosi, F.; Pásztor, M. An infrared study of CO chemisorption on the topology of supported rhodium. J. Phys. Chem. 1985, 89, 4789–4793. [Google Scholar] [CrossRef]
- Prime, M. Infrared study of CO chemisorption on zeolite and alumina supported rhodium. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1978, 74, 2570–2580. [Google Scholar]
- Rice, C.A.; Worley, S.D.; Curtis, C.W.; Guin, J.A.; Tarrer, A.R. The oxidation state of dispersed Rh on Al2O3. J. Chem. Phys. 1981, 74, 6487–6497. [Google Scholar] [CrossRef]
- Varga, E.; Pusztai, P.; Oszkó, A.; Baán, K.; Erdőhelyi, A.; Kónya, Z.; Kiss, J. Stability and Temperature-induced Agglomerization of Rh Nanoparticles Supported by CeO2. Langmuir 2016, 32, 2761–2770. [Google Scholar] [CrossRef]
- Kibis, L.S.; Svintsitskiy, D.A.; Derevyannikova, E.A.; Kardash, T.Y.; Slavinskaya, E.M.; Svetlichnyi, V.A.; Boronin, A.I. From high dispersed Rh3+ to nanoclusters and nanoparticles: Probing the low-temperature NO+CO activity of Rh-doped CeO2 catalysts. Appl. Surf. Sci. 2019, 493, 1055–1066. [Google Scholar] [CrossRef]
- Idriss, H.; Llorca, J. Low Temperature Infrared Study of Carbon Monoxide Adsorption on Rh/CeO2. Catalysts 2019, 9, 598. [Google Scholar] [CrossRef]
- Henry, C.R. Surface studies of supported model catalysts. Surf. Sci. Rep. 1988, 31, 231–233. [Google Scholar] [CrossRef]
- Solymosi, F. Importance of the electric properties of supports in the carrier effect. Catal. Rev. 1967, 1, 233–255. [Google Scholar] [CrossRef]
- Sasahara, A.; Pang, C.L.; Onishi, H. Probe Microscope Observation of Platinum Atoms Deposited on the TiO2(110)-(1 × 1) Surface. J. Phys. Chem. B 2006, 110, 13453–13457. [Google Scholar] [CrossRef] [PubMed]
- Sasahara, A.; Pang, C.L.; Onishi, H. Local Work Function of Pt Clusters Vacuum-Deposited on a TiO2 Surface. J. Phys. Chem. B 2006, 110, 17584–17588. [Google Scholar] [CrossRef] [PubMed]
- Kiss, J.; Németh, R.; Koós, Á.; Raskó, J. Characterization of Au-Rh/TiO2 Bimetallic Nanocatalysts by CO and CH3CN Adsorption: XPS, TEM and FTIR Measurements. J. Nanosci. Nanotech. 2009, 9, 3828–3836. [Google Scholar] [CrossRef]
- Oszkó, A.; Pótári, G.; Erdőhelyi, A.; Kukovecz, Á.; Kónya, Z.; Kiricsi, I.; Kiss, J. Structure of Au-Rh bimetallic system formed on titanate nanowires and nanotubes. Vacuum 2011, 85, 1114–1119. [Google Scholar] [CrossRef]
- Kiss, J.; Óvári, L.; Oszkó, A.; Pótári, G.; Tóth, M.; Baán, K.; Erdőhelyi, A. Structure and reactivity of Au-Rh bimetallic clusters on titanate nanowires, nanotubes and TiO2(110). Catal. Today 2012, 181, 163–170. [Google Scholar] [CrossRef]
- Tóth, M.; Kiss, J.; Oszkó, A.; Pótári, G.; László, B.; Erdőhelyi, A. Hydrogenation of Carbon Dioxide on Rh, Au and Au-Rh Bimetallic Clusters Supported on Titanate Nanotubes, Nanowires and TiO2. Top. Catal. 2012, 55, 747–756. [Google Scholar] [CrossRef]
- Solymosi, F.; Bánsági, T.; Erdőhelyi, A. Infrared study of the surface interaction between H2 and CO2 over rhodium on various supports. J. Chem. Soc. Faraday Trans. 1981, 77, 2645–2657. [Google Scholar] [CrossRef]
- Inoune, T.; Iizuka, T.; Tanabe, K. Hydrogenation of carbon dioxide and carbon monoxide over supported rhodium catalysts under 10 bar pressure. Appl. Catal. 1989, 46, 1–9. [Google Scholar] [CrossRef]
- Novák, E.; Fodor, K.; Szailer, T.; Oszkó, A.; Erdőhelyi, A. CO2 Hydrogenation on Rh/TiO2 Previously Reduced at Different Temperatures. Top. Catal. 2002, 20, 107–117. [Google Scholar] [CrossRef]
- Ruiz-Gracia, J.R.; Fierro-Gonzales, J.C.; Handy, B.E.; Hinojosa-Reyes, L.; De Haro Del Rio, D.A.; Lucio-Ortiz, C.J.; Valle-Cervantes, S.; Flores-Escamilla, G.A. An In Situ Infrared Study of CO2 Hydrogenation to formic Acid by Using Rhodium Supported on Titanate Nanotubes as Catalysts. ChemistrySelect 2019, 4, 4206–4216. [Google Scholar] [CrossRef]
- Óvári, L.; Bugyi, L.; Majzik, Zs.; Berko, A.; Kiss, J. Surface Structure and Composition of Au-Rh Bimetallic nanoclusters on TiO2(110): A LEIS and STM study. J. Phys. Chem. C 2008, 112, 18011–18016. [Google Scholar] [CrossRef]
- Óvári, L.; Berkó, A.; Balázs, N.; Majzik, Zs.; Kiss, J. Formation of Rh−Au Core−Shell Nanoparticles on TiO2(110) Surface Studied by STM and LEIS. Langmuir 2010, 26, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Palotás, K.; Óvári, L.; Vári, G.; Gubó, R.; Farkas, A.P.; Kiss, J.; Berkó, A.; Kónya, Z. Au-Rh surface structures on Rh(111): DFT-insights to the formation of an ordered surface alloy. J. Phys. Chem. C 2018, 122, 22435–22447. [Google Scholar] [CrossRef]
- Tenny, S.A.; Ratliff, J.S.; Roberts, C.C.; He, W.; Ammal, S.C.; Heyden, A.; Chen, D.A. Adsorbate-Induced Changes in the Surface Composition of Bimetallic Clusters: Pt-Au on TiO2(110). J. Phys. Chem. C 2010, 114, 21652–21663. [Google Scholar] [CrossRef]
- Gao, F.; Wang, Y.; Goodman, D.W. Reaction Kinetics and Polarization-Modulation Infrared Reflection Absorption Spectroscopy (PM-IRAS) Investigation of CO Oxidation over Supported Pd-Au Alloy Catalysts. J. Phys. Chem. C 2010, 114, 4036–4043. [Google Scholar] [CrossRef]
- Ozturk, O.; Park., J.B.; Ma, S.; Ratliff, J.S.; Zhou, J.; Mullins, D.R.; Chen, D.A. Probing the interactions of Pt, Rh and bimetallic Pt-Rh clusters with the TiO2(110) support. Surf. Sci. 2007, 601, 3099–3113. [Google Scholar] [CrossRef]
- Ferrando, R.; Jellinek, J.; Johnson, R.I. Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef]
- Kecskés, T.; Raskó, J.; Kiss, J. FTIR and mass spectrometric study of HCOOH interaction with TiO2 supported Rh and Au catalysts. Appl. Catal. A: General 2004, 268, 9–16. [Google Scholar]
- Wang, X.; Hong, Y.; Shi, H.; Szanyi, J. Kitetic modelling and transient DRIFTS-MS studies of CO2 methanation over Ru/Al2O3 catalysts. J. Catal. 2016, 343, 185–195. [Google Scholar] [CrossRef]
- Wang, X.; Shi, H.; Kwak, J.H.; Szanyi, J. Mechanism of CO2 hydrogenation on Pt/Al2O3 Catalysts. J. ACS Catal. 2015, 5, 6337–6349. [Google Scholar] [CrossRef]
- Kecskés, T.; Raskó, J.; Kiss, J. FTIR and mass spectrometric studies on the interaction of formaldehyde with TiO2 supported Pt and Au catalysts. Appl. Catal. A Gen. 2004, 273, 55–62. [Google Scholar] [CrossRef]
- Sápi, A.; Halasi, G.; Kiss, J.; Dobó, D.G.; Juhász, K.L.; Kolcsár, V.J.; Ferencz, Z.; Vári, G.; Matolin, V.; Erdőhelyi, A.; et al. In Situ DRIFTS and NAP-XPS Exploration of the Complexity of CO2 Hydrogenation over Size-Controlled Pt Nanoparticles Supported on Mesoporous NiO. J. Phys. Chem. C 2018, 122, 5553–5565. [Google Scholar] [CrossRef]
- Wang, X.; Shi, H.; Szanyi, J. Controlling selectives in CO2 reduction through mechanistic understanding. Nat. Commun. 2017, 8, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Baltrusaitis, J.; Schuttlefield, J.; Zeitler, E.; Grassian, V.H. Carbon dioxide adsorption on oxide nanoparticle surfaces. Chem. Eng. J. 2011, 170, 471–481. [Google Scholar] [CrossRef]
- Raskó, J.; Kecskés, T.; Kiss, J. Adsorption and reaction of formaldehyde on TiO2-supported Rh catalysts studied by FTIR and mass spectrometry. J. Catal. 2004, 226, 183–191. [Google Scholar] [CrossRef]
- Simanouchi, T. Tables of Molecular Vibrational Frequeces Consolidated, Volume 1; National Bureau of Standards: Washington, DC, USA, 1972. [Google Scholar]
- Ichikawa, M.; Fukushima, T. Infrared studies of metal additive effects on carbon monoxide chemisorption modes on silicon dioxide-supported rhodium-manganese-titanium and iron catalysts. J. Phys. Chem. 1985, 89, 1564–1567. [Google Scholar] [CrossRef]
- Stevenson, S.A.; Lisistsyn, A.; Knözinger, H. Adsorption of carbon monoxide on manganese-promoted rhodium/silica catalysts as studied by infrared spectroscopy. J. Phys. Chem. 1990, 94, 1576–1581. [Google Scholar] [CrossRef]
- Chuang, S.S.C.; Stevens, R.W.; Khatri, R. Mechanism of C2+ oxygenate synthesis on Rh catalysts. Top. Catal. 2005, 32, 225–232. [Google Scholar] [CrossRef]
- Sápi, A.; Kashaboina, U.; Baán, K.; Perez Gomez, J.; Szenti, I.; Halasi, Gy.; Kiss, J.; Nagy, B.; Varga, T.; Kukovecz, Á.; Kónya, Z. Synergetic of Pt nanoparticles and H-ZSM-5 zeolites for efficient CO2 activation: Role of interfacial sites in high activity. Front. Mate. 2019, 6, 127. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of synergy between Pt and oxide support. J. Catal. 2016, 343, 115–120. [Google Scholar] [CrossRef]
- Fisher, A.I.; Bell, A.T. A Comparative Study of CO and CO2 Hydrogenation over Rh/SiO2. J. Catal. 1996, 162, 54–65. [Google Scholar] [CrossRef]
- Yu, K.P.; Yu, W.Y.; Kuo, M.C.; Liou, Y.C.; Chien, S.H. Pt/titanate-nanotubes; A potential catalyst for CO2 hydrogenation. Appl. Catal. B 2008, 281, 112–118. [Google Scholar] [CrossRef]
- Farkas, A.P.; Solymosi, F. Activation and reaction of CO2 on K-promoted Au(111) surface. J. Phys. Chem. C 2009, 113, 19930–19936. [Google Scholar] [CrossRef]
- Byron, S.R.J.; Muruganandam, I.; Murthy, S.S. A review of the water gas shift reaction kinetics. Int. J. Chem. React. Eng. 2010, 8. [Google Scholar] [CrossRef]
- Gokhale, A.A.; Dumesic, J.A.; Mavrikakis, M.M. On the mechanism of low temperature water gas shift reaction on copper. J. Am. Chem. Soc. 2008, 130, 1402–1414. [Google Scholar] [CrossRef]
- Soria, M.A.; Perez, P.; Carabineiro, S.A.C.; Hodar, F.J.M.; Mendes, A.; Madeira, L.M. Effect of the preparation method on the catalytic activity and stability of Au/Fe2O3 catalysts in the low-temperature water–gas shift reaction. Appl.Catal. A Gen. 2014, 470, 45–55. [Google Scholar] [CrossRef]
- Lloyd, L.; Ridler, D.E.; Twigg, M.V. The Water-Gas Shift Reaction. In Catalyst Handbook, 2nd ed.; Frome, Chapter; Twigg, M.V., Ed.; Manson Publishing Ltd.: London, UK, 1996; Volume 6, p. 283. [Google Scholar]
- Ladebeck, J.R.; Wagner, J.P. Catalyst development for water-gas shift. In Handbook of Fuel Cell-Fundamentals, Technology and Applications; Wolf, V., Arnold, L., Hubert, A.G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2003; Volume 3, pp. 190–201. [Google Scholar]
- Li, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Low-temperature water-gas shift reaction over Cu- and Ni-loaded cerium oxide catalysts. Appl. Catal. B Envir. 2000, 27, 179–191. [Google Scholar] [CrossRef]
- Li, J.; Yoon, H.; Oh, H.T.; Washman, E.D. SrCe0.7Zr0.2Eu0.1O3-based hydrogen transport water gas shift reactor. Int. J. Hydrog. Energy 2012, 37, 16006–16012. [Google Scholar] [CrossRef]
- Grabow, L.C.; Gokhale, A.A.; Evans, S.T.; Dumesic, A.; Mavrikakis, M. Mechanism of the water gas shift reaction on Pt: First Principles, Experiments, and microkinetic modeling. J. Phys. Chem. C 2008, 112, 4608–4617. [Google Scholar] [CrossRef]
- Ovesen, C.V.; Clausen, B.S.; Hammershoi, B.S.; Steffensen, G.; Askgaard, T.; Chorkendorff, I.; Norskov, J.K.; Rasmussen, P.B.; Stolze, P.; Taylor, P.A. Microkinetic analysis of the water gas reaction under industrial conditions. J. Catal. 1996, 158, 170–180. [Google Scholar] [CrossRef]
- Kölbel, H.; Bhasttocharga, K.K. Synthese hochschmelzender Paraffine an Ruthenium-Kontakten. Just. Leibigs Ann. Chem. 1958, 618, 67–71. [Google Scholar] [CrossRef]
- Niwa, M.; Lunsford, L.H. The catalityc reactions of CO and H2O over supported rhodium. J. Catal. 1982, 75, 302–313. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdőhelyi, A.; Tombácz, I. Methane synthesis in the H2O+CO reaction over titania supported Rh and Rh-Pt catalysts. Appl. Catal. 1985, 14, 65–67. [Google Scholar] [CrossRef]
- Diagne, C.; Idriss, H.; Kiennemann, A. Hydrogen production by ethanol reforming over Rh/CeO2-ZrO2 catalysts. Catal. Commun. 2002, 3, 565–571. [Google Scholar] [CrossRef]
- Aupretre, F.; Descorme, C.; Duprez, D. Bio-ethanol catalytic steam reforming over supported metal catalysts. Catal. Commun. 2002, 3, 263–267. [Google Scholar] [CrossRef]
- Liguras, D.K.; Kondarides, D.J.; Verykios, X.E. Production of hydrogen for fuel cells by steam-reforming of ethanol over supported noble metal catalysts. Appl. Catal. B Environ. 2003, 43, 345–354. [Google Scholar] [CrossRef]
- Raskó, J.; Hancz, A.; Erdőhelyi, A. Surface species and gas phase products in steam reforming of ethanol on TiO2 and Rh/TiO2. Appl. Catal. A Gen. 2004, 269, 13–25. [Google Scholar] [CrossRef]
- Erdőhelyi, A.; Raskó, J.; Kecskés, T.; Tóth, M.; Dömök, M.; Baán, K. Hydrogen formation in ethanol steam reforming on supported noblemetal catalysts. Catal. Today 2006, 116, 367–376. [Google Scholar] [CrossRef]
- Ferencz, Zs.; Erdőhelyi, A.; Baán, K.; Oszkó, A.; Óvári, L.; Kónya, Z.; Papp, C.; Steinrück, H.-P.; Kiss, J. Effects of Support and Rh Additive on Co-Based Catalysts in the Ethanol Steam Reforming Reaction. ACS Catal. 2014, 4, 1205–1218. [Google Scholar] [CrossRef]
- Idriss, H.; Seebauer, E.G. Reactions of ethanol over metal oxide. J. Mol. Catal. A 2000, 152, 201–212. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Murdoch, M.; Waterhouse, G.I.N.; Metson, J.B.; Keane, M.A.; Llorca, J.; Idriss, H. Photoreaction of ethanol on Au/TiO2 anatase: Comparing the micro to nanoparticlesize activities of the support for hydrogen production. J. Photochem. Photobiol. A Chem. 2010, 216, 250–255. [Google Scholar] [CrossRef]
- Papageorgopoulos, D.C.; Ge, Q.; King, D.A. Synchronous Thermal Desorption and Decomposition of Ethanol on Rh{111}. J. Phys. Chem. 1995, 99, 17645–17649. [Google Scholar] [CrossRef]
- Houtman, C.J.; Barteau, M.A. Divergent Pathways of Acetaldehyde and Ethanol Decarbonylation on the Rh(111) Surface. J. Catal. 1991, 130, 528–546. [Google Scholar] [CrossRef]
- de Lima, S.M.; da Cruz, I.O.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Steam reforming, partal oxidation, and oxidative steam reforming of ethanol over Pt/CeZrO2 catalyst. J. Catal. 2008, 257, 356–368. [Google Scholar] [CrossRef]
- Tóth, M.; Varga, E.; Baán, K.; Oszkó, A.; Kiss, J.; Erdőhelyi, A. Partial oxidation of ethanol on supported Rh catalysts: Effect of the oxide support. J. Mol. Catal. A Chem. 2016, 411, 377–387. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdőhelyi, A.; Kocsis, M. Surface interaction between H2 and CO2 on Rh/Al2O3 studied by adsorption and infrared spectroscopic measurements. J. Catal. 1980, 65, 428–436. [Google Scholar] [CrossRef]
- Di Cosimo, J.I.; Diez, V.K.; Xu, M.; Iglesia, E.; Apesteguia, C.R. Structure and Surface and Catalytic Properties of Mg-Al Basic Oxides. J. Catal. 1998, 178, 499–510. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annealing Temperature (°C) | Assignment | Binding Energy (eV) | FWHM a (eV) | Surface Atomic Ratio Ti3+/Ti4+ | Surface Atomic Ratio O/Ti |
|---|---|---|---|---|---|
| 110 | O 1s | 530.8 | 1.3 | 0.026 | 2.48 |
| Ti3+ 2p3/2 | 457.5 | 1.2 | |||
| Ti4+ 2p3/2 | 459.1 | 1.2 | |||
| 200 | O 1s | 530.8 | 1.2 | 0.048 | 2.17 |
| Ti3+ 2p3/2 | 457.8 | 1.2 | |||
| Ti4+ 2p3/2 | 459.2 | 1.1 | |||
| 300 | O 1s | 530.8 | 1.2 | 0.046 | 1.96 |
| Ti3+ 2p3/2 | 458.1 | 1.4 | |||
| Ti4+ 2p3/2 | 459.4 | 1.1 | |||
| 400 | O 1s | 530.9 | 1.2 | 0.045 | 1.89 |
| Ti3+ 2p3/2 | 458.1 | 1.4 | |||
| Ti4+ 2p3/2 | 459.4 | 1.1 | |||
| 500 | O 1s | 530.8 | 1.2 | 0.060 | 1.97 |
| Ti3+ 2p3/2 | 458.1 | 1.6 | |||
| Ti4+ 2p3/2 | 459.4 | 1.1 |
| Catalyst | Amount of Adsorbed H2 μmol/g | Conversion % | CH4 Formation Rate μmol/gs | Turnover Number s−1 x 10−3 | Ea kJ/mol | Σ C μmol/g | ||
|---|---|---|---|---|---|---|---|---|
| in 5 min | in 80 min | in 5 min | in 80 min | in 80 min | in 80 min | |||
| Rh/TiO2 | 7.9 | 6.9 | 6.7 | 4.9 | 4.4 | 278 | 98.3 | 78.8 |
| Rh/TiONW | 7.5 | 8.9 | 4.5 | 6.6 | 3.2 | 213 | 96.5 | 121.5 |
| Rh/TiONT | 4.1 | 1.4 | 1 | 0.8 | 0.5 | 61 | 88.4 | 132.0 |
| Au-Rh/TiO2 | 2.4 | 3.3 | 2.5 | 2.2 | 1.5 | 312 | 81.3 | 38.9 |
| Au-Rh/TiONW | 5.0 | 1.5 | 1.3 | 1.1 | 0.9 | 90 | 85.3 | 98.6 |
| Au-Rh/TiONT | 2.5 | 0.4 | 0.4 | 0.2 | 0.1 | 20 | 98.8 | 215.7 |
| Au/TiO2 | 0 | 0.0006 | 0.0002 | 3.7 × 10−4 | 1 × 10−4 | - | - | 17.2 |
| Au/TiONW | 0 | 0.005 | 0.09 | 3.5 × 10−3 | 6 × 10−4 | - | - | - |
| Au/TiONT | 0 | 036 | 0.098 | 8.3 × 10−4 | 2.1 × 10−4 | - | - | 3.0 |
| D % | K % | WCO2 nmol/gs | TOF CO2 s−1 | |
|---|---|---|---|---|
| 1% Rh/TiO2 | 36 | 11.4 | 1078.1 | 30.8*10-3 |
| 1% Rh/TiONT | 10 | 3.4 | 295.6 | 30.4*10-3 |
| 1% Rh/TiONW | 29 | 4.7 | 456.8 | 16.2*10-3 |
| 1% (Rh+Au)TiONW | 33 | 1.9 | 176.8 | 5.5*10-3 |
| Conversion % | Selectivity % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C2H4 | C2H4O | H2 | (C2H5)O | |||||||
| min | 5. | 105. | 5. | 105. | 5. | 105. | 5. | 105. | 5. | 105. |
| Rh/TiO2 | 93 | 37 | 6.9 | 1.8 | 77.8 | 95.7 | 13.6 | 5.8 | 7 | 0.1 |
| Rh/TiONW | 44 | 36 | 0.4 | 0.2 | 86.6 | 94.7 | 12.5 | 10.8 | 3.7 | 0.6 |
| Rh/TiONT | 61 | 17 | 0.4 | 0.3 | 78.0 | 91.3 | 25.0 | 10.6 | 1.0 | 3.6 |
| TiONW | 1 | 7.1 | 3.2 | 3.4 | 66.7 | 66.1 | 5.0 | 0 | 26.0 | 28.0 |
| TiONT | 28.7 | 11.7 | 23 | 0.8 | 93 | 96.0 | 6.0 | 0 | 1.0 | 2.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss, J.; Sápi, A.; Tóth, M.; Kukovecz, Á.; Kónya, Z. Rh-induced Support Transformation and Rh Incorporation in Titanate Structures and Their Influence on Catalytic Activity. Catalysts 2020, 10, 212. https://doi.org/10.3390/catal10020212
Kiss J, Sápi A, Tóth M, Kukovecz Á, Kónya Z. Rh-induced Support Transformation and Rh Incorporation in Titanate Structures and Their Influence on Catalytic Activity. Catalysts. 2020; 10(2):212. https://doi.org/10.3390/catal10020212
Chicago/Turabian StyleKiss, János, András Sápi, Mariann Tóth, Ákos Kukovecz, and Zoltán Kónya. 2020. "Rh-induced Support Transformation and Rh Incorporation in Titanate Structures and Their Influence on Catalytic Activity" Catalysts 10, no. 2: 212. https://doi.org/10.3390/catal10020212
APA StyleKiss, J., Sápi, A., Tóth, M., Kukovecz, Á., & Kónya, Z. (2020). Rh-induced Support Transformation and Rh Incorporation in Titanate Structures and Their Influence on Catalytic Activity. Catalysts, 10(2), 212. https://doi.org/10.3390/catal10020212