A CO2-Mediated Conjugate Cyanide Addition to Chalcones


Abstract
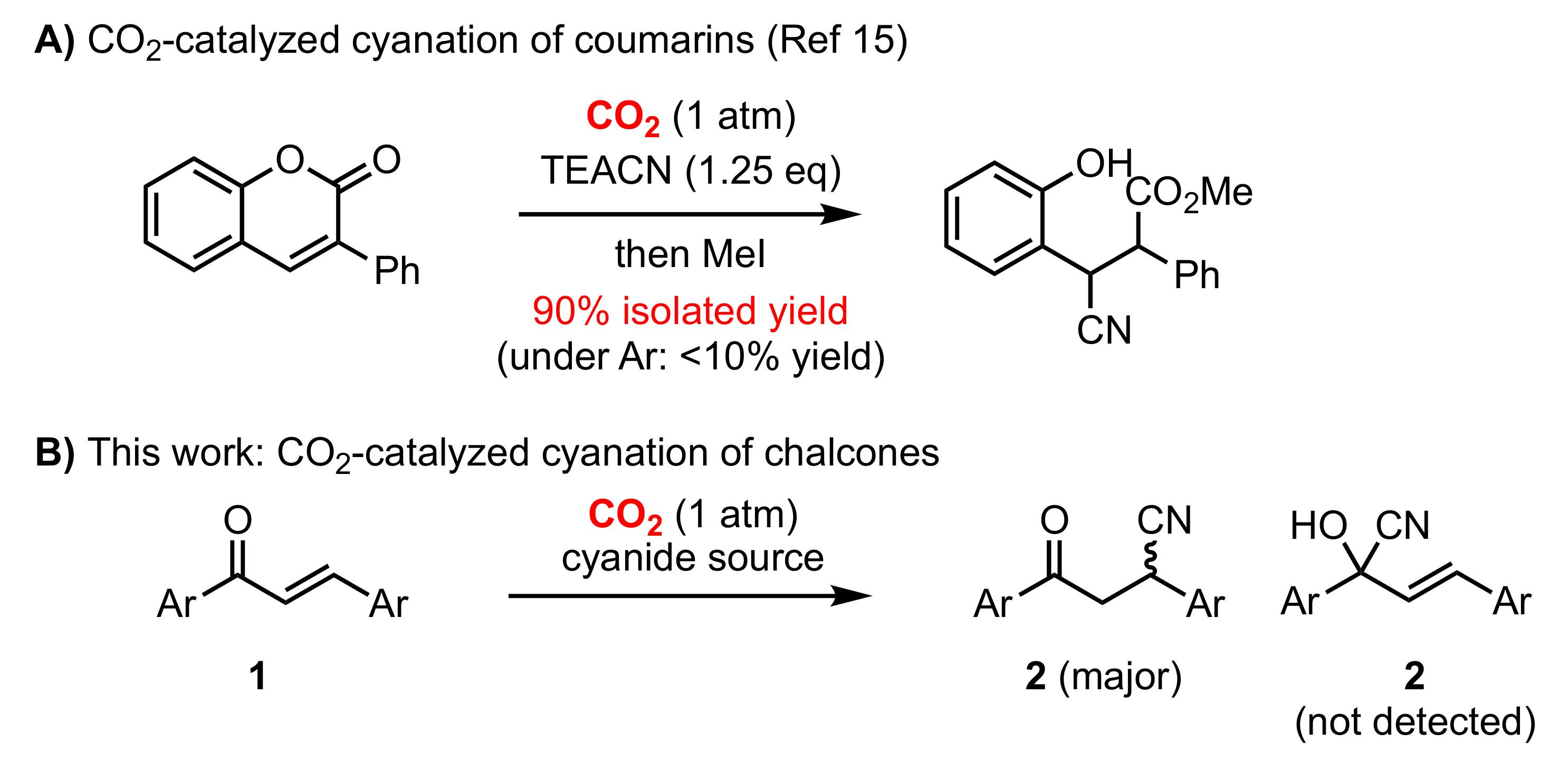
1. Introduction
2. Results
2.1. Optimization
2.2. Substrate Scope
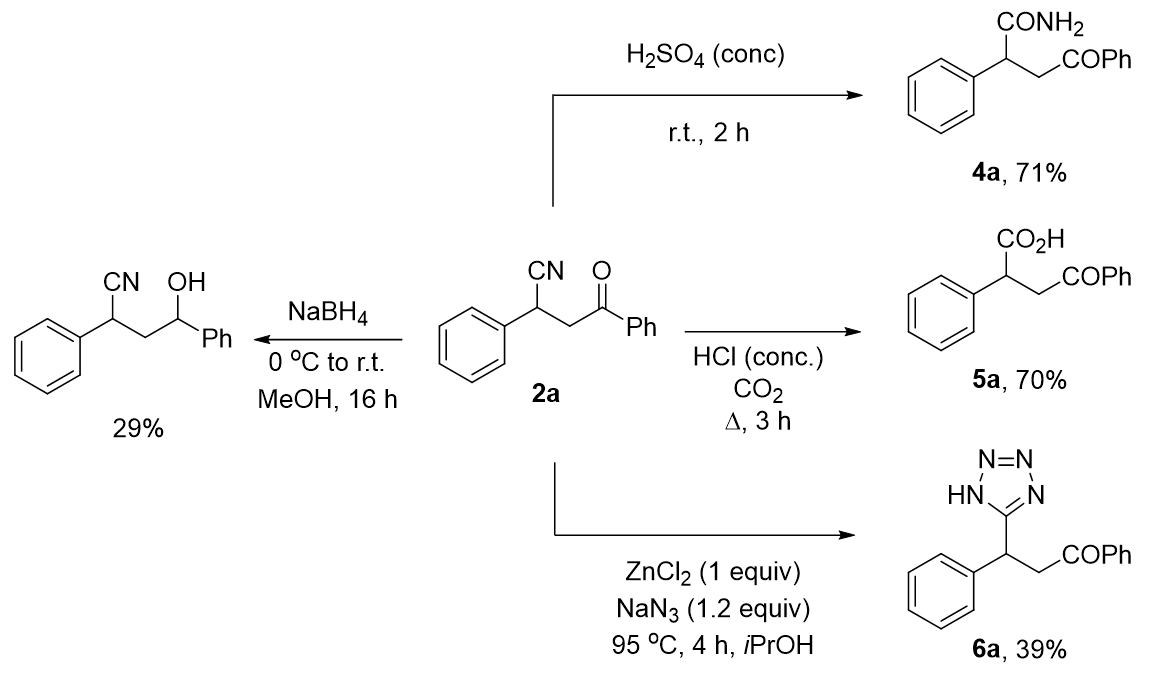
2.3. Functionalization of Cyanated Chalcones
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lee, J.-W. Toward ideal carbon dioxide functionalization. Chem. Sci. 2019, 10, 3905–3926. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Chand-Thakuri, P.; Young, M.C. Carbon Dioxide-Mediated C(sp2)–H Arylation of Primary and Secondary Benzylamines. J. Am. Chem. Soc. 2019, 141, 7980–7989. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Liu, D.; Young, M.C. Carbon Dioxide-Mediated C(sp3)–H Arylation of Amine Substrates. J. Am. Chem. Soc. 2018, 140, 6818–6822. [Google Scholar] [CrossRef]
- White, D.A. Cyanocarboxylation of activated olefins. J. Chem. Soc. Perkin Trans. 1976, 1, 1926–1930. [Google Scholar] [CrossRef]
- Leow, D.; Li, G.; Mei, T.-S.; Yu, J.-Q. Activation of remote meta-C–H bonds assisted by an end-on template. Nature 2012, 486, 518–522. [Google Scholar] [CrossRef]
- Dai, H.-X.; Li, G.; Zhang, X.-G.; Stepan, A.F.; Yu, J.-Q. Pd(II)-Catalyzed ortho- or meta-C–H Olefination of Phenol Derivatives. J. Am. Chem. Soc. 2013, 135, 7567–7571. [Google Scholar] [CrossRef]
- Bornschein, C.; Werkmeister, S.; Wendt, B.; Jiao, H.; Alberico, E.; Baumann, W.; Junge, H.; Junge, K.; Beller, M. Mild and selective hydrogenation of aromatic and aliphatic (di)nitriles with a well-defined iron pincer complex. Nat. Commun. 2014, 5, 4111. [Google Scholar] [CrossRef]
- Ahmed, T.J.; Knapp, S.M.M.; Tyler, D.R. Frontiers in catalytic nitrile hydration: Nitrile and cyanohydrin hydration catalyzed by homogeneous organometallic complexes. Coord. Chem. Rev. 2011, 255, 949–974. [Google Scholar] [CrossRef]
- Chu, X.-Q.; Ge, D.; Shen, Z.-L.; Loh, T.-P. Recent Advances in Radical-Initiated C(sp3)–H Bond Oxidative Functionalization of Alkyl Nitriles. ACS Catal. 2018, 8, 258–271. [Google Scholar] [CrossRef]
- Mattalia, J.-M.R. The reductive decyanation reaction: An overview and recent developments. Beilstein J. Org. Chem. 2017, 13, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D. Methods of Reactivity Umpolung. Angew. Chem. Int. Ed. 1979, 18, 239–258. [Google Scholar] [CrossRef]
- Tolman, C.A.; McKinney, R.J.; Seidel, W.C.; Druliner, J.D.; Stevens, W.R. Homogeneous Nickel-Catalyzed Olefin Hydrocyanation. In Advances in Catalysis; Eley, D.D., Pines, H., Weisz, P.B., Eds.; Academic Press Inc.: Cambridge, MA, USA, 1985; Volume 33, pp. 1–46. [Google Scholar]
- Friedman, L.; Shechter, H. Preparation of Nitriles from Halides and Sodium Cyanide. An Advantageous Nucleophilic Displacement in Dimethyl Sulfoxide1a. J. Org. Chem. 1960, 25, 877–879. [Google Scholar] [CrossRef]
- Roy, T.; Kim, M.J.; Yang, Y.; Kim, S.; Kang, G.; Ren, X.; Kadziola, A.; Lee, H.-Y.; Baik, M.-H.; Lee, J.-W. Carbon Dioxide-Catalyzed Stereoselective Cyanation Reaction. ACS Catal. 2019, 9, 6006–6011. [Google Scholar] [CrossRef]
- Li, Z.-F.; Li, Q.; Ren, L.-Q.; Li, Q.-H.; Peng, Y.-G.; Liu, T.-L. Cyano-borrowing reaction: Nickel-catalyzed direct conversion of cyanohydrins and aldehydes/ketones to β-cyano ketone. Chem. Sci. 2019, 10, 5787–5792. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Fujii, A.; Nakano, Y.; Sakaguchi, S.; Ishii, Y. Acetylcyanation of Aldehydes with Acetone Cyanohydrin and Isopropenyl Acetate by Cp*2Sm(thf)2. J. Org. Chem. 1999, 64, 4214–4216. [Google Scholar] [CrossRef]
- Ramesh, S.; Lalitha, A. Scandium(III) Triflate Catalyzed 1,4-Addition of Cyano Group to Enones Using Tetraethylammonium Cyanide as the Cyanide Source. Acta Chim. Slov. 2013, 60, 689–694. [Google Scholar]
- Guo, S.; Mi, X. Tetraarylphosphonium inner-salts (TAPIS) as both Lewis base catalyst and phase tag. Tetrahedron Lett. 2017, 58, 2881–2884. [Google Scholar] [CrossRef]
- Yang, J.; Shen, Y.; Chen, F.-X. Highly Efficient Cs2CO3-Catalyzed 1,4-Addition of Me3SiCN to Enones with Water as the Additive. Synthesis 2010, 2010, 1325–1333. [Google Scholar] [CrossRef]
- Ciller, J.A.; Seoane, C.; Soto, J.L. Synthesis of Heterocyclic Compounds, XXXVIII. Five-membered Heterocycles by Cyclization of 3-Benzoyl-4-oxobutanenitriles. Liebigs Ann. Chem. 1985, 1985, 51–57. [Google Scholar] [CrossRef]
- Allen, C.F.H.; Kimball, R.K. α-Phenyl-β-Benzoylpropionitrile. Org. Synth. 1930, 10, 80. [Google Scholar]
- Bellinger, T.J.; Harvin, T.; Pickens-Flynn, T.B.; Austin, N.; Whitaker, S.H.; Tang Yuk Tutein, M.L.C.; Hukins, D.T.; Deese, N.; Guo, F. Conjugate Addition of Grignard Reagents to Thiochromones Catalyzed by Copper Salts: A Unified Approach to Both 2-Alkylthiochroman-4-One and Thioflavanone. Molecules 2020, 25, 2128. [Google Scholar] [CrossRef] [PubMed]
- Davis, R. Notes: Condensation of Aromatic Aldehydes with Methyl Aryl Ketones and Sodium Cyanide. J. Org. Chem. 1959, 24, 880–882. [Google Scholar] [CrossRef]
- Vorona, S.; Artamonova, T.; Zevatskii, Y.; Myznikov, L. An Improved Protocol for the Preparation of 5-Substituted Tetrazoles from Organic Thiocyanates and Nitriles. Synthesis 2014, 46, 781–786. [Google Scholar] [CrossRef]
- Schilling, W.; Das, S. CO2-catalyzed/promoted transformation of organic functional groups. Tetrahedron Lett. 2018, 59, 3821–3828. [Google Scholar] [CrossRef]
- Lee, J.-W.; Juhl, M.; Petersen, A.R. CO2-Enabled Cyanohydrin Synthesis and Facile Iterative Homologation Reactions. Chem. Eur. J. 2020. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Lee, J.-W. A CO2-Catalyzed Transamidation Reaction. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Bode, J.W. On the Role of CO2 in NHC-Catalyzed Oxidation of Aldehydes. Org. Lett. 2011, 13, 2422–2425. [Google Scholar] [CrossRef]
- Li, H.; Wu, H.; Yu, Z.; Zhang, H.; Yang, S. CO(2)-Enabled Biomass Fractionation/Depolymerization: A Highly Versatile Pre-Step for Downstream Processing. ChemSusChem 2020, 13, 3565–3582. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}

| Entry. | Deviation from the Standard Reaction Conditions | Yield (%) b |
|---|---|---|
| 1 | Under N2 instead of CO2 | trace |
| 2 | 1.1 equiv. of NEt4CN | 67% |
| 3 | 3 equiv. of NEt4CN | 81% |
| 4 | KCN (2 equiv) instead of NEt4CN | 19% |
| 5 | KCN (3.3 equiv) instead of NEt4CN + 2 equiv of NMe4Cl | 72% |
| 6 | DCM as a solvent | 20% |
| 7 | DMF as a solvent | 35% |
| 8 | DMSO as a solvent | 65% |
| 9 | un-optimal solvents (EtOH, Et2O, TFH, dioxane) under CO2/N2 | n.r. |

| Entry | Chalcone, 1 | Time/h | Product, 2 | Yield/% c |
|---|---|---|---|---|
| 1 |  | 7.5 |  | 73 |
| 2 |  | 2.5 |  | 63 |
| 3 |  | 2 |  | 13 b |
| 4 |  | 2 |  | 59 |
| 5 |  | 7.5 |  | 64 |
| 6 |  | 2 |  | 20 b |
| 7 |  | 18 |  | 15 d |
| 8 |  | 18 |  | 37 |
| 9 |  | 18 |  | 74 |
| 10 |  | 18 |  | 23 |
| 11 |  | 18 |  | 41 |
| 12 |  | 18 |  | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dotzauer, S.; Hadaf, G.B.; Kamounah, F.S.; Kadziola, A.; Lee, J.-W. A CO2-Mediated Conjugate Cyanide Addition to Chalcones. Catalysts 2020, 10, 1481. https://doi.org/10.3390/catal10121481
Dotzauer S, Hadaf GB, Kamounah FS, Kadziola A, Lee J-W. A CO2-Mediated Conjugate Cyanide Addition to Chalcones. Catalysts. 2020; 10(12):1481. https://doi.org/10.3390/catal10121481
Chicago/Turabian StyleDotzauer, Simon, Gul Barg Hadaf, Fadhil S. Kamounah, Anders Kadziola, and Ji-Woong Lee. 2020. "A CO2-Mediated Conjugate Cyanide Addition to Chalcones" Catalysts 10, no. 12: 1481. https://doi.org/10.3390/catal10121481
APA StyleDotzauer, S., Hadaf, G. B., Kamounah, F. S., Kadziola, A., & Lee, J.-W. (2020). A CO2-Mediated Conjugate Cyanide Addition to Chalcones. Catalysts, 10(12), 1481. https://doi.org/10.3390/catal10121481