Direct Electrochemical Enzyme Electron Transfer on Electrodes Modified by Self-Assembled Molecular Monolayers



Abstract
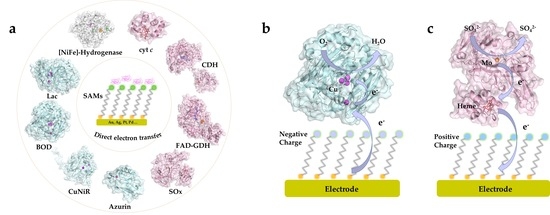
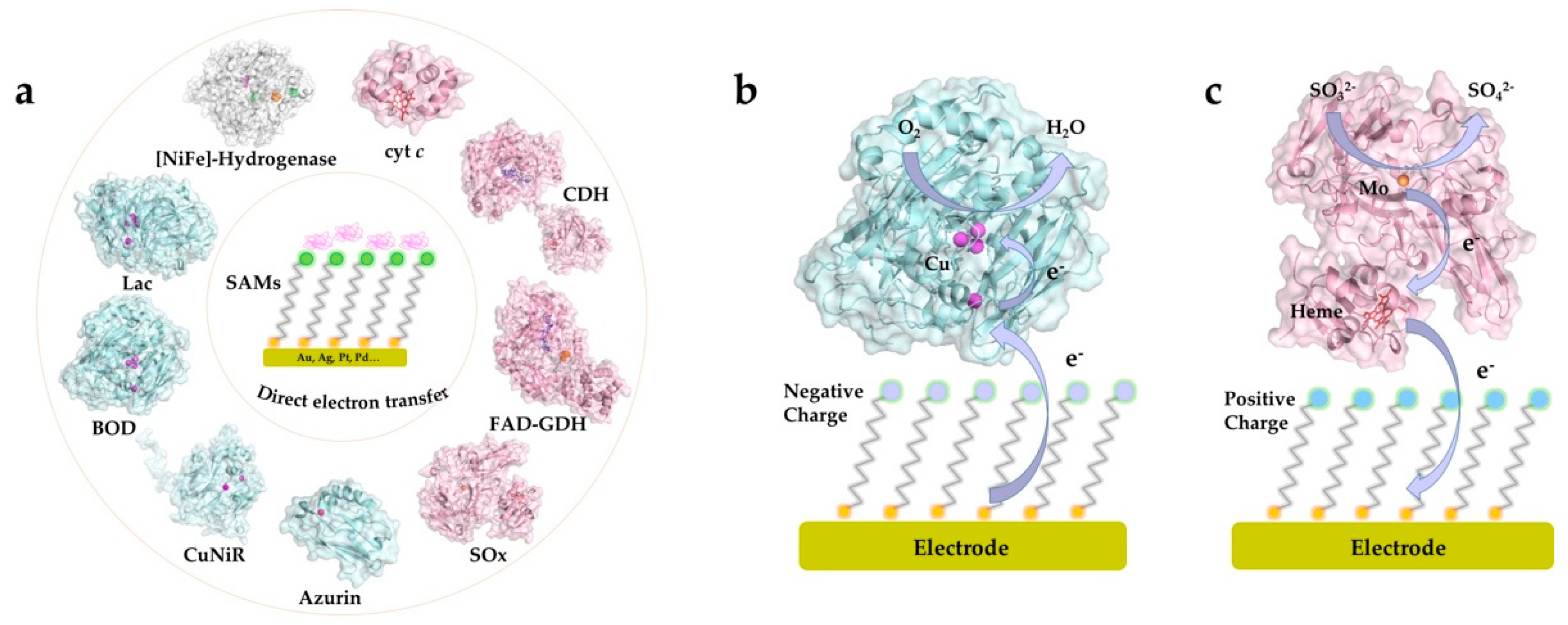
{kind=link}
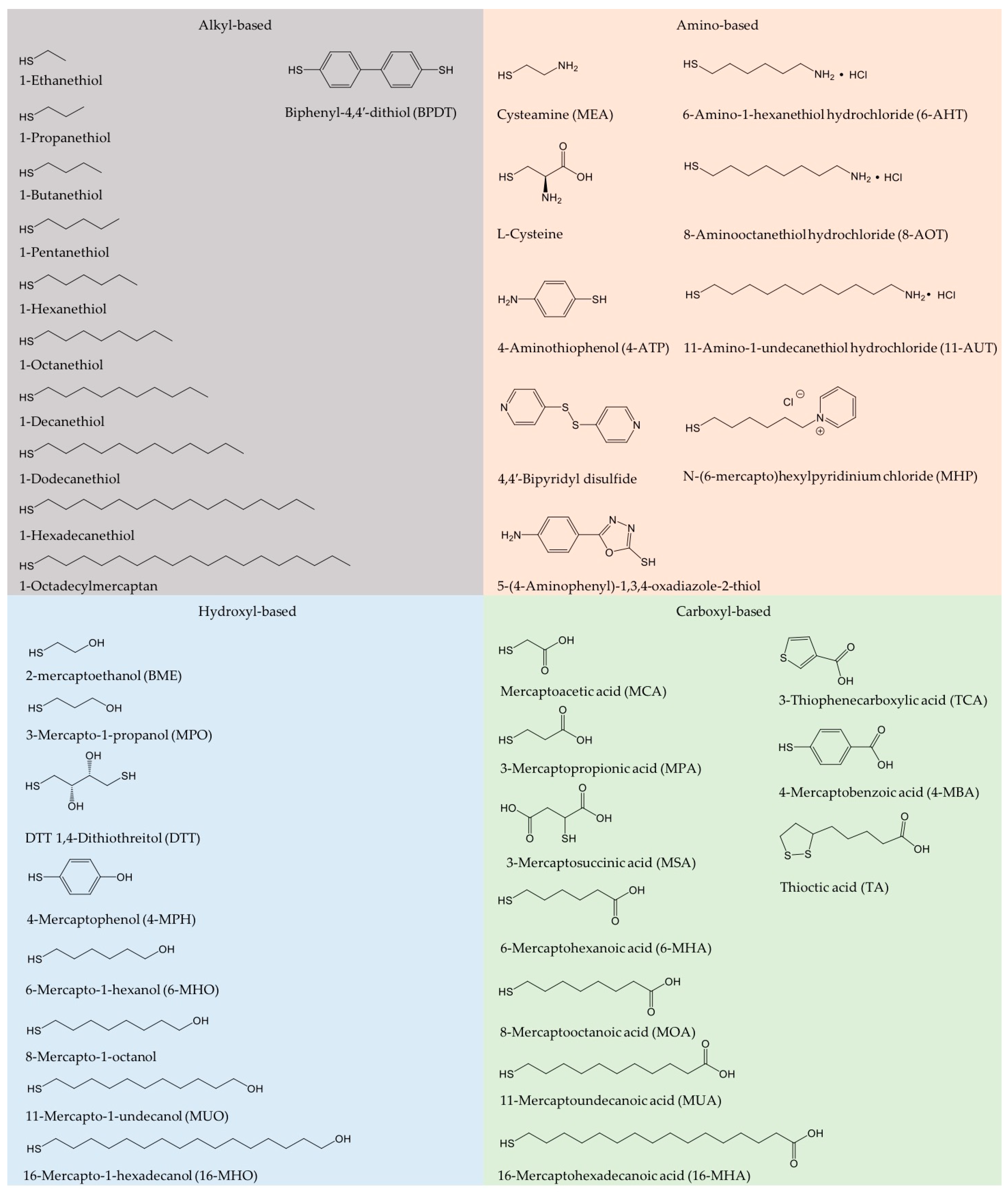{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Preparation and Characterization of SAM-Based Au- and Other Electrodes
2.1. Preparation of SAMs
2.2. Characterization Methods
2.2.1. Electrochemistry
2.2.2. Microscopy
2.2.3. Spectroscopy
3. SAMs and Electrochemical DET of Redox Proteins/Enzymes
3.1. Heme-Containing Proteins
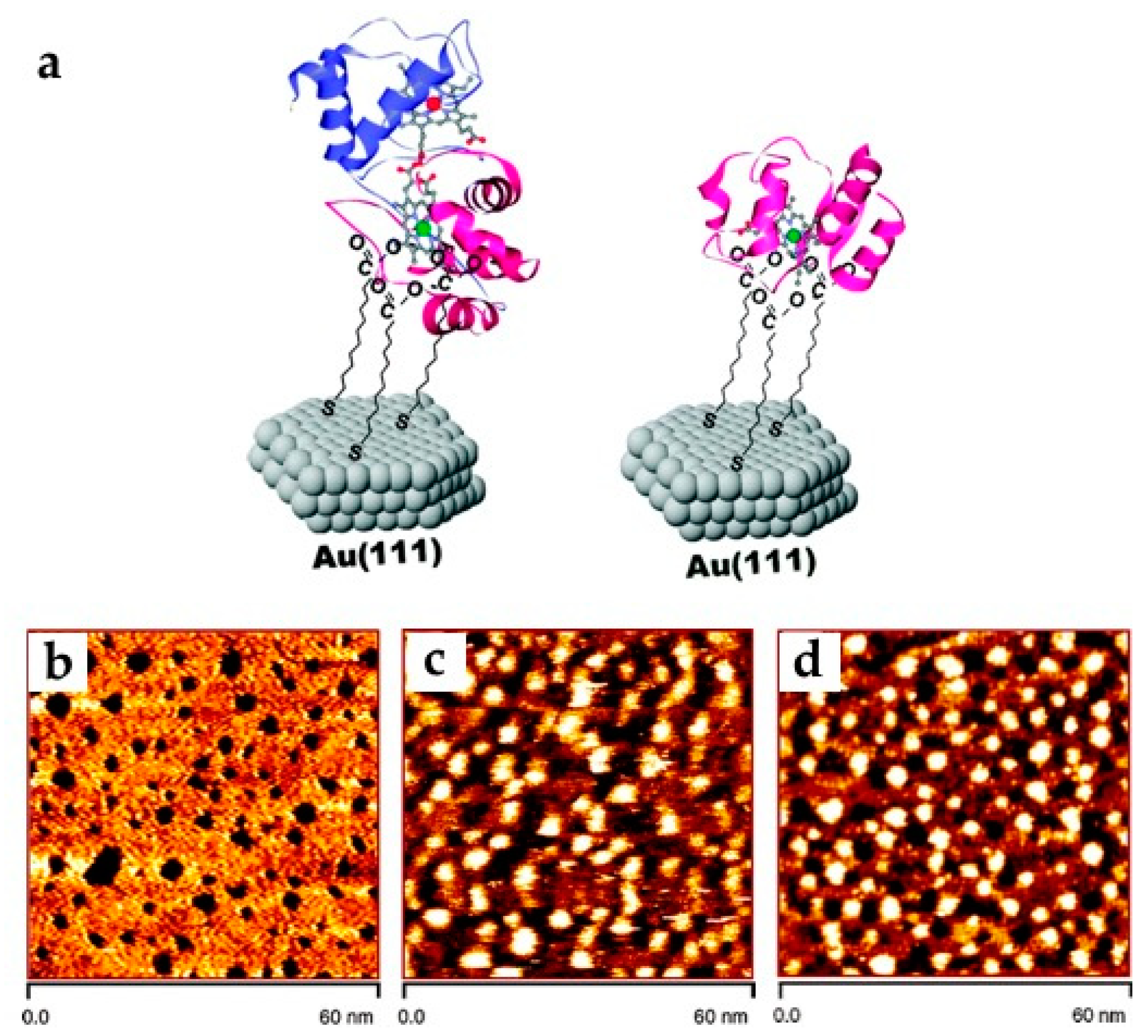
3.1.1. Cytochrome c
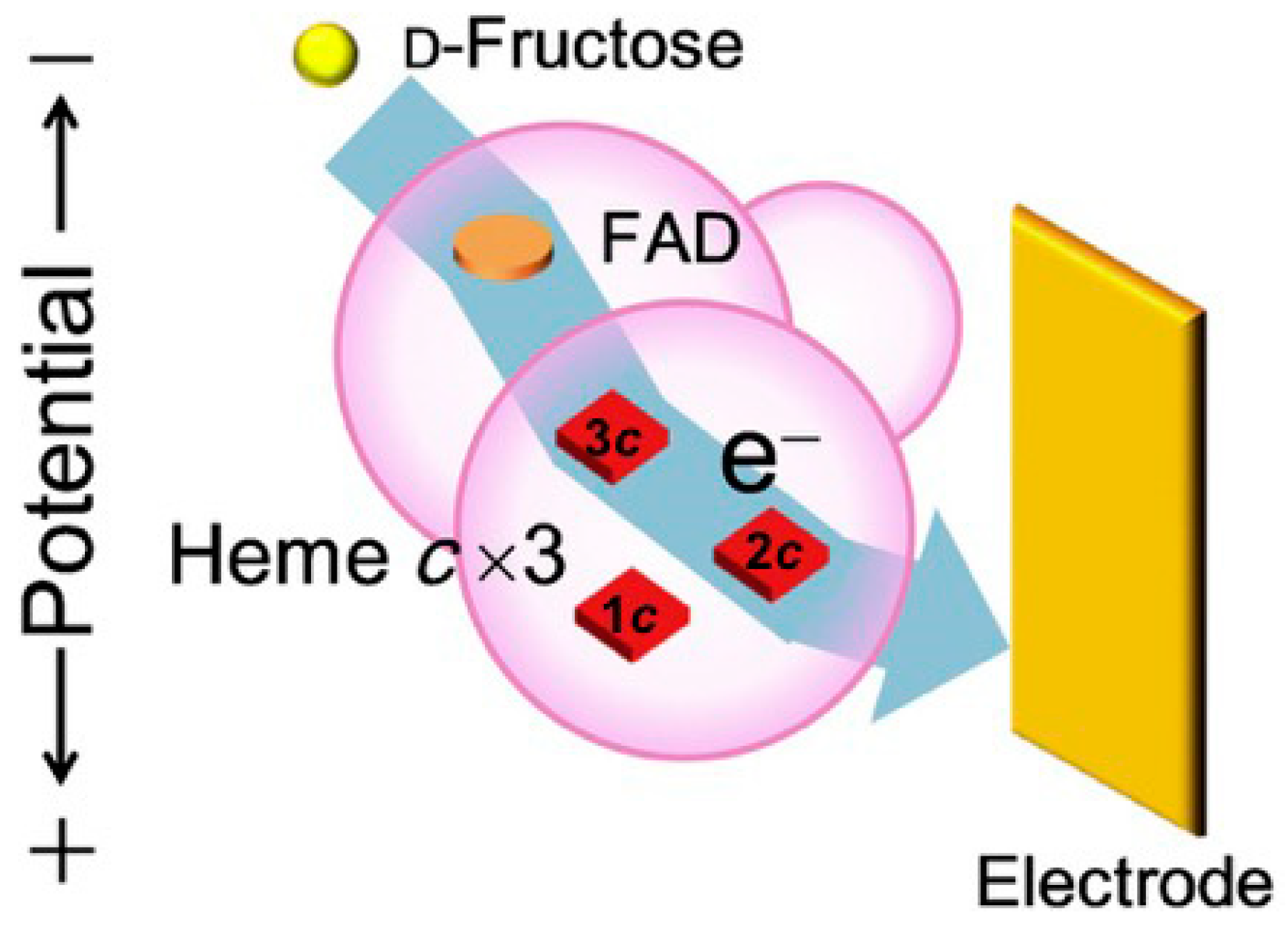
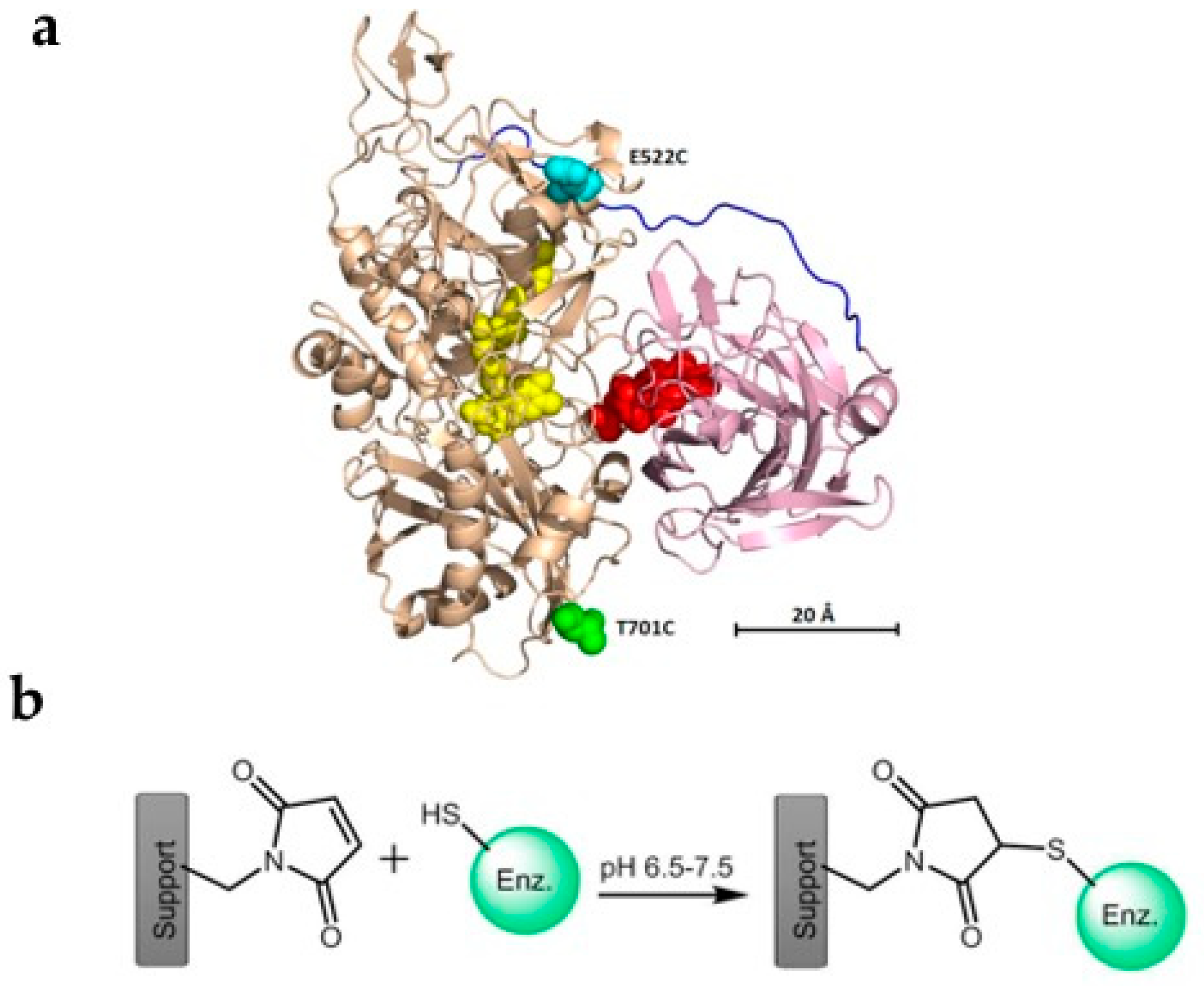
3.1.2. Fructose Dehydrogenase
3.1.3. FAD-Dependent Glucose Dehydrogenase
3.1.4. Sulfite Oxidase
3.2. Blue Copper Proteins
3.2.1. Azurin
3.2.2. Copper Nitrite Reductase
3.2.3. Bilirubin Oxidase
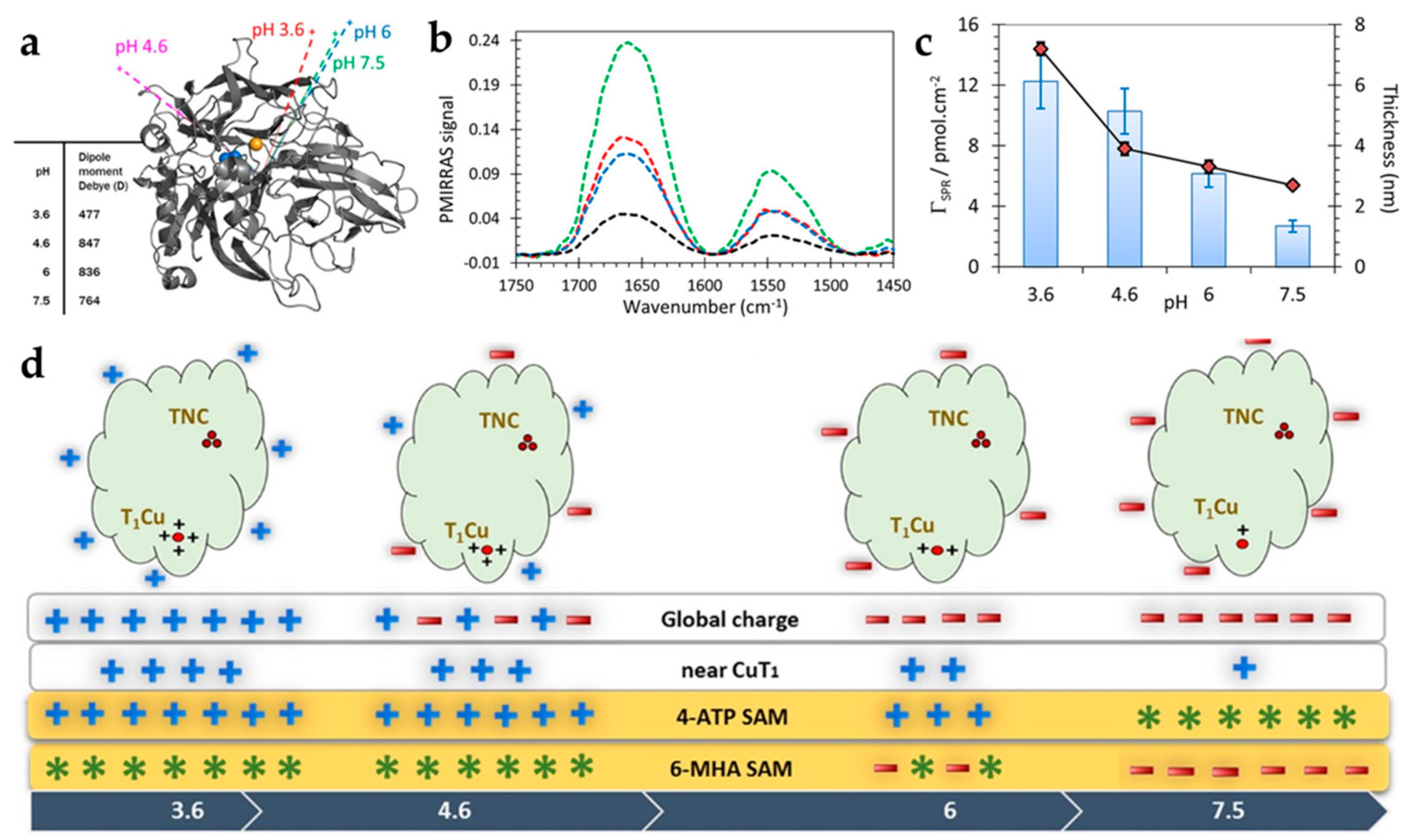
3.2.4. Laccase
3.3. [FeS]-Cluster Hydrogenases
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1170. [Google Scholar] [CrossRef] [PubMed]
- Nöll, T.; Nöll, G. Strategies for “wiring” redox-active proteins to electrodes and applications in biosensors, biofuel cells, and nanotechnology. Chem. Soc. Rev. 2011, 40, 3564–3576. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P. Porous Gold: A New Frontier for Enzyme-Based Electrodes. Nanomaterials 2020, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Chi, Q.; Ford, M.J.; Halder, A.; Hush, N.S.; Reimers, J.R.; Ulstrup, J. Sulfur ligand mediated electrochemistry of gold surfaces and nanoparticles: What, how, and why. Curr. Opin. Electrochem. 2017, 1, 7–15. [Google Scholar] [CrossRef]
- Ulman, A. Formation and structure of self-assembled monolayers. Chem. Rev. 1996, 96, 1533–1554. [Google Scholar] [CrossRef]
- Zhang, J.; Kuznetsov, A.M.; Medvedev, I.G.; Chi, Q.; Albrecht, T.; Jensen, P.S.; Ulstrup, J. Single-molecule electron transfer in electrochemical environments. Chem. Rev. 2008, 108, 2737–2791. [Google Scholar] [CrossRef]
- Bigelow, W.C.; Pickett, D.L.; Zisman, W.A. Oleophobic monolayers. I. Films adsorbed from solution in non-polar liquids. J. Colloid Sci. 1946, 1, 513–538. [Google Scholar] [CrossRef]
- Nuzzo, R.G.; Allara, D.L. Adsorption of Bifunctional Organic Disulfides on Gold Surfaces. J. Am. Chem. Soc. 1983, 105, 4481–4483. [Google Scholar] [CrossRef]
- Muskal, N.; Mandler, D. Thiol self-assembled monolayers on mercury surfaces: The adsorption and electrochemistry of v-mercaptoalkanoic acids. Electrochim. Acta 1999, 45, 537–548. [Google Scholar] [CrossRef]
- Williams, J.A.; Gorman, C.B. Alkanethiol Reductive Desorption from Self-Assembled Monolayers on Gold, Platinum, and Palladium Substrates. J. Phys. Chem. C 2007, 111, 12804–12810. [Google Scholar] [CrossRef]
- Hoque, E.; Derose, J.A.; Houriet, R.; Hoffmann, P.; Mathieu, H.J.; Fe, E.P. Stable Perfluorosilane Self-Assembled Monolayers on Copper Oxide Surfaces: Evidence of Siloxy-Copper Bond Formation. Chem. Mater. 2007, 19, 798–804. [Google Scholar] [CrossRef]
- Fischer, S.; Papageorgiou, A.C.; Marschall, M.; Reichert, J.; Diller, K.; Klappenberger, F.; Allegretti, F.; Nefedov, A.; Wöll, C.; Barth, J.V. L-Cysteine on Ag(111): A combined STM and X-ray spectroscopy study of anchorage and deprotonation. J. Phys. Chem. C 2012, 116, 20356–20362. [Google Scholar] [CrossRef]
- Alejandra, M.; Addato, F.; Rubert, A.A.; Benítez, G.A.; Fonticelli, M.H.; Carrasco, J.; Carro, P.; Salvarezza, R.C. Alkanethiol Adsorption on Platinum: Chain Length Effects on the Quality of Self-Assembled Monolayers. J. Phys. Chem. C 2011, 115, 17788–17798. [Google Scholar] [CrossRef]
- Feliciano-ramos, I.; Caban-acevedo, M.; Scibioh, M.A.; Cabrera, C.R. Self-assembled monolayers of L-cysteine on palladium electrodes. J. Electroanal. Chem. 2010, 650, 98–104. [Google Scholar] [CrossRef]
- Gooding, J.J.; Ciampi, S. The molecular level modification of surfaces: From self-assembled monolayers to complex molecular assemblies. Chem. Soc. Rev. 2011, 40, 2704–2718. [Google Scholar] [CrossRef]
- Reimers, J.R.; Ford, M.J.; Marcuccio, S.M.; Ulstrup, J.; Hush, N.S. Competition of van der Waals and chemical forces on gold-sulfur surfaces and nanoparticles. Nat. Rev. Chem. 2017, 1, 1–13. [Google Scholar] [CrossRef]
- Reimers, J.R.; Ford, M.J.; Halder, A.; Ulstrup, J.; Hush, N.S. Gold surfaces and nanoparticles are protected by Au(0)-thiyl species and are destroyed when Au(I)-thiolates form. Proc. Natl. Acad. Sci. USA 2016, 113, E1424–E1433. [Google Scholar] [CrossRef]
- Tang, J.; Yan, X.; Engelbrekt, C.; Ulstrup, J.; Magner, E.; Xiao, X.; Zhang, J. Development of graphene-based enzymatic biofuel cells: A minireview. Bioelectrochemistry 2020, 134, 107537. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Clement, R.; Bourassin, N.; Baaden, M.; de Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Lojou, E. Controlling redox enzyme orientation at planar electrodes. Catalysts 2018, 8, 192. [Google Scholar] [CrossRef]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253. [Google Scholar] [CrossRef]
- Mano, N.; De Poulpiquet, A. O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev. 2018, 118, 2392–2468. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nature 1992, 355, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Kittl, R.; Ludwig, R.; Gorton, L. Direct Electron Transfer from the FAD Cofactor of Cellobiose Dehydrogenase to Electrodes. ACS Catal. 2016, 6, 555–563. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.S.; Reginald, S.S.; Baek, S.; Lee, E.M.; Choi, I.G.; Chang, I.S. Biosensing and electrochemical properties of flavin adenine dinucleotide (FAD)-Dependent glucose dehydrogenase (GDH) fused to a gold binding peptide. Biosens. Bioelectron. 2020, 165, 112427. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The Immobilization of Fructose Dehydrogenase on Nanoporous Gold Electrodes for the Detection of Fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Gutierrez-Sanchez, C.; Ciaccafava, A.; Blanchard, P.Y.; Monsalve, K.; Giudici-Orticoni, M.T.; Lecomte, S.; Lojou, E. Efficiency of Enzymatic O2 Reduction by Myrothecium verrucaria Bilirubin Oxidase Probed by Surface Plasmon Resonance, PMIRRAS, and Electrochemistry. ACS Catal. 2016, 6, 5482–5492. [Google Scholar] [CrossRef]
- Di Bari, C.; Shleev, S.; De Lacey, A.L.; Pita, M. Laccase-modified gold nanorods for electrocatalytic reduction of oxygen. Bioelectrochemistry 2016, 107, 30–36. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.; Conghaile, P.; Pita, M.; Ludwig, R.; Magner, E. Immobilization of Redox Enzymes on Nanoporous Gold Electrodes: Applications in Biofuel Cells. ChemPlusChem 2017, 82, 553–560. [Google Scholar] [CrossRef]
- Murata, K.; Kajiya, K.; Nakamura, N.; Ohno, H. Direct electrochemistry of bilirubin oxidase on three-dimensional gold nanoparticle electrodes and its application in a biofuel cell. Energy Environ. Sci. 2009, 2, 1280–1285. [Google Scholar] [CrossRef]
- Adachi, T.; Kaida, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Bioelectrocatalytic performance of D-fructose dehydrogenase. Bioelectrochemistry 2019, 129, 1–9. [Google Scholar] [CrossRef]
- Xiao, X.; Si, P.; Magner, E. An overview of dealloyed nanoporous gold in bioelectrochemistry. Bioelectrochemistry 2016, 109, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Xia, H.Q.; Wu, R.; Bai, L.; Yan, L.; Magner, E.; Cosnier, S.; Lojou, E.; Zhu, Z.; Liu, A. Tackling the Challenges of Enzymatic (Bio)Fuel Cells. Chem. Rev. 2019, 119, 9509–9558. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236. [Google Scholar] [CrossRef]
- Vaz-Dominguez, C.; Campuzano, S.; Rüdiger, O.; Pita, M.; Gorbacheva, M.; Shleev, S.; Fernandez, V.M.; De Lacey, A.L. Laccase electrode for direct electrocatalytic reduction of O2 to H2O with high-operational stability and resistance to chloride inhibition. Biosens. Bioelectron. 2008, 24, 531–537. [Google Scholar] [CrossRef]
- Lindgren, A.; Gorton, L.; Ruzgas, T.; Baminger, U.; Haltrich, D.; Schülein, M. Direct electron transfer of cellobiose dehydrogenase from various biological origins at gold and graphite electrodes. J. Electroanal. Chem. 2001, 496, 76–81. [Google Scholar] [CrossRef]
- Yan, Y.M.; Baravik, I.; Tel-Vered, R.; Willner, I. An ethanol/O2 biofuel cell based on an electropolymerized bilirubin oxidase/Pt nanoparticle bioelectrocatalytic O2-reduction cathode. Adv. Mater. 2009, 21, 4275–4279. [Google Scholar] [CrossRef]
- Schubert, K.; Goebel, G.; Lisdat, F. Bilirubin oxidase bound to multi-walled carbon nanotube-modified gold. Electrochim. Acta 2009, 54, 3033–3038. [Google Scholar] [CrossRef]
- Climent, V.; Zhang, J.; Friis, E.P.; Østergaard, L.H.; Ulstrup, J. Voltammetry and single-molecule in situ scanning tunneling microscopy of laccases and bilirubin oxidase in electrocatalytic dioxygen reduction on Au(111) single-crystal electrodes. J. Phys. Chem. C 2012, 116, 1232–1243. [Google Scholar] [CrossRef]
- Chi, Q.; Farver, O.; Ulstrup, J. Long-range protein electron transfer observed at the single-molecule level: In situ mapping of redox-gated tunneling resonance. Proc. Natl. Acad. Sci. USA 2005, 102, 16203–16208. [Google Scholar] [CrossRef]
- Xiao, X.; Li, H.; Wang, M.; Zhang, K.; Si, P. Examining the effects of self-assembled monolayers on nanoporous gold based amperometric glucose biosensors. Analyst 2013, 139, 488–494. [Google Scholar] [CrossRef]
- Murata, K.; Suzuki, M.; Kajiya, K.; Nakamura, N.; Ohno, H. High performance bioanode based on direct electron transfer of fructose dehydrogenase at gold nanoparticle-modified electrodes. Electrochem. Commun. 2009, 11, 668–671. [Google Scholar] [CrossRef]
- Tominaga, M.; Ohtani, M.; Taniguchi, I. Gold single-crystal electrode surface modified with self-assembled monolayers for electron tunneling with bilirubin oxidase. Phys. Chem. Chem. Phys. 2008, 10, 6928–6934. [Google Scholar] [CrossRef] [PubMed]
- Chi, Q.; Zhang, J.; Andersen, J.E.T.; Ulstrup, J. Ordered assembly and controlled electron transfer of the blue copper protein azurin at gold (111) single-crystal substrates. J. Phys. Chem. B 2001, 105, 4669–4679. [Google Scholar] [CrossRef]
- Kizling, M.; Dzwonek, M.; Więckowska, A.; Bilewicz, R. Size Does Matter—Mediation of Electron Transfer by Gold Clusters in Bioelectrocatalysis. ChemCatChem 2018, 10, 1988–1992. [Google Scholar] [CrossRef]
- Al-Lolage, F.A.; Meneghello, M.; Ma, S.; Ludwig, R.; Bartlett, P.N. A Flexible Method for the Stable, Covalent Immobilization of Enzymes at Electrode Surfaces. ChemElectroChem 2017, 4, 1528–1534. [Google Scholar] [CrossRef]
- Matsumura, H.; Ortiz, R.; Ludwig, R.; Igarashi, K.; Samejima, M.; Gorton, L. Direct electrochemistry of phanerochaete chrysosporium cellobiose dehydrogenase covalently attached onto gold nanoparticle modified solid gold electrodes. Langmuir 2012, 28, 10925–10933. [Google Scholar] [CrossRef]
- Wang, X.; Falk, M.; Ortiz, R.; Matsumura, H.; Bobacka, J.; Ludwig, R.; Bergelin, M.; Gorton, L.; Shleev, S. Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes. Biosens. Bioelectron. 2012, 31, 219–225. [Google Scholar] [CrossRef]
- Rauf, S.; Zhou, D.; Abell, C.; Klenerman, D.; Kang, D.J. Building three-dimensional nanostructures with active enzymes by surface templated layer-by-layer assembly. Chem. Commun. 2006, 1721–1723. [Google Scholar] [CrossRef]
- Xiao, X.; Siepenkoetter, T.; Conghaile, P.; Leech, D.; Magner, E. Nanoporous Gold-Based Biofuel Cells on Contact Lenses. ACS Appl. Mater. Interfaces 2018, 10, 7107–7116. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.; Belochapkine, S.; Magner, E. Nanoporous Gold Electrodes with Tuneable Pore Sizes for Bioelectrochemical Applications. Electroanalysis 2016, 28, 2415–2423. [Google Scholar] [CrossRef]
- Jensen, P.S.; Chi, Q.; Zhang, J.; Ulstrup, J. Long-Range interfacial electrochemical electron transfer of Pseudomonas aeruginosa azurin-gold nanoparticle hybrid systems. J. Phys. Chem. C 2009, 113, 13993–14000. [Google Scholar] [CrossRef]
- Xu, S.; Han, X. A novel method to construct a third-generation biosensor: Self-assembling gold nanoparticles on thiol-functionalized poly(styrene-co-acrylic acid) nanospheres. Biosens. Bioelectron. 2004, 19, 1117–1120. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Suzuki, M.; Nakamura, N.; Ohno, H. Direct evidence of electron flow via the heme c group for the direct electron transfer reaction of fructose dehydrogenase using a silver nanoparticle-modified electrode. Electrochem. Commun. 2009, 11, 1623–1626. [Google Scholar] [CrossRef]
- Gutiérrez-Sánchez, C.; Pita, M.; Vaz-Domínguez, C.; Shleev, S.; De Lacey, A.L. Gold nanoparticles as electronic bridges for laccase-based biocathodes. J. Am. Chem. Soc. 2012, 134, 17212–17220. [Google Scholar] [CrossRef]
- Meredith, M.T.; Minteer, S.D. Biofuel Cells: Enhanced Enzymatic Bioelectrocatalysis. Annu. Rev. Anal. Chem. 2012, 5, 157–179. [Google Scholar] [CrossRef]
- Gross, A.J.; Holzinger, M.; Cosnier, S. Buckypaper bioelectrodes: Emerging materials for implantable and wearable biofuel cells. Energy Environ. Sci. 2018, 11, 1670–1687. [Google Scholar] [CrossRef]
- Mazurenko, I.; de Poulpiquet, A.; Lojou, E. Recent developments in high surface area bioelectrodes for enzymatic fuel cells. Curr. Opin. Electrochem. 2017, 5, 74–84. [Google Scholar] [CrossRef]
- Mazurenko, I.; Hitaishi, V.P.; Lojou, E. Recent advances in surface chemistry of electrodes to promote direct enzymatic bioelectrocatalysis. Curr. Opin. Electrochem. 2020, 19, 113–121. [Google Scholar] [CrossRef]
- Chaki, N.K.; Vijayamohanan, K. Self-assembled monolayers as a tunable platform for biosensor applications. Biosens. Bioelectron. 2002, 17, 1–12. [Google Scholar] [CrossRef]
- Xiao, X.; Ulstrup, J.; Li, H.; Wang, M.; Zhang, J.; Si, P. Nanoporous gold assembly of glucose oxidase for electrochemical biosensing. Electrochim. Acta 2014, 130, 559–567. [Google Scholar] [CrossRef]
- Serleti, A.; Salaj-Kosla, U.; Magner, E. The spatial and sequential immobilisation of cytochrome c at adjacent electrodes. Chem. Commun. 2013, 49, 8395–8397. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meunier-Prest, R.; Legay, G.; Raveau, S.; Chiffot, N.; Finot, E. Potential-assisted deposition of mixed alkanethiol self-assembled monolayers. Electrochim. Acta 2010, 55, 2712–2720. [Google Scholar] [CrossRef]
- Bradbury, C.R.; Zhao, J.; Fermín, D.J. Distance-independent charge-transfer resistance at gold electrodes modified by thiol monolayers and metal nanoparticles. J. Phys. Chem. C 2008, 112, 10153–10160. [Google Scholar] [CrossRef]
- Chi, Q.; Zhang, J.; Arslan, T.; Borg, L.; Pedersen, G.W.; Christensen, H.E.M.; Nazmudtinov, R.R.; Ulstrup, J. Approach to interfacial and intramolecular electron transfer of the diheme protein cytochrome c4 assembled on Au(111) surfaces. J. Phys. Chem. B 2010, 114, 5617–5624. [Google Scholar] [CrossRef]
- Engelbrekt, C.; Nazmutdinov, R.R.; Zinkicheva, T.T.; Glukhov, D.V.; Yan, J.; Mao, B.; Ulstrup, J.; Zhang, J. Chemistry of cysteine assembly on Au(100): Electrochemistry, in situ STM and molecular modeling. Nanoscale 2019, 11, 17235–17251. [Google Scholar] [CrossRef]
- Zhang, J.; Welinder, A.C.; Chi, Q.; Ulstrup, J. Electrochemically controlled self-assembled monolayers characterized with molecular and sub-molecular resolution. Phys. Chem. Chem. Phys. 2011, 13, 5526–5545. [Google Scholar] [CrossRef]
- Hao, X.; Zhang, J.; Christensen, H.E.M.; Wang, H.; Ulstrup, J. Electrochemical single-molecule AFM of the redox metalloenzyme copper nitrite reductase in action. ChemPhysChem 2012, 13, 2919–2924. [Google Scholar] [CrossRef]
- Marmisollé, W.A.; Capdevila, D.A.; De La Llave, E.; Williams, F.J.; Murgida, D.H. Self-assembled monolayers of NH2-terminated thiolates: Order, pKa, and specific adsorption. Langmuir 2013, 29, 5351–5359. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Clément, R.; Quattrocchi, L.; Parent, P.; Duché, D.; Zuily, L.; Ilbert, M.; Lojou, E.; Mazurenko, I. Interplay between Orientation at Electrodes and Copper Activation of Thermus thermophilus Laccase for O2 Reduction. J. Am. Chem. Soc. 2020, 142, 1394–1405. [Google Scholar] [CrossRef]
- Tang, J.; Werchmeister, R.M.L.; Preda, L.; Huang, W.; Zheng, Z.; Leimkühler, S.; Wollenberger, U.; Xiao, X.; Engelbrekt, C.; Ulstrup, J.; et al. Three-Dimensional Sulfite Oxidase Bioanodes Based on Graphene Functionalized Carbon Paper for Sulfite/O2 Biofuel Cells. ACS Catal. 2019, 9, 6543–6554. [Google Scholar] [CrossRef]
- Ciaccafava, A.; Infossi, P.; Ilbert, M.; Guiral, M.; Lecomte, S.; Giudici-Orticoni, M.T.; Lojou, E. Electrochemistry, AFM, and PM-IRRA spectroscopy of immobilized hydrogenase: Role of a hydrophobic helix in enzyme orientation for efficient H2 oxidation. Angew. Chem. Int. Ed. 2012, 51, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Loew, N.; Scheller, F.W.; Wollenberger, U. Characterization of self-assembling of glucose dehydrogenase in mono- and multilayers on gold electrodes. Electroanalysis 2004, 16, 1149–1154. [Google Scholar] [CrossRef]
- Yehezkeli, O.; Tel-Vered, R.; Raichlin, S.; Willner, I. Nano-engineered flavin-dependent glucose dehydrogenase/gold nanoparticle-modified electrodes for glucose sensing and biofuel cell applications. ACS Nano 2011, 5, 2385–2391. [Google Scholar] [CrossRef] [PubMed]
- Madden, C.; Vaughn, M.D.; Díez-Pérez, I.; Brown, K.A.; King, P.W.; Gust, D.; Moore, A.L.; Moore, T.A. Catalytic turnover of [FeFe]-hydrogenase based on single-molecule imaging. J. Am. Chem. Soc. 2012, 134, 1577–1582. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Mazurenko, I.; Harb, M.; Clément, R.; Taris, M.; Castano, S.; Duché, D.; Lecomte, S.; Ilbert, M.; De Poulpiquet, A.; et al. Electrostatic-Driven Activity, Loading, Dynamics, and Stability of a Redox Enzyme on Functionalized-Gold Electrodes for Bioelectrocatalysis. ACS Catal. 2018, 8, 12004–12014. [Google Scholar] [CrossRef]
- Guo, L.H.; Allen, H.; Hill, O. Direct electrochemistry of proteins and enzymes. Adv. Inorg. Chem. 1991, 36, 341–375. [Google Scholar] [CrossRef]
- Albery, W.J.; Eddowes, M.J.; Allen, H.; Hill, O.; Hillman, A.R. Mechanism of the Reduction and Oxidation Reaction of Cytochrome c at a Modified Gold Electrode. J. Am. Chem. Soc. 1981, 103, 3904–3910. [Google Scholar] [CrossRef]
- Armstrong, F.A.; Hill, H.A.O.; Walton, N.J. Direct Electrochemistry of Redox Proteins. Acc. Chem. Res. 1988, 21, 407–413. [Google Scholar] [CrossRef]
- Armstrong, F.A.; Heering, H.A.; Hirst, J. Reaction of complex metalloproteins studied by protein-film voltammetry. Chem. Soc. Rev. 1997, 26, 169–179. [Google Scholar] [CrossRef]
- Ma, S.; Laurent, C.V.F.P.; Meneghello, M.; Tuoriniemi, J.; Oostenbrink, C.; Gorton, L.; Bartlett, P.N.; Ludwig, R. Direct electron-transfer anisotropy of a site-specifically immobilized cellobiose dehydrogenase. ACS Catal. 2019, 9, 7607–7615. [Google Scholar] [CrossRef]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. Highly Sensitive Membraneless Fructose Biosensor Based on Fructose Dehydrogenase Immobilized onto Aryl Thiol Modified Highly Porous Gold Electrode: Characterization and Application in Food Samples. Anal. Chem. 2018, 90, 12131–12136. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.J.; Nielsen, M.F.; Thuesen, M.H.; Ulstrup, J. Electrochemistry of cytochrome c4 from Pseudomonas stutzeri. J. Phys. Chem. B 1997, 101, 2430–2436. [Google Scholar] [CrossRef]
- Avila, A.; Gregory, B.W.; Niki, K.; Cotton, T.M. An electrochemical approach to investigate gated electron transfer using a physiological model system: Cytochrome C immobilized on carboxylic acid-terminated alkanethiol self-assembled monolayers on gold electrodes. J. Phys. Chem. B 2000, 104, 2759–2766. [Google Scholar] [CrossRef]
- Wei, J.; Liu, H.; Dick, A.R.; Yamamoto, H.; He, Y.; Waldeck, D.H. Direct wiring of cytochrome c’s heme unit to an electrode: Electrochemical studies. J. Am. Chem. Soc. 2002, 124, 9591–9599. [Google Scholar] [CrossRef] [PubMed]
- Collinson, M.; Bowden, E.F.; Tarlov, M.J. Voltammetry of Covalently Immobilized Cytochrome c on Self-Assembled Monolayer Electrodes. Langmuir 1992, 8, 1247–1250. [Google Scholar] [CrossRef]
- El Kasmi, A.; Wallace, J.M.; Bowden, E.F.; Binet, S.M.; Linderman, R.J. Controlling interfacial electron-transfer kinetics of cytochrome c with mixed self-assembled monolayers. J. Am. Chem. Soc. 1998, 120, 225–226. [Google Scholar] [CrossRef]
- Yue, H.; Waldeck, D.H.; Schrock, K.; Kirby, D.; Knorr, K.; Switzer, S.; Rosmus, J.; Clark, R.A. Multiple sites for electron tunneling between cytochrome c and mixed self-assembled monolayers. J. Phys. Chem. C 2008, 112, 2514–2521. [Google Scholar] [CrossRef]
- Scott, R.A.; Mauk, A.G. Cytochrome c: A Multidisciplinary Approach; Univ Science Books: Sausalito, CA, USA, 1996. [Google Scholar]
- Jensen, P.S.; Chi, Q.; Grumsen, F.B.; Abad, J.M.; Horsewell, A.; Schiffrin, D.J.; Ulstrup, J. Gold nanoparticle assisted assembly of a heme protein for enhancement of long-range interfacial electron transfer. J. Phys. Chem. C 2007, 111, 6124–6132. [Google Scholar] [CrossRef]
- Niki, K.; Hardy, W.R.; Hill, M.G.; Li, H.; Sprinkle, J.R.; Margoliash, E.; Fujita, K.; Tanimura, R.; Nakamura, N.; Ohno, H.; et al. Coupling to lysine-13 promotes electron tunneling through carboxylate-terminated alkanethiol self-assembled monolayers to cytochrome c. J. Phys. Chem. B 2003, 107, 9947–9949. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Heterogeneous electron transfer of cytochrome c on coated silver electrodes. Electric field effects on structure and redox potential. J. Phys. Chem. B 2001, 105, 1578–1586. [Google Scholar] [CrossRef]
- Shermukhamedov, A.S.; Nazmutdinov, R.R.; Zinkicheva, T.T.; Bronshtein, D.M.; Zhang, J.; Mao, B.; Tian, Z.; Yan, J.; Wu, D.-Y.; Ulstrup, J. Electronic Spillover from a Metallic Nanoparticle: Can Simple Electrochemical Electron Transfer Processes Be Catalyzed by Electronic Coupling of a Molecular Scale Gold Nanoparticle Simultaneously to the Redox Molecule and the Electrode? J. Am. Chem. Soc. 2020, 142, 10646–10658. [Google Scholar] [CrossRef] [PubMed]
- Chazalviel, J.-N.; Allongue, P. On the Origin of the Efficient Nanoparticle Mediated Electron Transfer across a Self-Assembled Monolayer. J. Am. Chem. Soc. 2010, 133, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Engelbrekt, C.; Sørensen, K.H.; Zhang, J.; Welinder, A.C.; Jensen, P.S.; Ulstrup, J. Green synthesis of gold nanoparticles with starch-glucose and application in bioelectrochemistry. J. Mater. Chem. 2009, 19, 7839–7847. [Google Scholar] [CrossRef]
- Della Pia, E.A.; Chi, Q.; Jones, D.D.; MacDonald, J.E.; Ulstrup, J.; Elliott, M. Single-molecule mapping of long-range electron transport for a cytochrome b562 variant. Nano Lett. 2011, 11, 176–182. [Google Scholar] [CrossRef]
- Dronov, R.; Kurth, D.G.; Möhwald, H.; Scheller, F.W.; Lisdat, F. Communication in a protein stack: Electron transfer between cytochrome c and bilirubin oxidase within a polyelectrolyte multilayer. Angew. Chem. Int. Ed. 2008, 47, 3000–3003. [Google Scholar] [CrossRef]
- Dronov, R.; Kurth, D.G.; Möhwald, H.; Spricigo, R.; Leimkühler, S.; Wollenberger, U.; Rajagopalan, K.V.; Scheller, F.W.; Lisdat, F. Layer-by-layer arrangement by protein-protein interaction of sulfite oxidase and cytochrome c catalyzing oxidation of sulfite. J. Am. Chem. Soc. 2008, 130, 1122–1123. [Google Scholar] [CrossRef]
- Feifel, S.C.; Kapp, A.; Lisdat, F. Electroactive nanobiomolecular architectures of laccase and cytochrome c on electrodes: Applying silica nanoparticles as artificial matrix. Langmuir 2014, 30, 5363–5367. [Google Scholar] [CrossRef]
- Beissenhirtz, M.K.; Scheller, F.W.; Stöcklein, W.F.M.; Kurth, D.G.; Möhwald, H.; Lisdat, F. Electroactive cytochrome c multilayers within a polyelectrolyte assembly. Angew. Chem. Int. Ed. 2004, 43, 4357–4360. [Google Scholar] [CrossRef]
- Ma, S.; Ludwig, R. Direct Electron Transfer of Enzymes Facilitated by Cytochromes. ChemElectroChem 2019, 6, 958–975. [Google Scholar] [CrossRef]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. Enhanced Direct Electron Transfer of Fructose Dehydrogenase Rationally Immobilized on a 2-Aminoanthracene Diazonium Cation Grafted Single-Walled Carbon Nanotube Based Electrode. ACS Catal. 2018, 8, 10279–10289. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L.; Antiochia, R. Direct electron transfer of dehydrogenases for development of 3rd generation biosensors and enzymatic fuel cells. Sensors 2018, 18, 1319. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. The influence of pH and divalent/monovalent cations on the internal electron transfer (IET), enzymatic activity, and structure of fructose dehydrogenase. Anal. Bioanal. Chem. 2018, 410, 3253–3264. [Google Scholar] [CrossRef] [PubMed]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Mutation of Heme c Axial Ligands in D-Fructose Dehydrogenase For Investigation of Electron Transfer Pathways and Reduction of Overpotential in Direct Electron Transfer-type Bioelectrocatalysis. Electrochem. Commun. 2016, 67, 43–46. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of a protein-engineered variant of D-fructose dehydrogenase for direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2017, 77, 112–115. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Function of C-terminal hydrophobic region in fructose dehydrogenase. Electrochim. Acta 2015, 176, 976–981. [Google Scholar] [CrossRef]
- Meredith, M.T.; Minson, M.; Hickey, D.; Artyushkova, K.; Glatzhofer, D.T.; Minteer, S.D. Anthracene-modified multi-walled carbon nanotubes as direct electron transfer scaffolds for enzymatic oxygen reduction. ACS Catal. 2011, 1, 1683–1690. [Google Scholar] [CrossRef]
- Ludwig, R.; Harreither, W.; Tasca, F.; Gorton, L. Cellobiose dehydrogenase: A versatile catalyst for electrochemical applications. ChemPhysChem 2010, 11, 2674–2697. [Google Scholar] [CrossRef]
- Harreither, W.; Nicholls, P.; Sygmund, C.; Gorton, L.; Ludwig, R. Investigation of the pH-dependent electron transfer mechanism of ascomycetous class II cellobiose dehydrogenases on electrodes. Langmuir 2012, 28, 6714–6723. [Google Scholar] [CrossRef]
- Scheiblbrandner, S.; Ludwig, R. Cellobiose dehydrogenase: Bioelectrochemical insights and applications. Bioelectrochemistry 2020, 131, 107345. [Google Scholar] [CrossRef]
- Lamberg, P.; Hamit-Eminovski, J.; Toscano, M.D.; Eicher-Lorka, O.; Niaura, G.; Arnebrant, T.; Shleev, S.; Ruzgas, T. Electrical activity of cellobiose dehydrogenase adsorbed on thiols: Influence of charge and hydrophobicity. Bioelectrochemistry 2017, 115, 26–32. [Google Scholar] [CrossRef]
- Tavahodi, M.; Ortiz, R.; Schulz, C.; Ekhtiari, A.; Ludwig, R.; Haghighi, B.; Gorton, L. Direct Electron Transfer of Cellobiose Dehydrogenase on Positively Charged Polyethyleneimine Gold Nanoparticles. ChemPlusChem 2017, 82, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Mazzei, F.; Favero, G.; Fusco, G.; Ludwig, R.; Gorton, L.; Antiochia, R. Improved DET communication between cellobiose dehydrogenase and a gold electrode modified with a rigid self-assembled monolayer and green metal nanoparticles: The role of an ordered nanostructuration. Biosens. Bioelectron. 2017, 88, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Krikstolaityte, V.; Lamberg, P.; Toscano, M.D.; Silow, M.; Eicher-Lorka, O.; Ramanavicius, A.; Niaura, G.; Abariute, L.; Ruzgas, T.; Shleev, S. Mediatorless carbohydrate/oxygen biofuel cells with improved cellobiose dehydrogenase based bioanode. Fuel Cells 2014, 14, 792–800. [Google Scholar] [CrossRef]
- Meneghello, M.; Al-Lolage, F.A.; Ma, S.; Ludwig, R.; Bartlett, P.N. Studying Direct Electron Transfer by Site-Directed Immobilization of Cellobiose Dehydrogenase. ChemElectroChem 2019, 6, 700–713. [Google Scholar] [CrossRef]
- Yamashita, Y.; Lee, I.; Loew, N.; Sode, K. Direct electron transfer (DET) mechanism of FAD dependent dehydrogenase complexes ∼from the elucidation of intra- and inter-molecular electron transfer pathway to the construction of engineered DET enzyme complexes∼. Curr. Opin. Electrochem. 2018, 12, 92–100. [Google Scholar] [CrossRef]
- Korani, A.; Salimi, A.; Karimi, B. Guanine/Ionic Liquid Derived Ordered Mesoporous Carbon Decorated with AuNPs as Efficient NADH Biosensor and Suitable Platform for Enzymes Immobilization and Biofuel Cell Design. Electroanalysis 2017, 29, 2646–2655. [Google Scholar] [CrossRef]
- Lee, I.; Loew, N.; Tsugawa, W.; Lin, C.E.; Probst, D.; La Belle, J.T.; Sode, K. The electrochemical behavior of a FAD dependent glucose dehydrogenase with direct electron transfer subunit by immobilization on self-assembled monolayers. Bioelectrochemistry 2018, 121, 1–6. [Google Scholar] [CrossRef]
- Xiao, X.; Conghaile, P.; Leech, D.; Ludwig, R.; Magner, E. An oxygen-independent and membrane-less glucose biobattery/supercapacitor hybrid device. Biosens. Bioelectron. 2017, 98, 421–427. [Google Scholar] [CrossRef]
- Ito, Y.; Okuda-Shimazaki, J.; Tsugawa, W.; Loew, N.; Shitanda, I.; Lin, C.E.; La Belle, J.; Sode, K. Third generation impedimetric sensor employing direct electron transfer type glucose dehydrogenase. Biosens. Bioelectron. 2019, 129, 189–197. [Google Scholar] [CrossRef]
- Ratautas, D.; Laurynenas, A.; Dagys, M.; Marcinkevičiene, L.; Meškys, R.; Kulys, J. High current, low redox potential mediatorless bioanode based on gold nanoparticles and glucose dehydrogenase from Ewingella americana. Electrochim. Acta 2016, 199, 254–260. [Google Scholar] [CrossRef]
- Kim, Y.P.; Park, S.J.; Lee, D.; Kim, H.S. Electrochemical glucose biosensor by electrostatic binding of PQQ-glucose dehydrogenase onto self-assembled monolayers on gold. J. Appl. Electrochem. 2012, 42, 383–390. [Google Scholar] [CrossRef]
- Ferapontova, E.E.; Ruzgas, T.; Gorton, L. Direct electron transfer of heme- and molybdopterin cofactor-containing chicken liver sulfite oxidase on alkanethiol-modified gold electrodes. Anal. Chem. 2003, 75, 4841–4850. [Google Scholar] [CrossRef] [PubMed]
- Sezer, M.; Spricigo, R.; Utesch, T.; Millo, D.; Leimkuehler, S.; Mroginski, M.A.; Wollenberger, U.; Hildebrandt, P.; Weidinger, I.M. Redox properties and catalytic activity of surface-bound human sulfite oxidase studied by a combined surface enhanced resonance Raman spectroscopic and electrochemical approach. Phys. Chem. Chem. Phys. 2010, 12, 7894–7903. [Google Scholar] [CrossRef] [PubMed]
- Spricigo, R.; Dronov, R.; Lisdat, F.; Leimkühler, S.; Scheller, F.W.; Wollenberger, U. Electrocatalytic sulfite biosensor with human sulfite oxidase co-immobilized with cytochrome c in a polyelectrolyte-containing multilayer. Anal. Bioanal. Chem. 2009, 393, 225–233. [Google Scholar] [CrossRef][Green Version]
- Utesch, T.; Sezer, M.; Weidinger, I.M.; Mroginski, M.A. Adsorption of sulfite oxidase on self-assembled monolayers from molecular dynamics simulations. Langmuir 2012, 28, 5761–5769. [Google Scholar] [CrossRef]
- Zeng, T.; Leimkühler, S.; Wollenberger, U.; Fourmond, V. Transient Catalytic Voltammetry of Sulfite Oxidase Reveals Rate Limiting Conformational Changes. J. Am. Chem. Soc. 2017, 139, 11559–11567. [Google Scholar] [CrossRef]
- Zeng, T.; Frasca, S.; Rumschöttel, J.; Koetz, J.; Leimkühler, S.; Wollenberger, U. Role of Conductive Nanoparticles in the Direct Unmediated Bioelectrocatalysis of Immobilized Sulfite Oxidase. Electroanalysis 2016, 28, 2303–2310. [Google Scholar] [CrossRef]
- Frasca, S.; Rojas, O.; Salewski, J.; Neumann, B.; Stiba, K.; Weidinger, I.M.; Tiersch, B.; Leimkühler, S.; Koetz, J.; Wollenberger, U. Human sulfite oxidase electrochemistry on gold nanoparticles modified electrode. Bioelectrochemistry 2012, 87, 33–41. [Google Scholar] [CrossRef]
- Yan, J.; Frøkjær, E.; Engebrekt, C.; Leimkühler, S.; Ulstrup, J.; Wollenberger, U.; Xiao, X.; Zhang, J. Voltammetry and Single-molecule in Situ Scanning Tunnelling Microscopy of the Redox Metalloenzyme Human Sulfite Oxidase. ChemElectroChem 2020. [Google Scholar] [CrossRef]
- Kalimuthu, P.; Belaidi, A.A.; Schwarz, G.; Bernhardt, P.V. Chitosan-Promoted Direct Electrochemistry of Human Sulfite Oxidase. J. Phys. Chem. B 2017, 121, 9149–9159. [Google Scholar] [CrossRef]
- Khoshtariya, D.E.; Dolidze, T.D.; Shushanyan, M.; Davis, K.L.; Waldeck, D.H.; Van Eldik, R. Fundamental signatures of short- And long-range electron transfer for the blue copper protein azurin at Au/SAM junctions. Proc. Natl. Acad. Sci. USA 2010, 107, 2757–2762. [Google Scholar] [CrossRef] [PubMed]
- Vargo, M.L.; Gulka, C.P.; Gerig, J.K.; Manieri, C.M.; Dattelbaum, J.D.; Marks, C.B.; Lawrence, N.T.; Trawick, M.L.; Leopold, M.C. Distance dependence of electron transfer kinetics for azurin protein adsorbed to monolayer protected nanoparticle film assemblies. Langmuir 2010, 26, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chi, Q.; Hansen, A.G.; Jensen, P.S.; Salvatore, P.; Ulstrup, J. Interfacial electrochemical electron transfer in biology—Towards the level of the single molecule. FEBS Lett. 2012, 586, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, A.; Facci, P. Electron transfer in nanobiodevices. Eur. Polym. J. 2016, 83, 450–466. [Google Scholar] [CrossRef]
- López-Martínez, M.; Artés, J.M.; Sarasso, V.; Carminati, M.; Díez-Pérez, I.; Sanz, F.; Gorostiza, P. Differential Electrochemical Conductance Imaging at the Nanoscale. Small 2017, 13, 1–7. [Google Scholar] [CrossRef]
- Chin, Q.; Zhang, J.; Nielsen, J.U.; Friis, E.P.; Chorkendorff, I.; Canters, G.W.; Andersen, J.E.T.; Ulstrup, J. Molecular monolayers and interfacial electron transfer of Pseudomonas aeruginosa azurin on Au(111). J. Am. Chem. Soc. 2000, 122, 4047–4055. [Google Scholar] [CrossRef]
- Romero-Muñiz, C.; Ortega, M.; Vilhena, J.G.; Diéz-Pérez, I.; Cuevas, J.C.; Pérez, R.; Zotti, L.A. Mechanical deformation and electronic structure of a blue copper azurin in a solid-state junction. Biomolecules 2019, 9, 506. [Google Scholar] [CrossRef]
- Armstrong, F.A.; Barlow, N.L.; Burn, P.L.; Hoke, K.R.; Jeuken, L.J.C.; Shenton, C.; Webster, G.R. Fast, long-range electron-transfer reactions of a ‘blue’ copper protein coupled non-covalently to an electrode through a stilbenyl thiolate monolayer. Chem. Commun. 2004, 4, 316–317. [Google Scholar] [CrossRef]
- Chi, Q.; Zhang, J.; Jensen, P.S.; Christensen, H.E.M.; Ulstrup, J. Long-range interfacial electron transfer of metalloproteins based on molecular wiring assemblies. Faraday Discuss. 2006, 131, 181–195. [Google Scholar] [CrossRef]
- Zhang, J.; Welinder, A.C.; Hansen, A.G.; Christensen, H.E.M.; Ulstrup, J. Catalytic Monolayer Voltammetry and In Situ Scanning Tunneling Microscopy of Copper Nitrite Reductase on Cysteamine-Modified Au(111) Electrodes. J. Phys. Chem. B 2003, 107, 12480–12484. [Google Scholar] [CrossRef]
- Welinder, A.C.; Zhang, J.; Hansen, A.G.; Moth-Poulsen, K.; Christensen, H.E.M.; Kuznetsov, A.M.; Bjørnholm, T.; Ulstrup, J. Voltammetry and Electrocatalysis of Achromobacter Xylosoxidans Copper Nitrite Reductase on Functionalized Au(111)-Electrode Surfaces. Z. Phys. Chem. 2007, 221, 1343–1378. [Google Scholar] [CrossRef]
- Tang, J.; Yan, X.; Huang, W.; Engelbrekt, C.; Duus, J.Ø.; Ulstrup, J.; Xiao, X.; Zhang, J. Bilirubin oxidase oriented on novel type three-dimensional biocathodes with reduced graphene aggregation for biocathode. Biosens. Bioelectron. 2020, 167. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Leech, D.; Zhang, J. An oxygen-reducing biocathode with “oxygen tanks”. Chem. Commun. 2020, 56, 9767–9770. [Google Scholar] [CrossRef] [PubMed]
- Lopez, F.; Siepenkoetter, T.; Xiao, X.; Magner, E.; Schuhmann, W.; Salaj-Kosla, U. Potential pulse-assisted immobilization of Myrothecium verrucaria bilirubin oxidase at planar and nanoporous gold electrodes. J. Electroanal. Chem. 2018, 812, 194–198. [Google Scholar] [CrossRef]
- Ramírez, P.; Mano, N.; Andreu, R.; Ruzgas, T.; Heller, A.; Gorton, L.; Shleev, S. Direct electron transfer from graphite and functionalized gold electrodes to T1 and T2/T3 copper centers of bilirubin oxidase. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1364–1369. [Google Scholar] [CrossRef]
- Lalaoui, N.; De Poulpiquet, A.; Haddad, R.; Le Goff, A.; Holzinger, M.; Gounel, S.; Mermoux, M.; Infossi, P.; Mano, N.; Lojou, E.; et al. A membraneless air-breathing hydrogen biofuel cell based on direct wiring of thermostable enzymes on carbon nanotube electrodes. Chem. Commun. 2015, 51, 7447–7450. [Google Scholar] [CrossRef]
- Al-Lolage, F.A.; Bartlett, P.N.; Gounel, S.; Staigre, P.; Mano, N. Site-Directed Immobilization of Bilirubin Oxidase for Electrocatalytic Oxygen Reduction. ACS Catal. 2019, 9, 2068–2078. [Google Scholar] [CrossRef]
- McArdle, T.; McNamara, T.P.; Fei, F.; Singh, K.; Blanford, C.F. Optimizing the Mass-Specific Activity of Bilirubin Oxidase Adlayers through Combined Electrochemical Quartz Crystal Microbalance and Dual Polarization Interferometry Analyses. ACS Appl. Mater. Interfaces 2015, 7, 25270–25280. [Google Scholar] [CrossRef]
- Gholami, F.; Navaee, A.; Salimi, A.; Ahmadi, R.; Korani, A.; Hallaj, R. Direct Enzymatic Glucose/O2 Biofuel Cell based on Poly-Thiophene Carboxylic Acid alongside Gold Nanostructures Substrates Derived through Bipolar Electrochemistry. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Yang, S.; Liu, J.; Quan, X.; Zhou, J. Bilirubin Oxidase Adsorption onto Charged Self-Assembled Monolayers: Insights from Multiscale Simulations. Langmuir 2018, 34, 9818–9828. [Google Scholar] [CrossRef]
- Pankratov, D.; Sundberg, R.; Suyatin, D.B.; Sotres, J.; Barrantes, A.; Ruzgas, T.; Maximov, I.; Shleev, S. The influence of nanoparticles on enzymatic bioelectrocatalysis. RSC Adv. 2014, 4, 38164–38168. [Google Scholar] [CrossRef]
- Cabrita, J.F.; Abrantes, L.M.; Viana, A.S. N-Hydroxysuccinimide-terminated self-assembled monolayers on gold for biomolecules immobilisation. Electrochim. Acta 2005, 50, 2117–2124. [Google Scholar] [CrossRef]
- Balland, V.; Hureau, C.; Cusano, A.M.; Liu, Y.; Tron, T.; Limoges, B. Oriented immobilization of a fully active monolayer of histidine-tagged recombinant laccase on modified gold electrodes. Chem. A Eur. J. 2008, 14, 7186–7192. [Google Scholar] [CrossRef] [PubMed]
- Pita, M.; Gutierrez-Sanchez, C.; Olea, D.; Velez, M.; Garcia-Diego, C.; Shleev, S.; Fernandez, V.M.; Lacey, A.L.D. High redox potential cathode based on laccase covalently attached to gold electrode. J. Phys. Chem. C 2011, 115, 13420–13428. [Google Scholar] [CrossRef]
- Shleev, S.; Christenson, A.; Serezhenkov, V.; Burbaev, D.; Yaropolov, A.; Gorton, L.; Ruzgas, T. Electrochemical redox transformations of T1 and T2 copper sites in native Trametes hirsuta laccase at gold electrode. Biochem. J. 2005, 385, 745–754. [Google Scholar] [CrossRef]
- Thorum, M.S.; Anderson, C.A.; Hatch, J.J.; Campbell, A.S.; Marshall, N.M.; Zimmerman, S.C.; Lu, Y.; Gewirth, A.A. Direct, electrocatalytic oxygen reduction by laccase on anthracene-2-methanethiol-modified gold. J. Phys. Chem. Lett. 2010, 1, 2251–2254. [Google Scholar] [CrossRef]
- Traunsteiner, C.; Sek, S.; Huber, V.; Valero-Vidal, C.; Kunze-Liebhäuser, J. Laccase immobilized on a mixed thiol monolayer on Au(111)—Structure-dependent activity towards oxygen reduction. Electrochim. Acta 2016, 213, 761–770. [Google Scholar] [CrossRef]
- Hakamada, M.; Takahashi, M.; Mabuchi, M. Enhanced thermal stability of laccase immobilized on monolayer-modified nanoporous Au. Mater. Lett. 2012, 66, 4–6. [Google Scholar] [CrossRef]
- Krassen, H.; Stripp, S.T.; Böhm, N.; Berkessel, A.; Happe, T.; Ataka, K.; Heberle, J. Tailor-made modification of a gold surface for the chemical binding of a high-activity [FeFe] hydrogenase. Eur. J. Inorg. Chem. 2011, 1138–1146. [Google Scholar] [CrossRef]
- Gutiérrez-Sanz, Ó.; Tapia, C.; Marques, M.C.; Zacarias, S.; Vélez, M.; Pereira, I.A.C.; De Lacey, A.L. Induction of a Proton Gradient across a Gold-Supported Biomimetic Membrane by Electroenzymatic H2 Oxidation. Angew. Chem. 2015, 127, 2722–2725. [Google Scholar] [CrossRef]
- Goris, T.; Wait, A.F.; Saggu, M.; Fritsch, J.; Heidary, N.; Stein, M.; Zebger, I.; Lendzian, F.; Armstrong, F.A.; Friedrich, B.; et al. A unique iron-sulfur cluster is crucial for oxygen tolerance of a [NiFe]-hydrogenase. Nat. Chem. Biol. 2011, 7, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; Wait, A.F.; Lenz, O.; Friedrich, B.; Armstrong, F.A. A kinetic and thermodynamic understanding of O2 tolerance in [NiFe]-hydrogenases. Proc. Natl. Acad. Sci. USA 2009, 106, 20681–20686. [Google Scholar] [CrossRef] [PubMed]
- Sezer, M.; Frielingsdorf, S.; Millo, D.; Heidary, N.; Utesch, T.; Mroginski, M.A.; Friedrich, B.; Hildebrandt, P.; Zebger, I.; Weidinger, I.M. Role of the HoxZ subunit in the electron transfer pathway of the membrane-bound [NiFe]-hydrogenase from Ralstonia eutropha immobilized on electrodes. J. Phys. Chem. B 2011, 115, 10368–10374. [Google Scholar] [CrossRef] [PubMed]
- Millo, D.; Pandelia, M.E.; Utesch, T.; Wisitruangsakul, N.; Mroginski, M.A.; Lubitz, W.; Hildebrandt, P.; Zebger, I. Spectroelectrochemical study of the [NiFe] hydrogenase from Desulfovibrio vulgaris miyazaki F in solution and immobilized on biocompatible gold surfaces. J. Phys. Chem. B 2009, 113, 15344–15351. [Google Scholar] [CrossRef]
- Léger, C.; Jones, A.K.; Albracht, S.P.J.; Armstrong, F.A. Effect of a dispersion of interfacial electron transfer rates on steady state catalytic electron transport in [NiFe]-hydrogenase and other enzymes. J. Phys. Chem. B 2002, 106, 13058–13063. [Google Scholar] [CrossRef]
- Gutiérrez-Sánchez, C.; Olea, D.; Marques, M.; Fernández, V.M.; Pereira, I.A.C.; Vélez, M.; Lacey, A.L.D. Oriented immobilization of a membrane-bound hydrogenase onto an electrode for direct electron transfer. Langmuir 2011, 27, 6449–6457. [Google Scholar] [CrossRef]
- Zhang, B.; Song, W.; Pang, P.; Lai, H.; Chen, Q.; Zhang, P.; Lindsay, S. Role of contacts in long-range protein conductance. Proc. Natl. Acad. Sci. USA 2019, 116, 5886–5891. [Google Scholar] [CrossRef]
- Lagunas, A.; Guerra-Castellano, A.; Nin-Hill, A.; Díaz-Moreno, I.; De la Rosa, M.A.; Samitier, J.; Rovira, C.; Gorostiza, P. Long distance electron transfer through the aqueous solution between redox partner proteins. Nat. Commun. 2018, 9, 3–9. [Google Scholar] [CrossRef]








Publisher‘s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, X.; Tang, J.; Tanner, D.; Ulstrup, J.; Xiao, X. Direct Electrochemical Enzyme Electron Transfer on Electrodes Modified by Self-Assembled Molecular Monolayers. Catalysts 2020, 10, 1458. https://doi.org/10.3390/catal10121458
Yan X, Tang J, Tanner D, Ulstrup J, Xiao X. Direct Electrochemical Enzyme Electron Transfer on Electrodes Modified by Self-Assembled Molecular Monolayers. Catalysts. 2020; 10(12):1458. https://doi.org/10.3390/catal10121458
Chicago/Turabian StyleYan, Xiaomei, Jing Tang, David Tanner, Jens Ulstrup, and Xinxin Xiao. 2020. "Direct Electrochemical Enzyme Electron Transfer on Electrodes Modified by Self-Assembled Molecular Monolayers" Catalysts 10, no. 12: 1458. https://doi.org/10.3390/catal10121458
APA StyleYan, X., Tang, J., Tanner, D., Ulstrup, J., & Xiao, X. (2020). Direct Electrochemical Enzyme Electron Transfer on Electrodes Modified by Self-Assembled Molecular Monolayers. Catalysts, 10(12), 1458. https://doi.org/10.3390/catal10121458