Recent Progress in Applications of Enzymatic Bioelectrocatalysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tuning of Nanostructured Electrodes and Protein-Engineering of Redox Enzymes
2.1. Electrode Nanomaterials
2.2. Protein-Engineering Approaches
2.2.1. Formal Potential Shift of Electrode-Active Sites
2.2.2. Downsizing
2.2.3. Surface Amino Acid Mutation
2.2.4. Fusion Protein
3. Novel Bioelectrochemical System
3.1. Biosupercapacitor
3.2. Bioelectrosynthesis
3.2.1. Dihydrogen Production
3.2.2. Formate Production
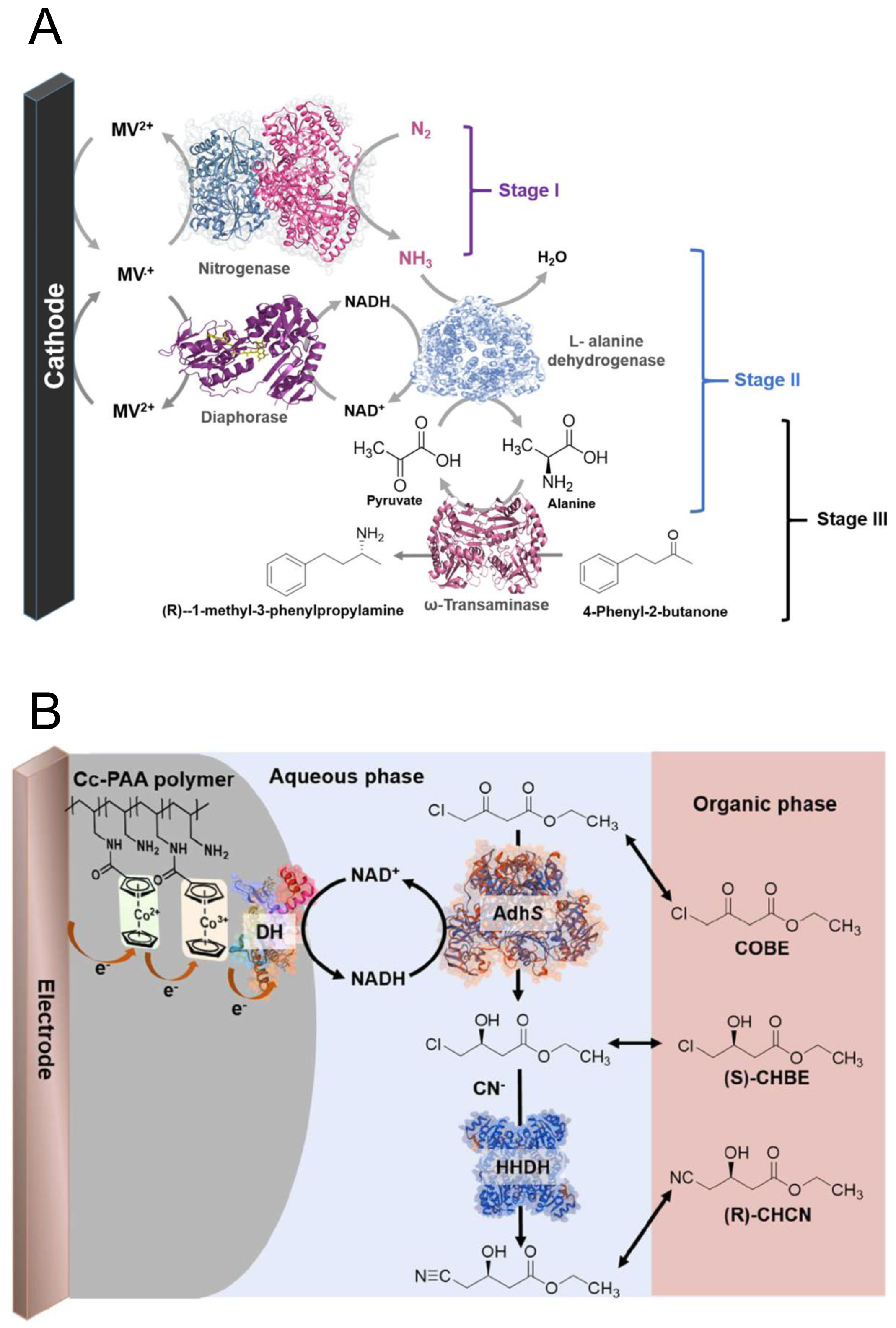
3.2.3. Ammonia Production
3.2.4. NAD(P)+-Dependent Bioelectrosynthesis
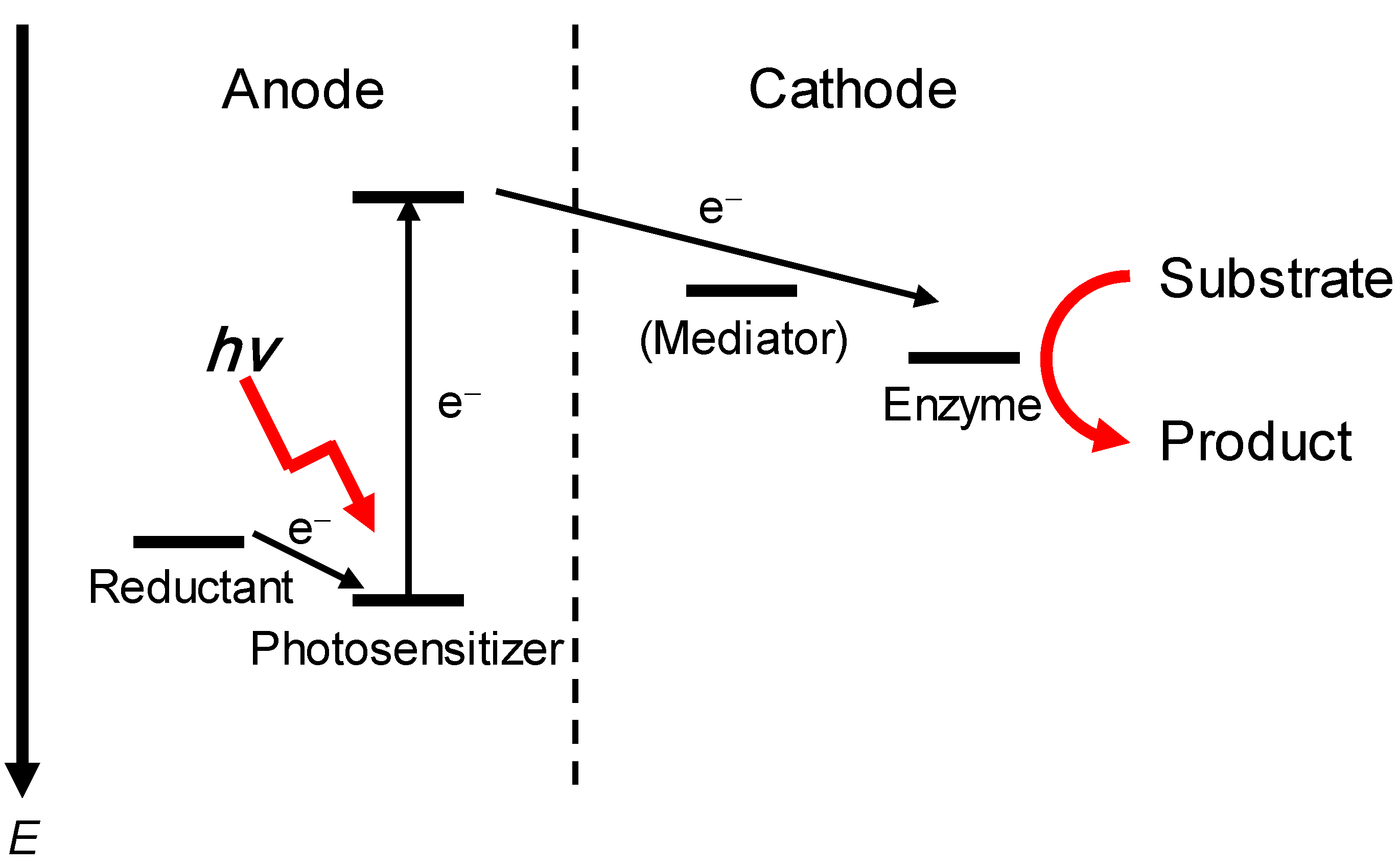
3.3. Photo-Bioelectrocatalysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bartlett, P.N. Bioelectrochemistry: Fundamentals, Experimental Techniques and Applications; John Wiley & Sons: Chichester, UK, 2008. [Google Scholar]
- Wilson, G.S.; Johnson, M.A. In-Vivo Electrochemistry: What Can We Learn about Living Systems? Chem. Rev. 2008, 108, 2462–2481. [Google Scholar] [CrossRef]
- Heller, A. Electrical connection of enzyme redox centers to electrodes. J. Phys. Chem. 1992, 96, 3579–3587. [Google Scholar] [CrossRef]
- Kano, K. Fundamentals and Applications of Redox Enzyme-functionalized Electrode Reactions. Electrochemistry 2019, 87, 301–311. [Google Scholar] [CrossRef]
- Scheller, F.; Kirstein, D.; Schubert, F.; Wollenberger, U.; Ollson, B.; Gorton, L.; Johansson, G. Enzyme electrodes and their application. Philos. Trans. R. Soc. B Biol. Sci. 1987, 316, 85–94. [Google Scholar] [CrossRef]
- Thevenot, D.R.; Tóth, K.; Durst, R.A.; Wilson, G.S. Electrochemical Biosensors: Recommended Definitions and Classification. Pure Appl. Chem. 1999, 71, 2333–2348. [Google Scholar] [CrossRef] [Green Version]
- Martinkova, P.; Kostelnik, A.; Valek, T.; Pohanska, M. Main streams in the Construction of Biosensors and Their Applications. Int. J. Electrochem. Sci. 2017, 12, 7386–7403. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L. Enzyme based amperometric biosensors. Curr. Opin. Electrochem. 2018, 10, 157–173. [Google Scholar] [CrossRef]
- Kucherenko, I.S.; Soldatkin, A.; Kucherenko, D.Y.; Soldatkina, O.V.; Dzyadevych, S.V. Advances in nanomaterial application in enzyme-based electrochemical biosensors: A review. Nanoscale Adv. 2019, 1, 4560–4577. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.H.; Lee, S.H.; Lee, U.J.; Fermin, C.D.; Kim, M. Immobilized Enzymes in Biosensor Applications. Materials 2019, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Pinyou, P.; Blay, V.; Muresan, L.M.; Noguer, T. Enzyme-modified electrodes for biosensors and biofuel cells. Mater. Horiz. 2019, 6, 1336–1358. [Google Scholar] [CrossRef]
- Bollella, P.; Katz, E. Enzyme-Based Biosensors: Tackling Electron Transfer Issues. Sensors 2020, 20, 3517. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.; Katz, E.; Willner, B. Electrical contact of redox enzyme layers associated with electrodes: Routes to amperometric biosensors. Electroanalysis 1997, 9, 965–977. [Google Scholar] [CrossRef]
- Scheller, F.; Schubert, F.; Pfeiffer, D.; Hintsche, R.; Dransfeld, I.; Renneberg, R.; Wollenberger, U.; Riedel, K.; Pavlova, M.; Kühn, M.; et al. Research and development of biosensors. A review. Analyst 1989, 114, 653–662. [Google Scholar] [CrossRef] [PubMed]
- De Poulpiquet, A.; Ranava, D.; Monsalve, K.; Giudici-Orticoni, M.-T.; Lojou, E. Biohydrogen for a New Generation of H2/O2Biofuel Cells: A Sustainable Energy Perspective. ChemElectroChem 2014, 1, 1724–1750. [Google Scholar] [CrossRef]
- Barton, S.C.; Gallaway, J.; Atanassov, P. Enzymatic Biofuel Cells for Implantable and Microscale Devices. Chem. Rev. 2004, 104, 4867–4886. [Google Scholar] [CrossRef]
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as Working or Inspirational Electrocatalysts for Fuel Cells and Electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef]
- Meredith, M.T.; Minteer, S.D. Biofuel Cells: Enhanced Enzymatic Bioelectrocatalysis. Annu. Rev. Anal. Chem. 2012, 5, 157–179. [Google Scholar] [CrossRef]
- Mazurenko, I.; De Poulpiquet, A.; Lojou, E. Recent developments in high surface area bioelectrodes for enzymatic fuel cells. Curr. Opin. Electrochem. 2017, 5, 74–84. [Google Scholar] [CrossRef]
- Mano, N.; De Poulpiquet, A. O2Reduction in Enzymatic Biofuel Cells. Chem. Rev. 2018, 118, 2392–2468. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Xia, H.-Q.; Wu, R.; Bai, L.; Yan, L.; Magner, E.; Cosnier, S.; Lojou, E.; Zhu, Z.; Liu, A. Tackling the Challenges of Enzymatic (Bio)Fuel Cells. Chem. Rev. 2019, 119, 9509–9558. [Google Scholar] [CrossRef]
- Shleev, S.; González-Arribas, E.; Falk, M. Biosupercapacitors. Curr. Opin. Electrochem. 2017, 5, 226–233. [Google Scholar] [CrossRef]
- Krieg, T.; Sydow, A.; Schröder, U.; Schrader, J.; Holtmann, D. Reactor concepts for bioelectrochemical syntheses and energy conversion. Trends Biotechnol. 2014, 32, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Paddock, R.M.; Bowden, E.F. Electrocatalytic reduction of hydrogen peroxide via direct electron transfer from pyrolytic graphite electrodes to irreversibly adsorbed cytochrome c peroxidase. J. Electroanal. Chem. Interfacial Electrochem. 1989, 260, 487–494. [Google Scholar] [CrossRef]
- Xia, H.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-type Bioelectrocatalysis of Peroxidase at Mesoporous Carbon Electrodes and Its Application for Glucose Determination Based on Bienzyme System. Anal. Sci. 2017, 33, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, T.; Wang, J.; Xia, H.; Wang, S.; Yu, X. Direct Electrochemistry and Electrocatalysis of Horseradish Peroxidase Immobilized in a DNA/Chitosan-Fe3O4 Magnetic Nanoparticle Bio-Complex Film. Materials 2014, 7, 1069–1083. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Kano, K. Nanostructured Porous Electrodes by the Anodization of Gold for an Application as Scaffolds in Direct-electron-transfer-type Bioelectrocatalysis. Anal. Sci. 2018, 34, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Siritanaratkul, B.; Megarity, C.F.; Roberts, T.G.; Samuels, T.O.M.; Winkler, M.; Warner, J.H.; Happe, T.; Armstrong, F.A. Transfer of photosynthetic NADP+/NADPH recycling activity to a porous metal oxide for highly specific, electrochemically-driven organic synthesis. Chem. Sci. 2017, 8, 4579–4586. [Google Scholar] [CrossRef] [Green Version]
- Yehezkeli, O.; Raichlin, S.; Tel-Vered, R.; Kesselman, E.; Danino, D.; Willner, I. Biocatalytic Implant of Pt Nanoclusters into Glucose Oxidase: A Method to Electrically Wire the Enzyme and to Transform It from an Oxidase to a Hydrogenase. J. Phys. Chem. Lett. 2010, 1, 2816–2819. [Google Scholar] [CrossRef]
- Trifonov, A.; Stemmer, A.; Tel-Vered, R. Enzymatic self-wiring in nanopores and its application in direct electron transfer biofuel cells. Nanoscale Adv. 2019, 1, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Fujii, T.; Honda, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct electron transfer-type bioelectrocatalysis of FAD-dependent glucose dehydrogenase using porous gold electrodes and enzymatically implanted platinum nanoclusters. Bioelectrochemistry 2020, 133, 107457. [Google Scholar] [CrossRef]
- Léger, C.; Bertrand, P. Direct Electrochemistry of Redox Enzymes as a Tool for Mechanistic Studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236. [Google Scholar] [CrossRef] [Green Version]
- Kitazumi, Y.; Shirai, O.; Kano, K. Catalyst Materials for Bioelectrochemical Systems: Fundamentals and Applications; ACS Publications: Washington, DC, USA, 2020; pp. 147–163. [Google Scholar]
- Gorton, L.; Jönsson-Pettersson, G.; Csöregi, E.; Johansson, K.; Domínguez, E.; Marko-Varga, G. Amperometric biosensors based on an apparent direct electron transfer between electrodes and immobilized peroxidases. Plenary lecture. Analyst 1992, 117, 1235–1241. [Google Scholar] [CrossRef]
- Degani, Y.; Heller, A. Electrical communication between redox centers of glucose oxidase and electrodes via electrostatically and covalently bound redox polymers. J. Am. Chem. Soc. 1989, 111, 2357–2358. [Google Scholar] [CrossRef]
- Ohara, T.J.; Rajagopalan, R.; Heller, A. Glucose Electrodes Based on Cross-Linked (Os(bpy)2Cl)+/2+ Complexed Poly(1-vinylimidazole) Films. Anal. Chem. 1993, 65, 3512–3517. [Google Scholar] [CrossRef]
- Timur, S.; Yigzaw, Y.; Gorton, L. Electrical wiring of pyranose oxidase with osmium redox polymers. Sens. Actuators B Chem. 2006, 113, 684–691. [Google Scholar] [CrossRef]
- Nikitina, O.; Shleev, S.; Gayda, G.; Demkiv, O.; Gonchar, M.; Gorton, L.; Csöregi, E.; Nistor, M. Bi-enzyme biosensor based on NAD+- and glutathione-dependent recombinant formaldehyde dehydrogenase and diaphorase for formaldehyde assay. Sens. Actuators B Chem. 2007, 125, 1–9. [Google Scholar] [CrossRef]
- Nieh, C.-H.; Kitazumi, Y.; Shirai, O.; Yamamoto, M.; Kano, K. Potentiometric coulometry based on charge accumulation with a peroxidase/osmium polymer-immobilized electrode for sensitive determination of hydrogen peroxide. Electrochem. Commun. 2013, 33, 135–137. [Google Scholar] [CrossRef]
- Alsaoub, S.; Conzuelo, F.; Gounel, S.; Mano, N.; Schuhmann, W.; Ruff, A. Introducing Pseudocapavitive Bioelectrodes into a Biofuel Cell/Biosupercapacitor Hybrid Device for Optimized Open Circuit Voltage. ChemElectroChem 2019, 6, 2080–2087. [Google Scholar] [CrossRef]
- Tsujimura, S.; Takeuchi, S. Toward an Ideal Platform Structure Based on MgO-Templated Carbon for Flavin Adenine Dinucleotide-Dependent Glucose Dehydrogenase-Os Polymer-Hydrogel Electrodes. Electrochim. Acta 2020, 343, 136110. [Google Scholar] [CrossRef]
- Xiao, X.; Conghaile, P.Ó.; Leech, D.; Magner, E. Use of Polymer Coatings to Enhance the Response of Redox-Polymer-Mediated Electrodes. ChemElectroChem 2019, 6, 1344–1349. [Google Scholar] [CrossRef] [Green Version]
- Čėnas, N.K.; Pocius, A.K.; Kulys, J.J. Electron Exchange between Flavin- and Heme-Containing Enzymes and Electrodes Modified by Redox Polymers. Bioelectrochem. Bioenerg. 1983, 11, 61–73. [Google Scholar] [CrossRef]
- Nieh, C.-H.; Kitazumi, Y.; Shirai, O.; Kano, K. Sensitive d-amino acid biosensor based on oxidase/peroxidase system mediated by pentacyanoferrate-bound polymer. Biosens. Bioelectron. 2013, 47, 350–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieh, C.-H.; Tsujimura, S.; Shirai, O.; Kano, K. Amperometric biosensor based on reductive H2O2 detection using pentacyanoferrate-bound polymer for creatinine determination. Anal. Chim. Acta 2013, 767, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Merchant, S.A.; Meredith, M.T.; Tran, T.O.; Brunski, D.B.; Johnson, M.B.; Glatzhofer, D.T.; Schmidtke, D.W. Effect of Mediator Spacing on Electrochemical and Enzymatic Response of Ferrocene Redox Polymers. J. Phys. Chem. C 2010, 114, 11627–11634. [Google Scholar] [CrossRef]
- Tran, T.O.; Lammert, E.G.; Chen, J.; Merchant, S.A.; Brunski, D.B.; Keay, J.C.; Johnson, M.B.; Glatzhofer, D.T.; Schmidtke, D.W. Incorporation of Single-Walled Carbon Nanotubes into Ferrocene-Modified Linear Polyethylenimine Redox Polymer Films. Langmuir 2011, 27, 6201–6210. [Google Scholar] [CrossRef]
- Milton, R.D.; Abdellaoui, S.; Khadka, N.; Dean, D.R.; Leech, D.; Seefeldt, L.C.; Minteer, S.D. Nitrogenase bioelectrocatalysis: Heterogeneous ammonia and hydrogen production by MoFe protein. Energy Environ. Sci. 2016, 9, 2550–2554. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Yuan, M.; Cai, R.; Lim, K.; Minteer, S.D. Nitrogenase Bioelectrocatalysis: ATP-Independent Ammonia Production Using a Redox Polymer/MoFe Protein System. ACS Catal. 2020, 10, 6854–6861. [Google Scholar] [CrossRef]
- Eng, L.H.; Elmgren, M.; Komlos, P.; Nordling, M.; Lindquist, S.-E.; Neujahr, H.Y. Viologen-Based Redox Polymer for Contacting the Low-Potential Redox Enzyme Hydrogenase at an Electrode Surface. J. Phys. Chem. 1994, 98, 7068–7072. [Google Scholar] [CrossRef]
- Shiraiwa, S.; So, K.; Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Reactivation of Standard (NiFe)-Hydrogenase and Bioelectrochemical Catalysis of Proton Reduction and Hydrogen Oxidation in a Mediated-Electron-Transfer System. Bioelectrochemistry 2018, 123, 156–161. [Google Scholar] [CrossRef]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Kano, K. Bioelectrocatalytic formate oxidation and carbon dioxide reduction at high current density and low overpotential with tungsten-containing formate dehydrogenase and mediators. Electrochem. Commun. 2016, 65, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. High-Power Formate/Dioxygen Biofuel Cell Based on Mediated Electron Transfer Type Bioelectrocatalysis. ACS Catal. 2017, 7, 5668–5673. [Google Scholar] [CrossRef]
- Kitazumi, Y.; Shirai, O.; Yamamoto, M.; Kano, K. Numerical simulation of diffuse double layer around microporous electrodes based on the Poisson–Boltzmann equation. Electrochim. Acta 2013, 112, 171–175. [Google Scholar] [CrossRef]
- Serafin, V.; Hernández, P.; Agüí, L.; Yáñez-Sedeño, P.; Pingarrón, J.M. Electrochemical Biosensor for Creatinine Based on the Immobilization of Creatininase, Creatinase and Sarcosine Oxidase onto a Ferrocene/Horseradish Peroxidase/Gold Nanoparticles/Multi-Walled Carbon Nanotubes/Teflon Composite Electrode. Electrochim. Acta 2013, 97, 175–183. [Google Scholar] [CrossRef]
- Holzinger, M.; Le Goff, A.; Cosnier, S. Carbon nanotube/enzyme biofuel cells. Electrochim. Acta 2012, 82, 179–190. [Google Scholar] [CrossRef]
- Tominaga, M.; Sasaki, A.; Togami, M. Laccase Bioelectrocatalyst at a Steroid-Type Biosurfactant-Modified Carbon Nanotube Interface. Anal. Chem. 2015, 87, 5417–5421. [Google Scholar] [CrossRef]
- Tsujimura, S.; Nishina, A.; Kamitaka, Y.; Kano, K. Coulometric d-Fructose Biosensor Based on Direct Electron Transfer Using D-Fructose Dehydrogenase. Anal. Chem. 2009, 81, 9383–9387. [Google Scholar] [CrossRef]
- Flexer, V.; Durand, F.; Tsujimura, S.; Mano, N. Efficient Direct Electron Transfer of PQQ-glucose Dehydrogenase on Carbon Cryogel Electrodes at Neutral pH. Anal. Chem. 2011, 83, 5721–5727. [Google Scholar] [CrossRef]
- Hamano, Y.; Tsujimura, S.; Shirai, O.; Kano, K. Micro-cubic monolithic carbon cryogel electrode for direct electron transfer reaction of fructose dehydrogenase. Bioelectrochemistry 2012, 88, 114–117. [Google Scholar] [CrossRef]
- Murata, K.; Akatsuka, W.; Tsujimura, S. Bioelectrocatalytic Oxidation of Glucose on MgO-templated Mesoporous Carbon-modified Electrode. Chem. Lett. 2014, 43, 928–930. [Google Scholar] [CrossRef]
- Mazurenko, I.; Clément, R.; Byrne-Kodjabachian, D.; de Poulpiquet, A.; Tsujimura, S.; Lojou, E. Pore Size Effect of MgO-Templated Carbon on Enzymatic H2 Oxidation by the Hyperthermophilic Hydrogenase from Aquifex aeolicus. J. Electroanal. Chem. 2018, 812, 221–226. [Google Scholar] [CrossRef]
- Takahashi, Y.; Wanibuchi, M.; Kitazumi, Y.; Shirai, O.; Kano, K. Improved direct electron transfer-type bioelectrocatalysis of bilirubin oxidase using porous gold electrodes. J. Electroanal. Chem. 2019, 843, 47–53. [Google Scholar] [CrossRef]
- Mie, Y.; Yasutake, Y.; Ikegami, M.; Tamura, T. Anodized gold surface enables mediator-free and low-overpotential electrochemical oxidation of NADH: A facile method for the development of an NAD+-dependent enzyme biosensor. Sens. Actuators B Chem. 2019, 288, 512–518. [Google Scholar] [CrossRef]
- Miyata, M.; Kitazumi, Y.; Shirai, O.; Kataoka, K.; Kano, K. Diffusion-limited biosensing of dissolved oxygen by direct electron transfer-type bioelectrocatalysis of multi-copper oxidases immobilized on porous gold microelectrodes. J. Electroanal. Chem. 2020, 860, 113895. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.; Belochapkine, S.; Magner, E. Nanoporous Gold Electrodes with Tuneable Pore Sizes for Bioelectrochemical Applications. Electroanalysis 2016, 28, 2415–2423. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The Immobilization of Fructose Dehydrogenase on Nanoporous Gold Electrodes for the Detection of Fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Xiao, X.; Siepenkoetter, T.; Conghaile, P.Ó.; Leech, D.; Magner, E. Nanoporous Gold-Based Biofuel Cells on Contact Lenses. ACS Appl. Mater. Interfaces 2018, 10, 7107–7116. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.T.; Lau, C.; Brozik, S.; Atanassov, P.; Banta, S. Engineering of Glucose Oxidase for Direct Electron Transfer via Site-Specific Gold Nanoparticle Conjugation. J. Am. Chem. Soc. 2011, 133, 19262–19265. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Sánchez, C.; Pita, M.; Vaz-Domínguez, C.; Shleev, S.; De Lacey, A.L. Gold Nanoparticles as Electronic Bridges for Laccase-Based Biocathodes. J. Am. Chem. Soc. 2012, 134, 17212–17220. [Google Scholar] [CrossRef]
- Suzuki, M.; Murata, K.; Nakamura, N.; Ohno, H. The Effect of Particle Size on the Direct Electron Transfer Reactions of Metalloproteins Using Au Nanoparticle-Modified Electrodes. Electrochemistry 2012, 80, 337–339. [Google Scholar] [CrossRef] [Green Version]
- Monsalve, K.; Roger, M.; Gutierrez-Sanchez, C.; Ilbert, M.; Nitsche, S.; Byrne-Kodjabachian, D.; Marchi, V.; Lojou, E. Hydrogen bioelectrooxidation on gold nanoparticle-based electrodes modified by Aquifex aeolicus hydrogenase: Application to hydrogen/oxygen enzymatic biofuel cells. Bioelectrochemistry 2015, 106, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. Direct Electron Transfer-Type Four-Way Bioelectrocatalysis of CO2/Formate and NAD+/NADH Redox Couples by Tungsten-Containing Formate Dehydrogenase Adsorbed on Gold Nanoparticle-Embedded Mesoporous Carbon Electrodes Modified with 4-Mercaptopyridine. Electrochem. Commun. 2017, 84, 75–79. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Improved direct electron transfer-type bioelectrocatalysis of bilirubin oxidase using thiol-modified gold nanoparticles on mesoporous carbon electrode. J. Electroanal. Chem. 2019, 832, 158–164. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Mazurenko, I.; Murali, A.V.; De Poulpiquet, A.; Coustillier, G.; Delaporte, P.; Lojou, E. Nanosecond Laser–Fabricated Monolayer of Gold Nanoparticles on ITO for Bioelectrocatalysis. Front. Chem. 2020, 8, 431. [Google Scholar] [CrossRef]
- Kizling, M.; Dzwonek, M.; Więckowska, A.; Bilewicz, R. Gold nanoparticles in bioelectrocatalysis—The role of nanoparticle size. Curr. Opin. Electrochem. 2018, 12, 113–120. [Google Scholar] [CrossRef]
- Murata, K.; Suzuki, M.; Nakamura, N.; Ohno, H. Direct evidence of electron flow via the heme c group for the direct electron transfer reaction of fructose dehydrogenase using a silver nanoparticle-modified electrode. Electrochem. Commun. 2009, 11, 1623–1626. [Google Scholar] [CrossRef]
- Zhang, L.; Beaton, S.E.; Carr, S.B.; Armstrong, F.A. Direct visible light activation of a surface cysteine-engineered [NiFe]-hydrogenase by silver nanoclusters. Energy Environ. Sci. 2018, 11, 3342–3348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Can, M.; Ragsdale, S.W.; Armstrong, F. Fast and Selective Photoreduction of CO2 to CO Catalyzed by a Complex of Carbon Monoxide Dehydrogenase, TiO2, and Ag Nanoclusters. ACS Catal. 2018, 8, 2789–2795. [Google Scholar] [CrossRef]
- Nakamura, R.; Kamiya, K.; Hashimoto, K. Direct electron-transfer conduits constructed at the interface between multicopper oxidase and nanocrystalline semiconductive Fe oxides. Chem. Phys. Lett. 2010, 498, 307–311. [Google Scholar] [CrossRef]
- Kizling, M.; Rekorajska, A.; Krysinski, P.; Bilewicz, R. Magnetic-field-induced orientation of fructose dehydrogenase on iron oxide nanoparticles for enhanced direct electron transfer. Electrochem. Commun. 2018, 93, 66–70. [Google Scholar] [CrossRef]
- Rozniecka, E.; Jonsson-Niedziolka, M.; Sobczak, J.W.; Opallo, M. Mediatorless bioelectrocatalysis of dioxygen reduction at indium-doped tin oxide (ITO) and ITO nanoparticulate film electrodes. Electrochim. Acta 2011, 56, 8739–8745. [Google Scholar] [CrossRef]
- Willner, I.; Katz, E. Integration of Layered Redox Proteins and Conductive Supports for Bioelectronic Applications. Angew. Chem. Int. Ed. 2000, 39, 1180–1218. [Google Scholar] [CrossRef]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nat. Cell Biol. 1992, 355, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta BBA Rev. Bioenerg. 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture). Angew. Chem. Int. Ed. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Takeuchi, R.; Kitazumi, Y.; Shirai, O.; Kano, K. Significance of Mesoporous Electrodes for Noncatalytic Faradaic Process of Randomly Oriented Redox Proteins. J. Phys. Chem. C 2016, 120, 26270–26277. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Effects of Mesoporous Structures on Direct Electron Transfer-Type Bioelectrocatalysis: Facts and Simulation on a Three-Dimensional Model of Random Orientation of Enzymes. Electrochemistry 2017, 85, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Hitaishi, V.P.; Clement, R.; Bourassin, N.; Baaden, M.; De Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Sacquin-Mora, S. Controlling Redox Enzyme Orientation at Planar Electrodes. Catalysts 2018, 8, 192. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; So, K.; Kitazumi, Y.; Shirai, O.; Nishikawa, K.; Higuchi, Y.; Kano, K. Dual gas-diffusion membrane- and mediatorless dihydrogen/air-breathing biofuel cell operating at room temperature. J. Power Sources 2016, 335, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kawano, T.; Kataoka, K.; Kano, K. Effects of Elimination of α Helix Regions on Direct Electron Transfer-type Bioelectrocatalytic Properties of Copper Efflux Oxidase. Electrochemistry 2020, 88, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Kitazumi, Y.; Shirai, O.; Kano, K. Enhanced direct electron transfer-type bioelectrocatalysis of bilirubin oxidase on negatively charged aromatic compound-modified carbon electrode. J. Electroanal. Chem. 2016, 763, 104–109. [Google Scholar] [CrossRef]
- So, K.; Kawai, S.; Hamano, Y.; Kitazumi, Y.; Shirai, O.; Hibi, M.; Ogawa, J.; Kano, K. Improvement of a direct electron transfer-type fructose/dioxygen biofuel cell with a substrate-modified biocathode. Phys. Chem. Chem. Phys. 2014, 16, 4823–4829. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. Highly Sensitive Membraneless Fructose Biosensor Based on Fructose Dehydrogenase Immobilized onto Aryl Thiol Modified Highly Porous Gold Electrode: Characterization and Application in Food Samples. Anal. Chem. 2018, 90, 12131–12136. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Interaction between d-fructose dehydrogenase and methoxy-substituent-functionalized carbon surface to increase productive orientations. Electrochim. Acta 2016, 218, 41–46. [Google Scholar] [CrossRef]
- So, K.; Sakai, K.; Kano, K. Gas diffusion bioelectrodes. Curr. Opin. Electrochem. 2017, 5, 173–182. [Google Scholar] [CrossRef]
- Wong, T.S.; Schwaneberg, U. Protein engineering in bioelectrocatalysis. Curr. Opin. Biotechnol. 2003, 14, 590–596. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Borsari, M.; Cowan, J.A.; Ranieri, A.; Sola, M. Control of cytochrome C redox potential: Axial ligation and protein environment effects. J. Am. Chem. Soc. 2002, 124, 5315–5324. [Google Scholar] [CrossRef]
- Li, H.; Webb, S.P.; Ivanic, J.; Jensen, J.H. Determinants of the Relative Reduction Potentials of Type-1 Copper Sites in Proteins. J. Am. Chem. Soc. 2004, 126, 8010–8019. [Google Scholar] [CrossRef]
- Kawai, S.; Yakushi, T.; Matsushita, K.; Kitazumi, Y.; Shirai, O.; Kano, K. The electron transfer pathway in direct electrochemical communication of fructose dehydrogenase with electrodes. Electrochem. Commun. 2014, 38, 28–31. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Mutation of heme c axial ligands in d-fructose dehydrogenase for investigation of electron transfer pathways and reduction of overpotential in direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2016, 67, 43–46. [Google Scholar] [CrossRef]
- Adachi, T.; Kaida, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Bioelectrocatalytic performance of d-fructose dehydrogenase. Bioelectrochemistry 2019, 129, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kamitaka, Y.; Tsujimura, S.; Kataoka, K.; Sakurai, T.; Ikeda, T.; Kano, K. Effects of axial ligand mutation of the type I copper site in bilirubin oxidase on direct electron transfer-type bioelectrocatalytic reduction of dioxygen. J. Electroanal. Chem. 2007, 601, 119–124. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of a protein-engineered variant of d -fructose dehydrogenase for direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2017, 77, 112–115. [Google Scholar] [CrossRef]
- Kaida, Y.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Ultimate downsizing of d-fructose dehydrogenase for improving the performance of direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2019, 98, 101–105. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Protein-Engineering Improvement of Direct Electron Transfer-Type Bioelectrocatalytic Properties of d-Fructose Dehydrogenase. Electrochemistry 2019, 87, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Kaida, Y.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Discussion on Direct Electron Transfer-Type Bioelectrocatalysis of Downsized and Axial-Ligand Exchanged Variants of d-Fructose Dehydrogenase. Electrochemistry 2020, 88, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Hamamoto, K.; Kitazumi, Y.; Shirai, O.; Kano, K. Diffusion-Controlled Mediated Electron Transfer-Type Bioelectrocatalysis Using Ultrathin-Ring and Microband Electrodes as Ultimate Amperometric Glucose Sensors. Anal. Sci. 2017, 33, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Mazurenko, I.; Hitaishi, V.P.; Lojou, E. Recent advances in surface chemistry of electrodes to promote direct enzymatic bioelectrocatalysis. Curr. Opin. Electrochem. 2020, 19, 113–121. [Google Scholar] [CrossRef]
- Ferapontova, E.E.; Schmengler, K.; Börchers, T.; Ruzgas, T.; Gorton, L. Effect of cysteine mutations on direct electron transfer of horseradish peroxidase on gold. Biosens. Bioelectron. 2002, 17, 953–963. [Google Scholar] [CrossRef]
- Bollella, P.; Ludwig, R.; Gorton, L. Cellobiose dehydrogenase: Insights on the nanostructuration of electrodes for improved development of biosensors and biofuel cells. Appl. Mater. Today 2017, 9, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Algov, I.; Grushka, J.; Zarivach, R.; Alfonta, L. Highly Efficient Flavin–Adenine Dinucleotide Glucose Dehydrogenase Fused to a Minimal Cytochrome C Domain. J. Am. Chem. Soc. 2017, 139, 17217–17220. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Okuda-Shimazaki, J.; Mori, K.; Kojima, K.; Tsugawa, W.; Ikebukuro, K.; Lin, C.-E.; La Belle, J.T.; Yoshida, H.; Sode, K. Designer fungus FAD glucose dehydrogenase capable of direct electron transfer. Biosens. Bioelectron. 2019, 123, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Okuda, J.; Sode, K. PQQ glucose dehydrogenase with novel electron transfer ability. Biochem. Biophys. Res. Commun. 2004, 314, 793–797. [Google Scholar] [CrossRef]
- Gilardi, G.; Meharenna, Y.T.; Tsotsou, G.E.; Sadeghi, S.J.; Fairhead, M.; Giannini, S. Molecular Lego: Design of molecular assemblies of P450 enzymes for nanobiotechnology. Biosens. Bioelectron. 2002, 17, 133–145. [Google Scholar] [CrossRef]
- Hanashi, T.; Yamazaki, T.; Tsugawa, W.; Ferri, S.; Nakayama, D.; Tomiyama, M.; Ikebukuro, K.; Sode, K. BioCapacitor-A Novel Category of Biosensor. Biosens. Bioelectron. 2009, 24, 1837–1842. [Google Scholar] [CrossRef]
- Conzuelo, F.; Ruff, A.; Schuhmann, W. Self-powered bioelectrochemical devices. Curr. Opin. Electrochem. 2018, 12, 156–163. [Google Scholar] [CrossRef]
- Falk, M.; Shleev, S. Hybrid Dual-functioning Electrodes for Combined Ambient Energy Harvesting and Charge Strage: Towards Self-Powered Systems. Biosens. Bioelectron. 2019, 126, 275–291. [Google Scholar] [CrossRef]
- Agnès, C.; Holzinger, M.; Le Goff, A.; Reuillard, B.; Elouarzaki, K.; Tingry, S.; Cosnier, S. Supercapacitor/biofuel cell hybrids based on wired enzymes on carbon nanotube matrices: Autonomous reloading after high power pulses in neutral buffered glucose solutions. Energy Environ. Sci. 2014, 7, 1884–1888. [Google Scholar] [CrossRef]
- Pankratov, D.; Blum, Z.; Suyatin, D.B.; Popov, V.O.; Shleev, S. Self-Charging Electrochemical Biocapacitor. ChemElectroChem 2013, 1, 343–346. [Google Scholar] [CrossRef]
- Pankratov, D.; Blum, Z.; Shleev, S. Hybrid Electric Power Biodevices. ChemElectroChem 2014, 1, 1798–1807. [Google Scholar] [CrossRef]
- Xiao, X.; Magner, E. A quasi-solid-state and self-powered biosupercapacitor based on flexible nanoporous gold electrodes. Chem. Commun. 2018, 54, 5823–5826. [Google Scholar] [CrossRef] [PubMed]
- Pankratov, D.; Shen, F.; Ortiz, R.; Toscano, M.D.; Thormann, E.; Zhang, J.; Gorton, L.; Chi, Q. Fuel-independent and membrane-less self-charging biosupercapacitor. Chem. Commun. 2018, 54, 11801–11804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrowski, T.; González-Arribas, E.; Ludwig, R.; Toscano, M.D.; Shleev, S.; Schuhmann, W. Rechargeable, Flexible and Mediator-Free Biosupercapacitor Based on Transparent ITO Nanoparticle Modified Electrodes Acting in μM Glucose Containing Buffers. Biosens. Bioelectron. 2018, 101, 84–89. [Google Scholar] [CrossRef]
- Pankratov, D.; Conzuelo, F.; Pinyou, P.; Alsaoub, S.; Schuhmann, W.; Shleev, S. A Nernstian Biosupercapacitor. Angew. Chem. Int. Ed. 2016, 55, 15434–15438. [Google Scholar] [CrossRef]
- Alsaoub, S.; Ruff, A.; Conzuelo, F.; Ventosa, E.; Ludwig, R.; Shleev, S.; Schuhmann, W. An Intrinsic Self-Charging Biosupercapacitor Comprised of a High-Potential Bioanode and a Low-Potential Biocathode. ChemPlusChem 2017, 82, 576–583. [Google Scholar] [CrossRef]
- Zhao, F.; Bobrowski, T.; Ruff, A.; Hartmann, V.; Nowaczyk, M.M.; Rögner, M.; Conzuelo, F.; Schuhmann, W. A light-driven Nernstian biosupercapacitor. Electrochim. Acta 2019, 306, 660–666. [Google Scholar] [CrossRef]
- González-Arribas, E.; Falk, M.; Aleksejeva, O.; Bushnev, S.; Sebastián, P.; Feliu, J.M.; Shleev, S. A Conventional Symmetric Biosupercapacitor Based on Rusticyanin Modified Gold Electrodes. J. Electroanal. Chem. 2018, 816, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Pankratov, D.; Pankratova, G.; Toscano, M.D.; Zhang, J.; Ulstrup, J.; Chi, Q.; Gorton, L. Supercapacitor/Biofuel Cell Hybrid Device Employing Biomolecules for Energy Conversion and Charge Stroge. Bioelectrochemistry 2019, 128, 94–99. [Google Scholar] [CrossRef]
- Wu, R.; Ma, C.; Zhu, Z. Enzymatic electrosynthesis as an emerging electrochemical synthesis platform. Curr. Opin. Electrochem. 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Schmid, A.I.; Dordick, J.S.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt, B. Industrial biocatalysis today and tomorrow. Nat. Cell Biol. 2001, 409, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Zaks, A. Industrial biocatalysis. Curr. Opin. Chem. Biol. 2001, 5, 130–136. [Google Scholar] [CrossRef]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and Exploiting the Electrocatalytic Properties of Hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.; Hirst, J. Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc. Natl. Acad. Sci. USA 2011, 108, 14049–14054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, K.; Hsieh, B.-C.; Maruyama, A.; Kitazumi, Y.; Shirai, O.; Kano, K. Interconversion between formate and hydrogen carbonate by tungsten-containing formate dehydrogenase-catalyzed mediated bioelectrocatalysis. Sens. Bio Sens. Res. 2015, 5, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Reda, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 10654–10658. [Google Scholar] [CrossRef] [Green Version]
- Bassegoda, A.; Madden, C.; Wakerley, D.W.; Reisner, E.; Hirst, J. Reversible Interconversion of CO2 and Formate by a Molybdenum-Containing Formate Dehydrogenase. J. Am. Chem. Soc. 2014, 136, 15473–15476. [Google Scholar] [CrossRef] [Green Version]
- Takagi, K.; Kano, K.; Ikeda, T. Mediated bioelectrocatalysis based on NAD-related enzymes with reversible characteristics. J. Electroanal. Chem. 1998, 445, 211–219. [Google Scholar] [CrossRef]
- Megarity, C.F.; Siritanaratkul, B.; Heath, R.S.; Wan, L.; Morello, G.; Fitzpatrick, S.R.; Booth, R.L.; Sills, A.J.; Robertson, A.W.; Warner, J.H.; et al. Electrocatalytic Volleyball: Rapid Nanoconfined Nicotinamide Cycling for Organic Synthesis in Electrode Pores. Angew. Chem. Int. Ed. 2019, 58, 4948–4952. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Siritanaratkul, B.; Megarity, C.F.; Armstrong, F.A. Efficient Electrocatalytic CO2 Fixation by Nanoconfined Enzymes via a C3-to-C4 Reaction That Is Favored over H2 Production. ACS Catal. 2019, 9, 11255–11262. [Google Scholar] [CrossRef] [Green Version]
- Alkotaini, B.; Abdellaoui, S.; Hasan, K.; Grattieri, M.; Quah, T.; Cai, R.; Yuan, M.; Minteer, S.D. Sustainable Bioelectrosynthesis of the Bioplastic Polyhydroxybutyrate: Overcoming Substrate Requirement for NADH Regeneration. ACS Sustain. Chem. Eng. 2018, 6, 4909–4915. [Google Scholar] [CrossRef]
- Wan, L.; Megarity, C.F.; Siritanaratkul, B.; Armstrong, F. A hydrogen fuel cell for rapid, enzyme-catalysed organic synthesis with continuous monitoring. Chem. Commun. 2018, 54, 972–975. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Heath, R.S.; Siritanaratkul, B.; Megarity, C.F.; Sills, A.J.; Thompson, M.P.; Turner, N.J.; Armstrong, F. Enzyme-catalysed enantioselective oxidation of alcohols by air exploiting fast electrochemical nicotinamide cycling in electrode nanopores. Green Chem. 2019, 21, 4958–4963. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, O.; Woolerton, T.W.; Parkin, A.; Lukey, M.J.; Reisner, E.; Seravalli, J.; Pierce, E.; Ragsdale, S.W.; Sargent, F.; Armstrong, F.A. Water−Gas Shift Reaction Catalyzed by Redox Enzymes on Conducting Graphite Platelets. J. Am. Chem. Soc. 2009, 131, 14154–14155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of a bioelectrochemical formate generating system from carbon dioxide and dihydrogen. Electrochem. Commun. 2018, 97, 73–76. [Google Scholar] [CrossRef]
- Milton, R.D.; Cai, R.; Abdellaoui, S.; Leech, D.; De Lacey, A.L.; Pita, M.; Minteer, S.D. Bioelectrochemical Haber-Bosch Process: An Ammonia-Producing H2 /N2 Fuel Cell. Angew. Chem. Int. Ed. 2017, 56, 2680–2683. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Prater, M.B.; Cai, R.; Dong, F.; Chen, H.; Minteer, S.D. Bioelectrocatalytic Conversion from N2 to Chiral Amino Acids in a H2/α-Keto Acid Enzymatic Fuel Cell. J. Am. Chem. Soc. 2020, 142, 4028–4036. [Google Scholar] [CrossRef]
- Sakai, K.; Kitazumi, Y.; Shirai, O.; Takagi, K.; Kano, K. Efficient bioelectrocatalytic CO2 reduction on gas-diffusion-type biocathode with tungsten-containing formate dehydrogenase. Electrochem. Commun. 2016, 73, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, J.; Tsujimura, S.; Kano, K. Coulometric bioelectrocatalytic reactions based on NAD-dependent dehydrogenases in tricarboxylic acid cycle. Electrochim. Acta 2008, 54, 328–333. [Google Scholar] [CrossRef]
- Chen, H.; Cai, R.; Patel, J.; Dong, F.; Chen, H.; Minteer, S.D. Upgraded Bioelectrocatalytic N2 Fixation: From N2 to Chiral Amine Intermediates. J. Am. Chem. Soc. 2019, 141, 4963–4971. [Google Scholar] [CrossRef]
- Dong, F.; Chen, H.; Malapit, C.A.; Prater, M.B.; Li, M.; Yuan, M.; Lim, K.; Minteer, S.D. Biphasic Bioelectrocatalytic Synthesis of Chiral β-Hydroxy Nitriles. J. Am. Chem. Soc. 2020, 142, 8374–8382. [Google Scholar] [CrossRef] [PubMed]
- Hickey, D.P.; Cai, R.; Yang, Z.-Y.; Grunau, K.; Einsle, O.; Seefeldt, L.C.; Minteer, S.D. Establishing a Thermodynamic Landscape for the Active Site of Mo-Dependent Nitrogenase. J. Am. Chem. Soc. 2019, 141, 17150–17157. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.E. STRUCTURE: Nitrogenase Reveals Its Inner Secrets. Science 2002, 297, 1654–1655. [Google Scholar] [CrossRef] [PubMed]
- Milton, R.D.; Minteer, S.D. Enzymatic Bioelectrosynthetic Ammonia Production: Recent Electrochemistry of Nitrogenase, Nitrate Reductase, and Nitrite Reductase. ChemPlusChem 2016, 82, 513–521. [Google Scholar] [CrossRef]
- Cai, R.; Minteer, S.D. Nitrogenase Bioelectrocatalysis: From Understanding Electron-Transfer Mechanisms to Energy Applications. ACS Energy Lett. 2018, 3, 2736–2742. [Google Scholar] [CrossRef]
- Antiochia, R.; Gallina, A.; Lavagnini, I.; Magno, F. Kinetic and Thermodynamic Aspects of NAD-Related Enzyme-Linked Mediated Bioelectrocatalysis. Electroanalysis 2002, 14, 1256–1261. [Google Scholar] [CrossRef]
- Lobo, M.J.; Miranda, A.J.; Tuñón, P. Amperometric Biosensors Based on NAD(P)-Dependent Dehydrogenase Enzymes. Electroanalysis 1997, 9, 191–201. [Google Scholar] [CrossRef]
- Sokol, K.P.; Robinson, W.E.; Warnan, J.; Kornienko, N.; Nowaczyk, M.M.; Ruff, A.; Zhang, J.Z.; Reisner, E. Bias-free photoelectrochemical water splitting with photosystem II on a dye-sensitized photoanode wired to hydrogenase. Nat. Energy 2018, 3, 944–951. [Google Scholar] [CrossRef]
- Sokol, K.P.; Robinson, W.E.; Oliveira, A.R.; Warnan, J.; Nowaczyk, M.M.; Ruff, A.; Pereira, I.A.C.; Reisner, E. Photoreduction of CO2 with a Formate Dehydrogenase Driven by Photosystem II Using a Semi-artificial Z-Scheme Architecture. J. Am. Chem. Soc. 2018, 140, 16418–16422. [Google Scholar] [CrossRef] [Green Version]
- Riedel, M.; Wersig, J.; Ruff, A.; Schuhmann, W.; Zouni, A.; Lisdat, F. A Z-Scheme-Inspired Photobioelectrochemical H2O/O2 Cell with a 1V Open-Circuit Voltage Combining Photosystem II and PbS Quantum Dots. Angew. Chem. Int. Ed. 2019, 58, 801–805. [Google Scholar] [CrossRef]
- Adachi, T.; Kataoka, K.; Kitazumi, Y.; Shirai, O.; Kano, K. A Bio-solar Cell with Thylakoid Membranes and Bilirubin Oxidase. Chem. Lett. 2019, 48, 686–689. [Google Scholar] [CrossRef]
- Pankratov, D.; Pankratova, G.; Dyachkova, T.P.; Falkman, P.; Åkerlund, H.-E.; Toscano, M.D.; Chi, Q.; Gorton, L. Supercapacitive Biosolar Cell Driven by Direct Electron Transfer between Photosynthetic Membranes and CNT Networks with Enhanced Performance. ACS Energy Lett. 2017, 2, 2635–2639. [Google Scholar] [CrossRef]
- Rasmussen, M.; Wingersky, A.; Minteer, S.D. Improved Performance of a Thylakoid Bio-Solar Cell by Incorporation of Carbon Quantum Dots. ECS Electrochem. Lett. 2013, 3, H1–H3. [Google Scholar] [CrossRef]
- Efrati, A.; Tel-Vered, R.; Michaeli, R.; Nechushtai, R.; Willner, I. Cytochrome c-coupled photosystem I and photosystem II (PSI/PSII) photo-bioelectrochemical cells. Energy Environ. Sci. 2013, 6, 2950. [Google Scholar] [CrossRef]
- Kirchhofer, N.D.; Rasmussen, M.; Dahlquist, F.W.; Minteer, S.D.; Bazan, G.C. The photobioelectrochemical activity of thylakoid bioanodes is increased via photocurrent generation and improved contacts by membrane-intercalating conjugated oligoelectrolytes. Energy Environ. Sci. 2015, 8, 2698–2706. [Google Scholar] [CrossRef]
- Pankratova, G.; Pankratov, D.; Hasan, K.; Åkerlund, H.-E.; Albertsson, P.-Å.; Leech, D.; Shleev, S.; Gorton, L. Supercapacitive Photo-Bioanodes and Biosolar Cells: A Novel Approach for Solar Energy Harnessing. Adv. Energy Mater. 2017, 7, 1602285. [Google Scholar] [CrossRef]
- Yehezkeli, O.; Tel-Vered, R.; Wasserman, J.; Trifonov, A.; Michaeli, D.; Nechushtai, R.; Willner, I. Integrated photosystem II-based photo-bioelectrochemical cells. Nat. Commun. 2012, 3, 742. [Google Scholar] [CrossRef] [Green Version]
- Calkins, J.O.; Umasankar, Y.; O’Neill, H.; Ramasamy, R.P. High photo-electrochemical activity of thylakoid–carbon nanotube composites for photosynthetic energy conversion. Energy Environ. Sci. 2013, 6, 1891–1900. [Google Scholar] [CrossRef]
- Tsujimura, S.; Wadano, A.; Kano, K.; Ikeda, T. Photosynthetic bioelectrochemical cell utilizing cyanobacteria and water-generating oxidase. Enzym. Microb. Technol. 2001, 29, 225–231. [Google Scholar] [CrossRef]
- Mimcault, M.; Carpentier, R. Kinetics of Photocurrent Induction by a Thylakoid Containing Elctrochemical Cell. J. Electroanal. Chem. 1989, 276, 145–158. [Google Scholar] [CrossRef]
- Carpentier, R.; Lemieux, S.; Mimeault, M.; Purcell, M.; Goetze, D.C. A Photoelectrochemical Cell Using Immobilized Photosynthetic Membranes. J. Electroanal. Chem. Interfacial Electrochem. 1989, 276, 391–401. [Google Scholar] [CrossRef]
- González-Arribas, E.; Aleksejeva, O.; Bobrowski, T.; Toscano, M.D.; Gorton, L.; Schuhmann, W.; Shleev, S. Solar biosupercapacitor. Electrochem. Commun. 2017, 74, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.; Minteer, S.D. Investigating the Mechanism of Thylakoid Direct Electron Transfer for Photocurrent Generation. Electrochim. Acta 2014, 126, 68–73. [Google Scholar] [CrossRef]
- Saboe, P.O.; Conte, E.; Chan, S.; Feroz, H.; Ferlez, B.; Farell, M.; Poyton, M.F.; Sines, I.T.; Yan, H.; Bazan, G.C.; et al. Biomimetic wiring and stabilization of photosynthetic membrane proteins with block copolymer interfaces. J. Mater. Chem. A 2016, 4, 15457–15463. [Google Scholar] [CrossRef]
- Kanso, H.; Pankratova, G.; Bollella, P.; Leech, D.; Hernandez, D.; Gorton, L. Sunlight photocurrent generation from thylakoid membranes on gold nanoparticle modified screen-printed electrodes. J. Electroanal. Chem. 2018, 816, 259–264. [Google Scholar] [CrossRef]
- Hamidi, H.; Hasan, K.; Emek, S.C.; Dilgin, Y.; Åkerlund, H.-E.; Albertsson, P.-. Åke; Leech, D.; Gorton, L. Photocurrent Generation from Thylakoid Membranes on Osmium-Redox-Polymer-Modified Electrodes. ChemSusChem 2015, 8, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Hasan, K.; Milton, R.D.; Grattieri, M.; Wang, T.; Stephanz, M.; Minteer, S.D. Photobioelectrocatalysis of Intact Chloroplasts for Solar Energy Conversion. ACS Catal. 2017, 7, 2257–2265. [Google Scholar] [CrossRef]
- Takeuchi, R.; Suzuki, A.; Sakai, K.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of photo-driven bioanodes using thylakoid membranes and multi-walled carbon nanotubes. Bioelectrochemistry 2018, 122, 158–163. [Google Scholar] [CrossRef]
- Hasan, K.; Dilgin, Y.; Emek, S.C.; Tavahodi, M.; Åkerlund, H.-E.; Albertsson, P.-Å.; Gorton, L. Photoelectrochemical Communication between Thylakoid Membranes and Gold Electrodes through Different Quinone Derivatives. ChemElectroChem 2014, 1, 131–139. [Google Scholar] [CrossRef]
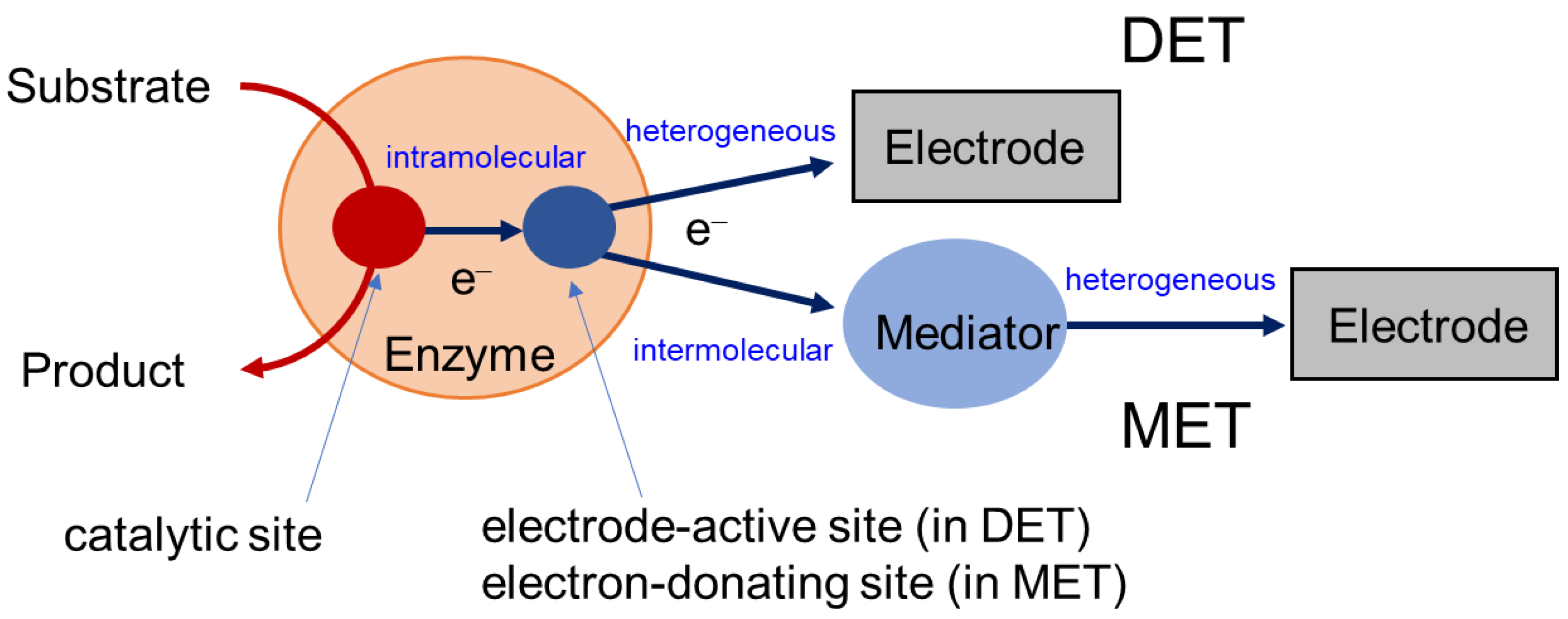
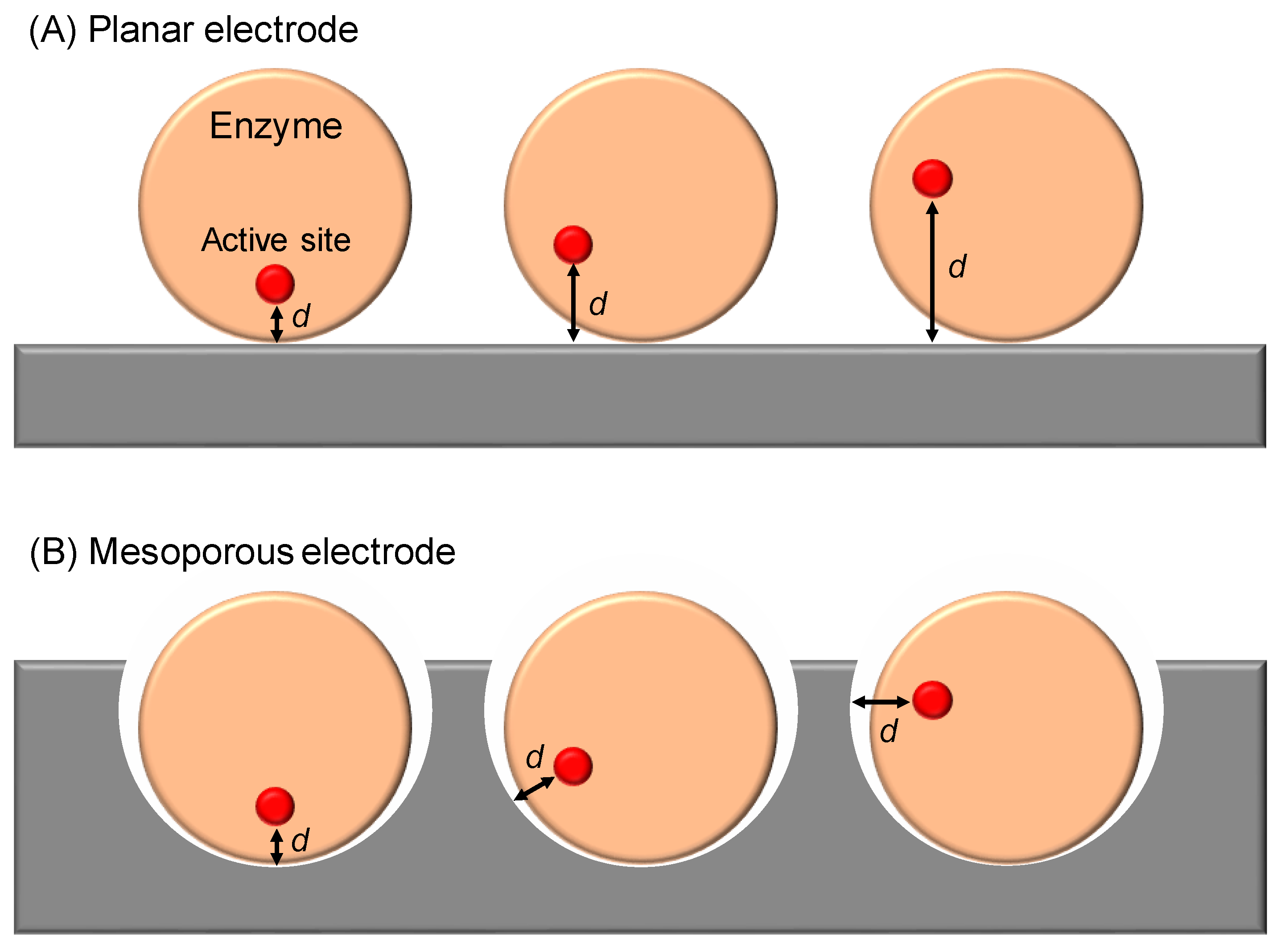
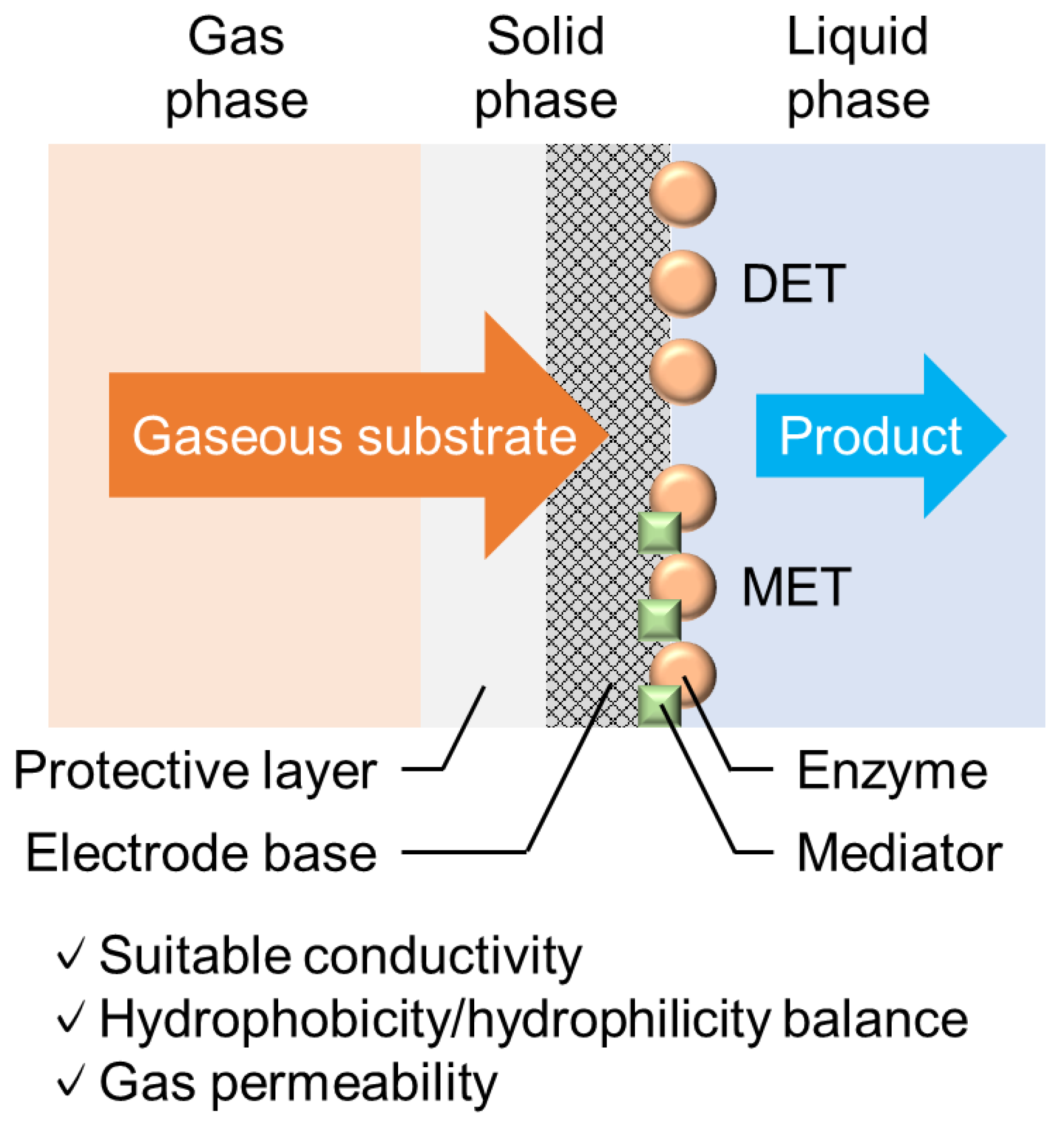
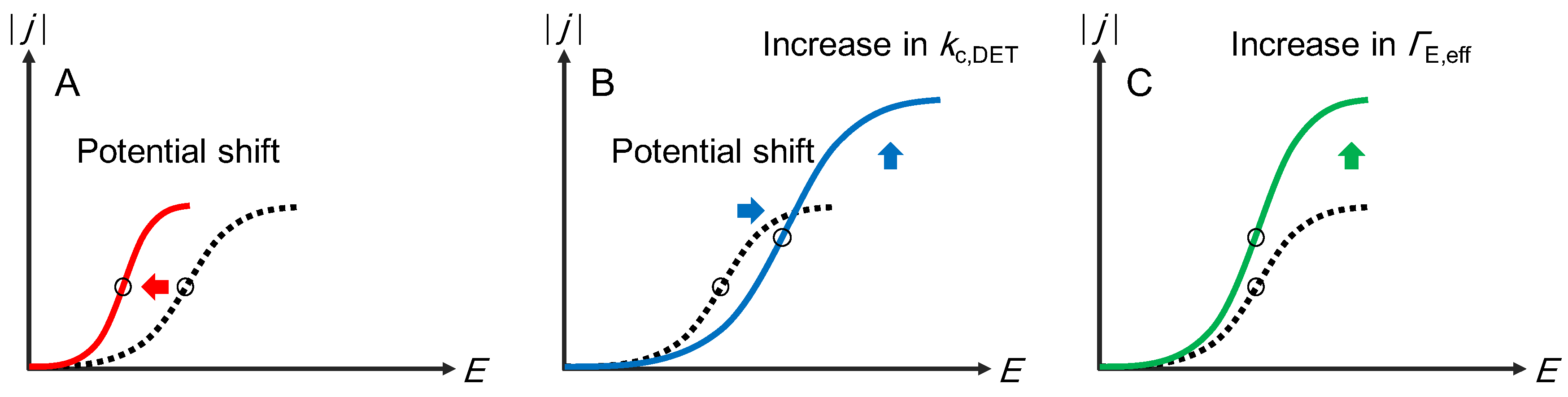
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Recent Progress in Applications of Enzymatic Bioelectrocatalysis. Catalysts 2020, 10, 1413. https://doi.org/10.3390/catal10121413
Adachi T, Kitazumi Y, Shirai O, Kano K. Recent Progress in Applications of Enzymatic Bioelectrocatalysis. Catalysts. 2020; 10(12):1413. https://doi.org/10.3390/catal10121413
Chicago/Turabian StyleAdachi, Taiki, Yuki Kitazumi, Osamu Shirai, and Kenji Kano. 2020. "Recent Progress in Applications of Enzymatic Bioelectrocatalysis" Catalysts 10, no. 12: 1413. https://doi.org/10.3390/catal10121413
APA StyleAdachi, T., Kitazumi, Y., Shirai, O., & Kano, K. (2020). Recent Progress in Applications of Enzymatic Bioelectrocatalysis. Catalysts, 10(12), 1413. https://doi.org/10.3390/catal10121413