Physical and Chemical Synthesis of Au/CeO2 Nanoparticle Catalysts for Room Temperature CO Oxidation: A Comparative Study



Abstract
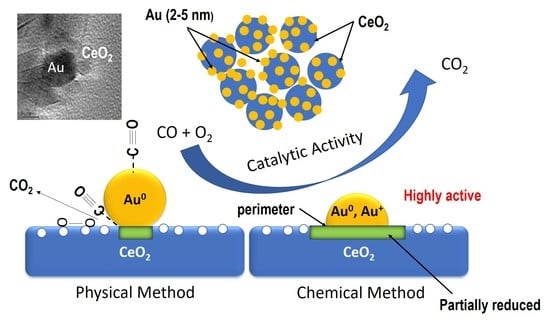
1. Introduction
2. Results and Discussion
2.1. Characterization of As-Synthesized (Fresh) Au/CeO2 Nanoparticle Catalyst
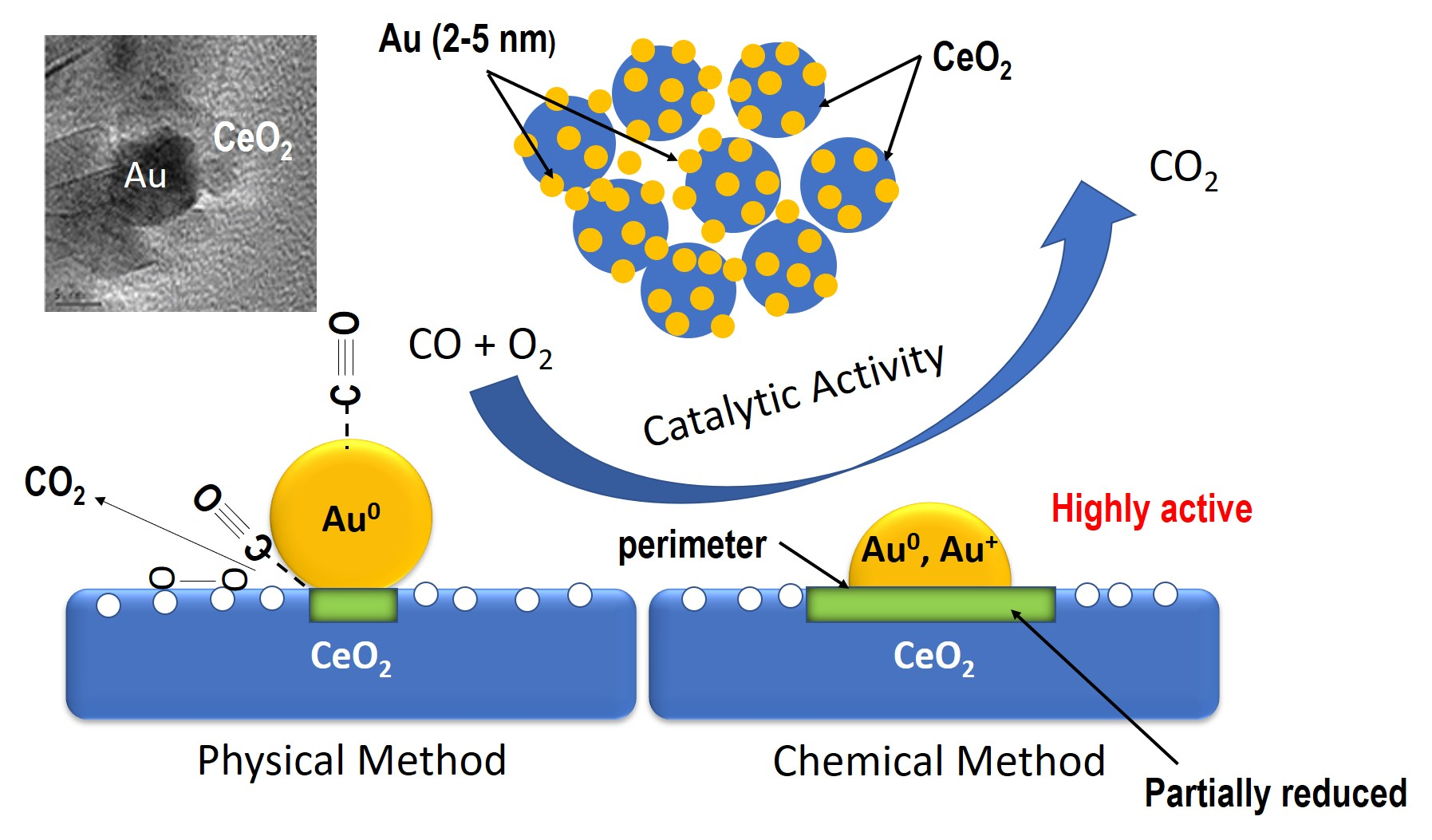
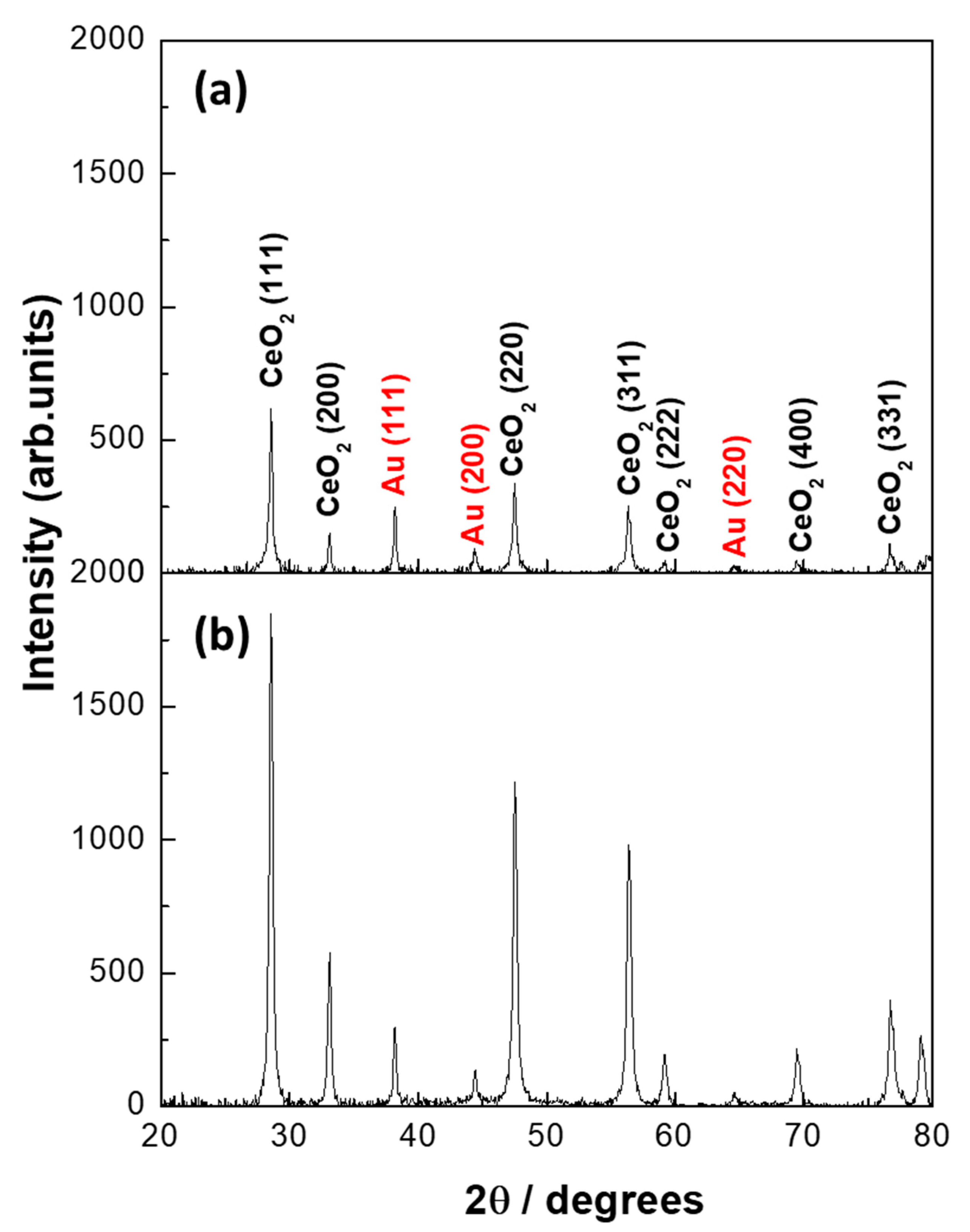
2.1.1. Inductive Coupled Plasma (ICP), BET Surface Area, and X-Ray Diffraction (XRD) of As-Synthesized Au/CeO2 (Fresh) Samples
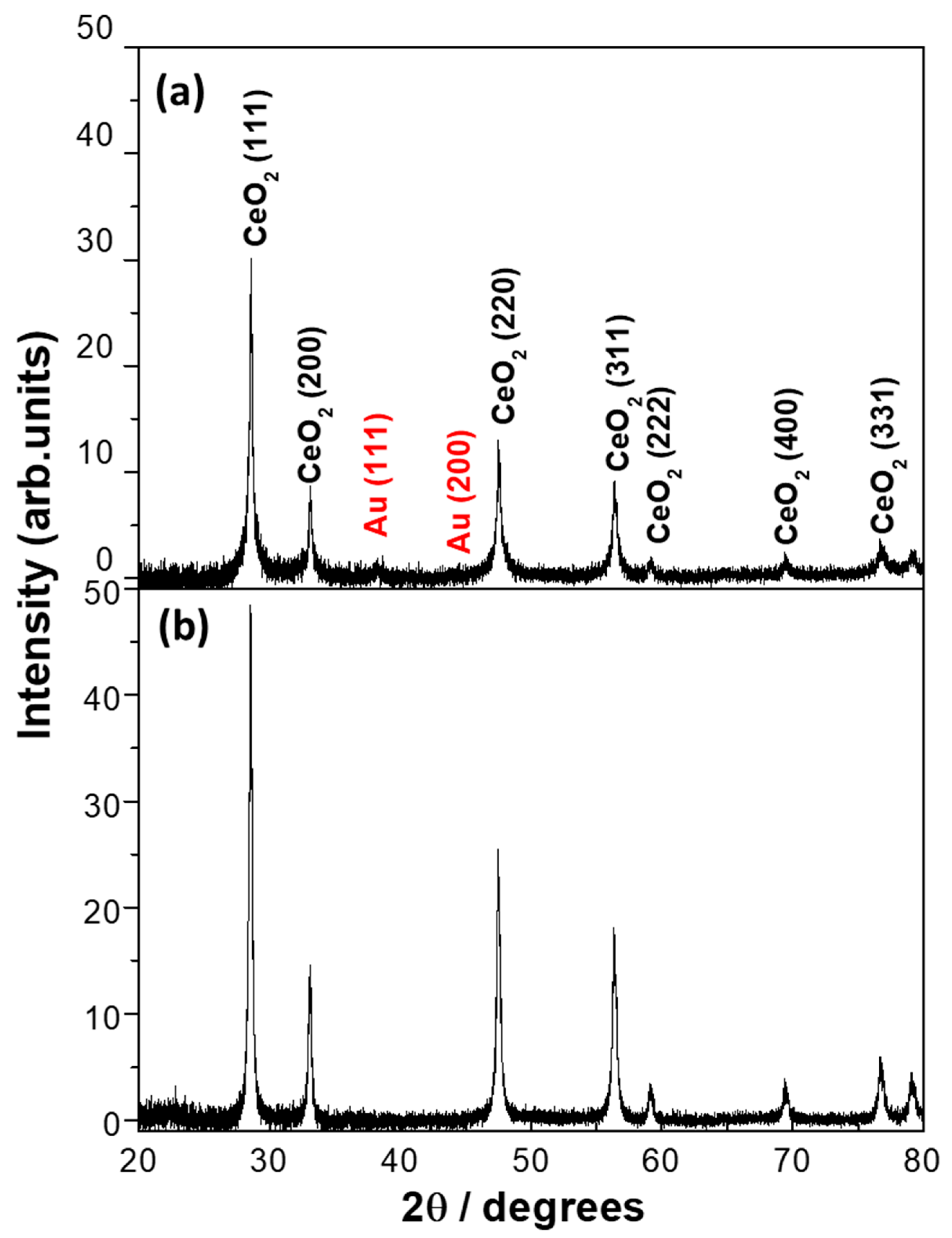
2.1.2. Scanning Electron Microscopy (SEM) of Fresh Au/CeO2
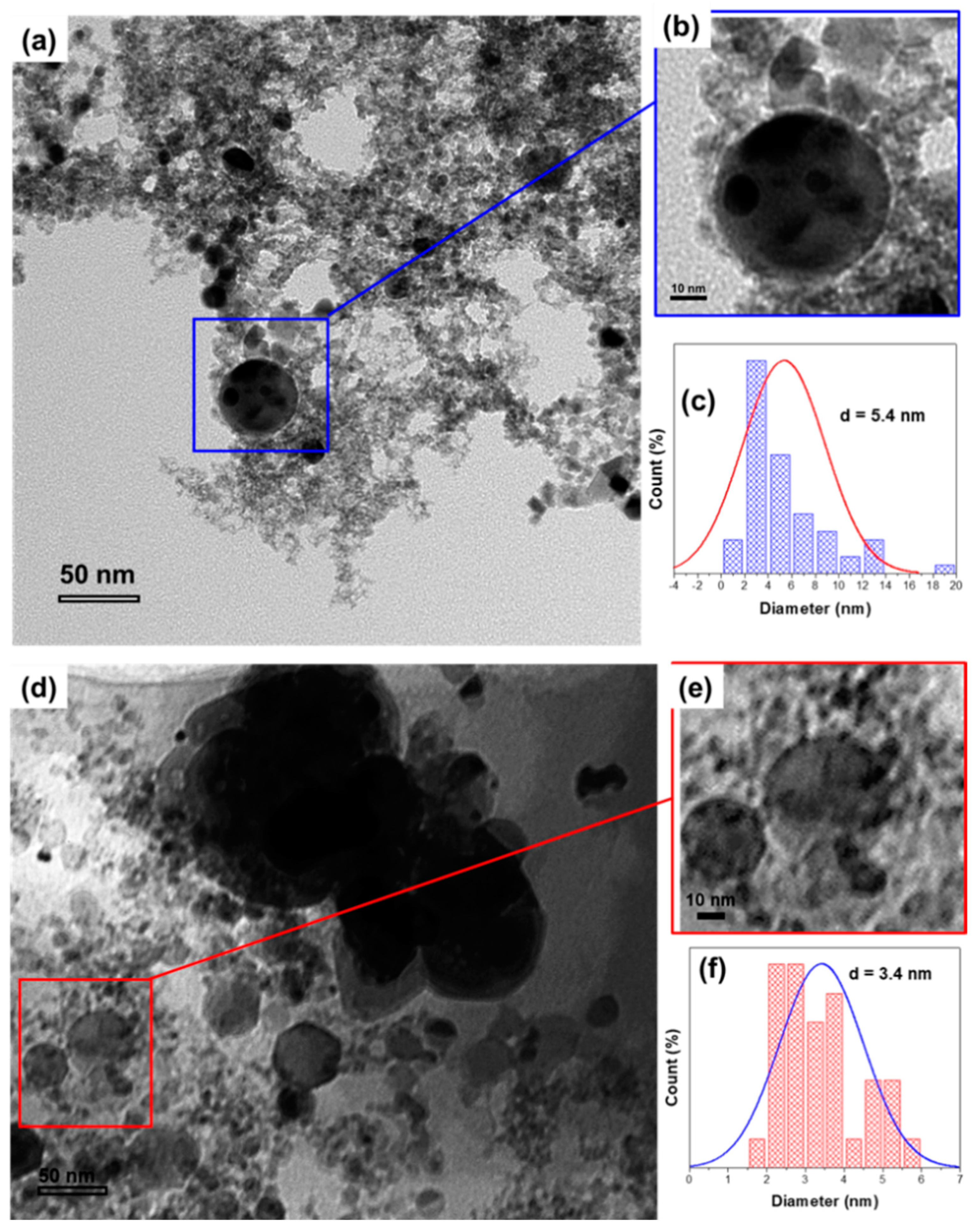
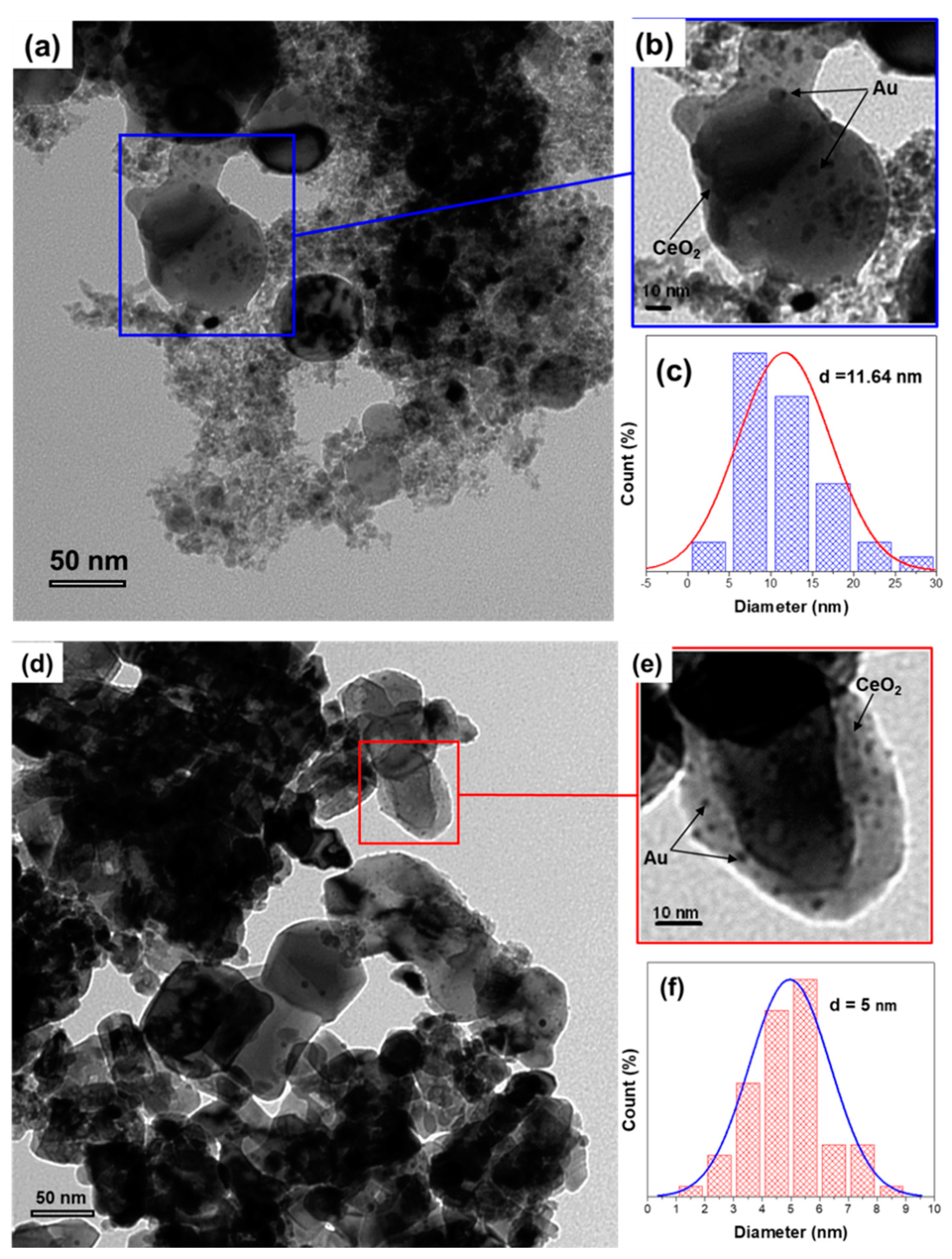
2.1.3. Transmission Electron Microscopy (TEM) of Fresh Au/CeO2
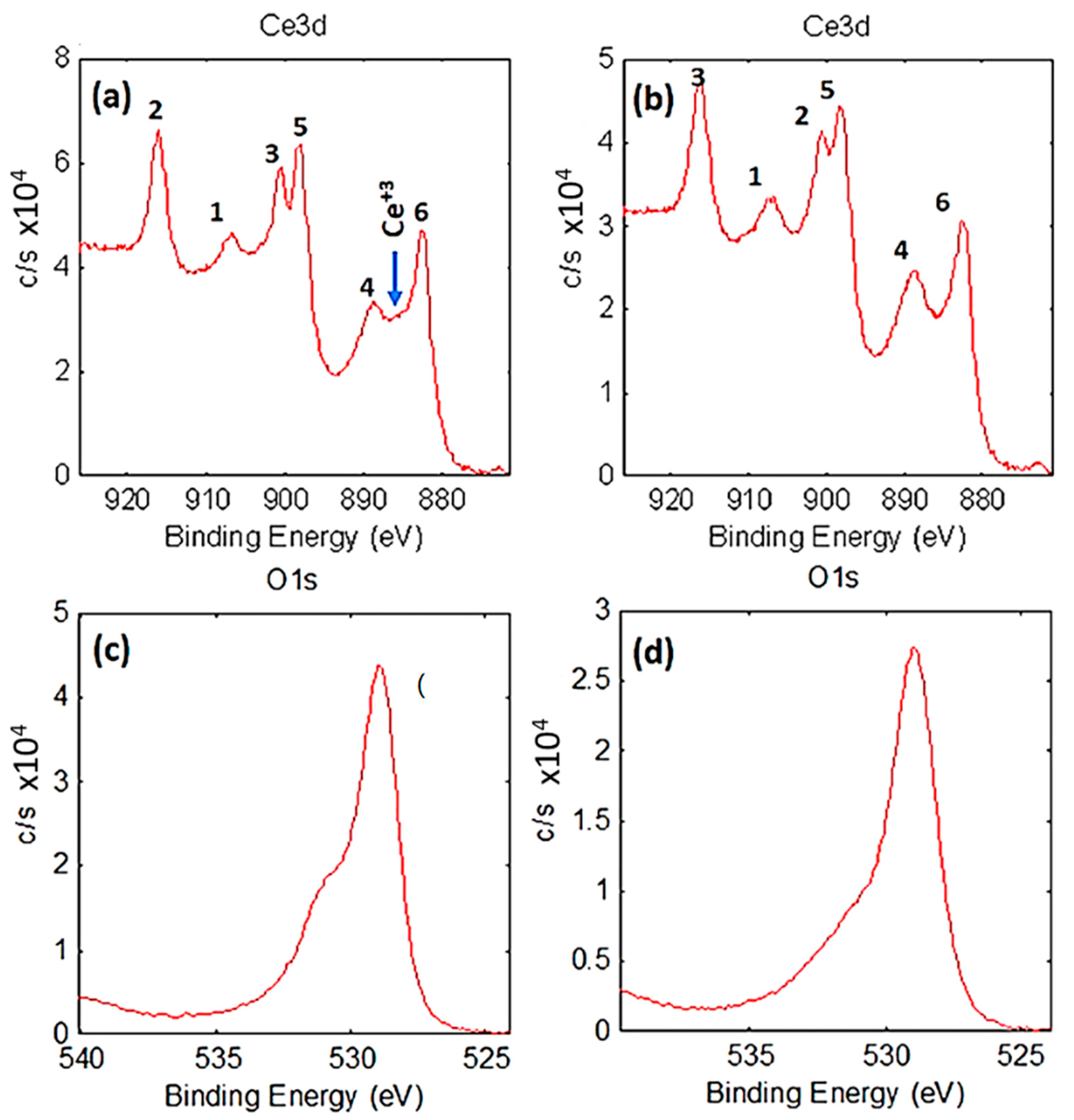
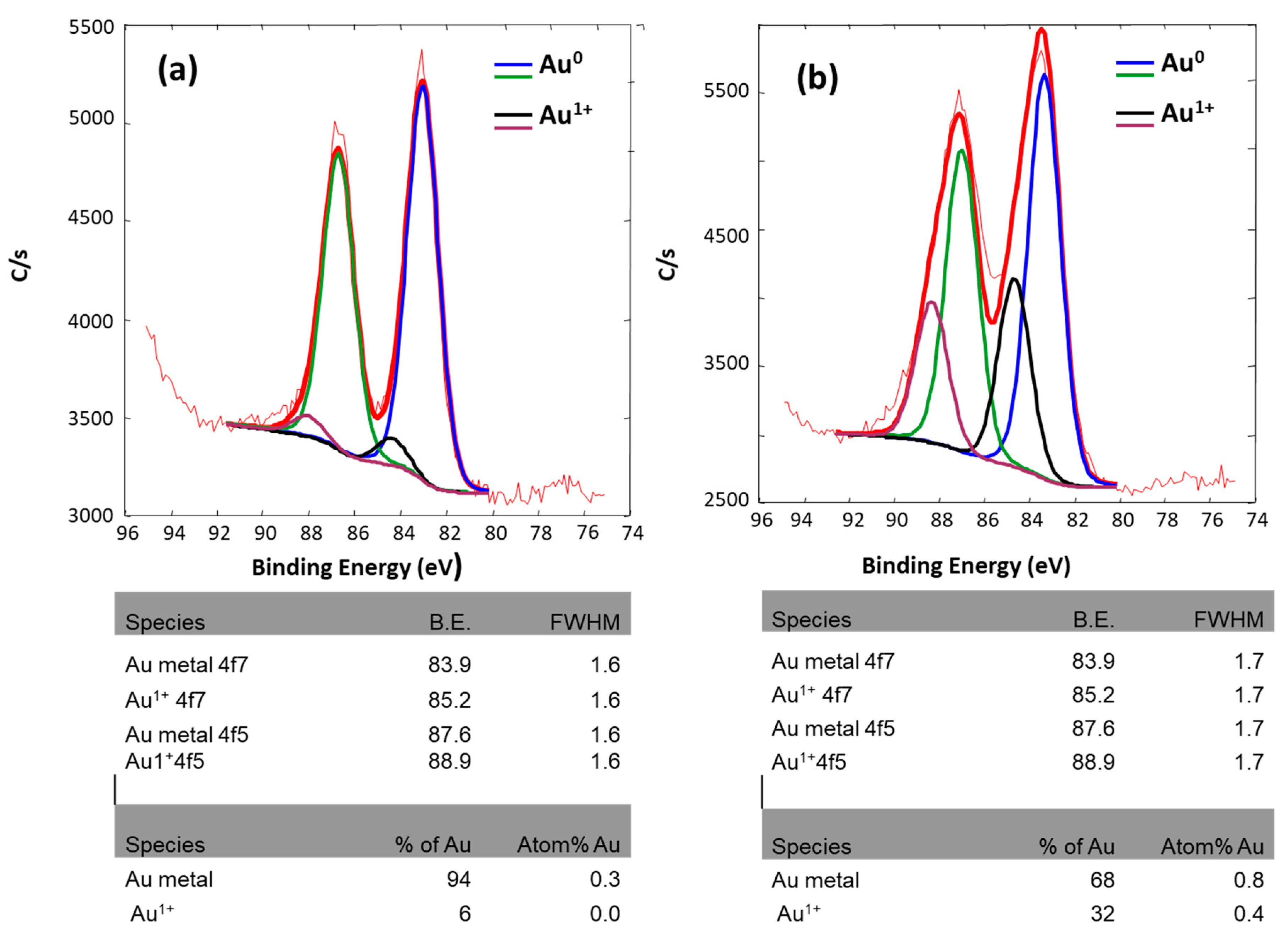
2.1.4. Chemical and Electronic States: X-Ray Photoelectron Spectroscopy (XPS) of As-Synthesized Au/CeO2
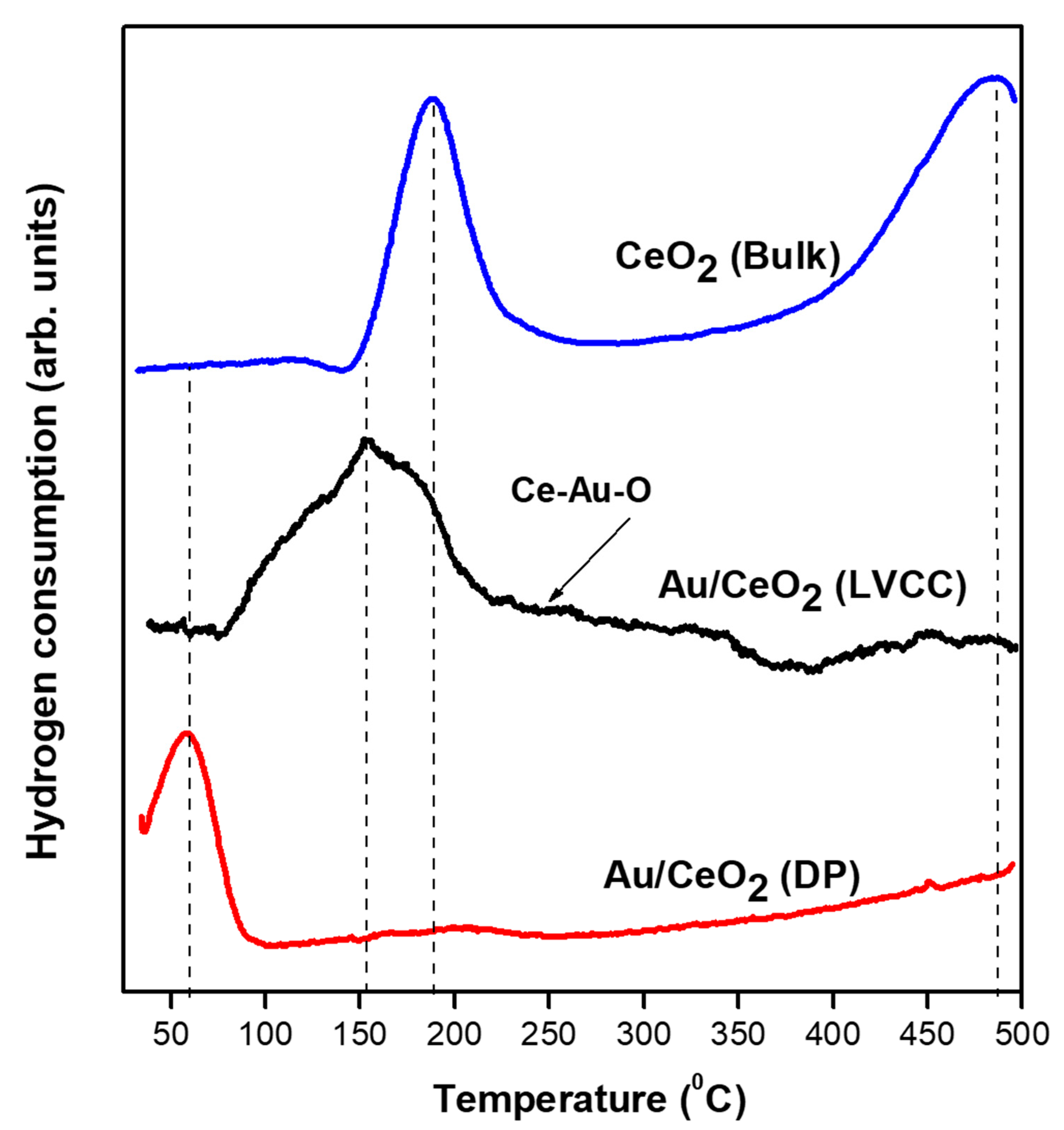
2.1.5. Temperature Programmed Reduction (H2-TPR) of Fresh Au/CeO2
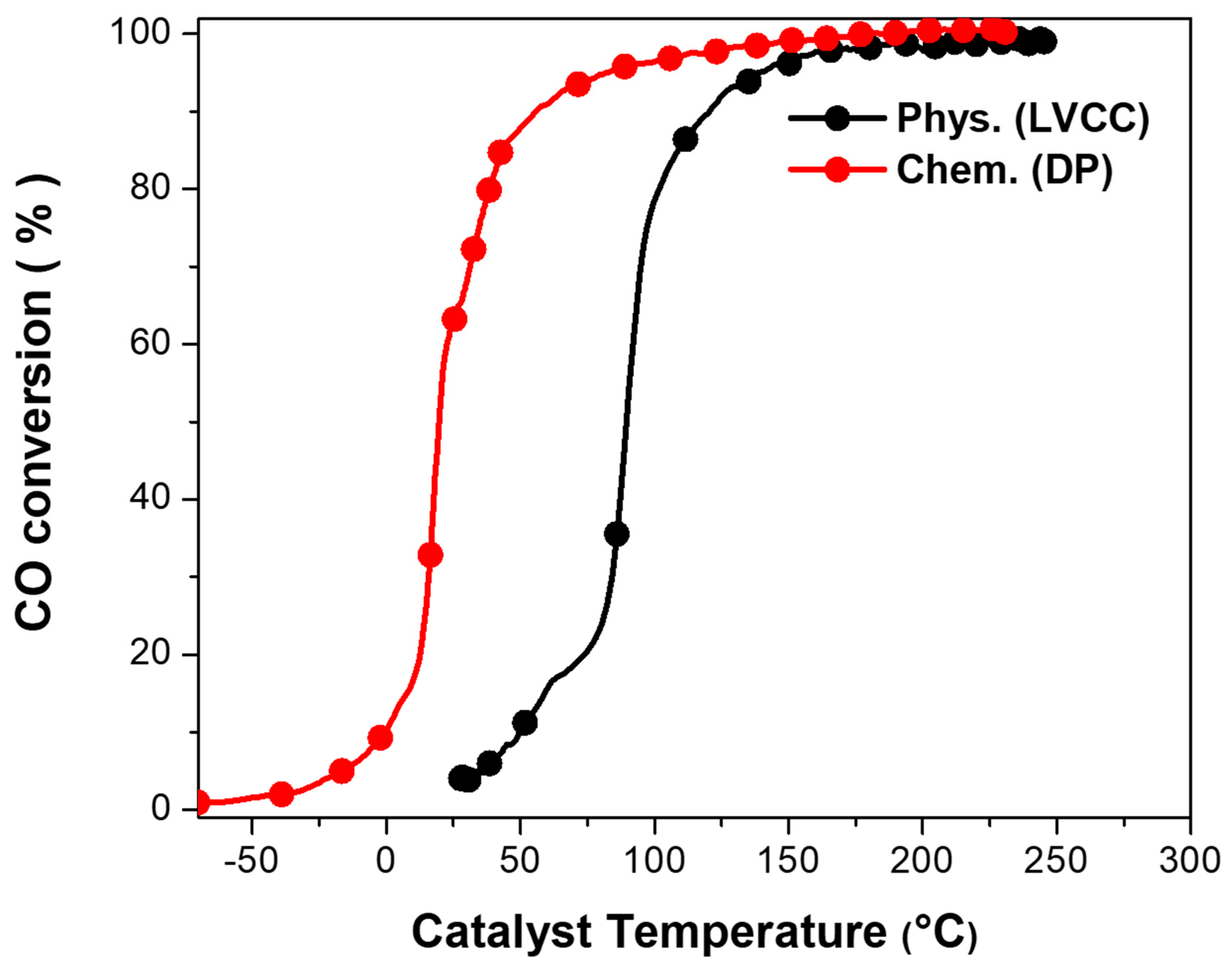
2.2. Catalytic CO Oxidation Measurement
2.3. Characterization of Au/CeO2 Nanoparticle Catalyst after Heat Treatment in CO/O2 Mixture (Second Light-Off)
3. Methods
3.1. Experimental Method
3.2. Catalyst Preparation
3.2.1. Physical Method (Laser Vaporization Controlled Condensation (LVCC))
3.2.2. Chemical Method (Deposition-Precipitation (DP))
3.3. Catalyst Characterization
3.4. Catalytic Activity Measurements
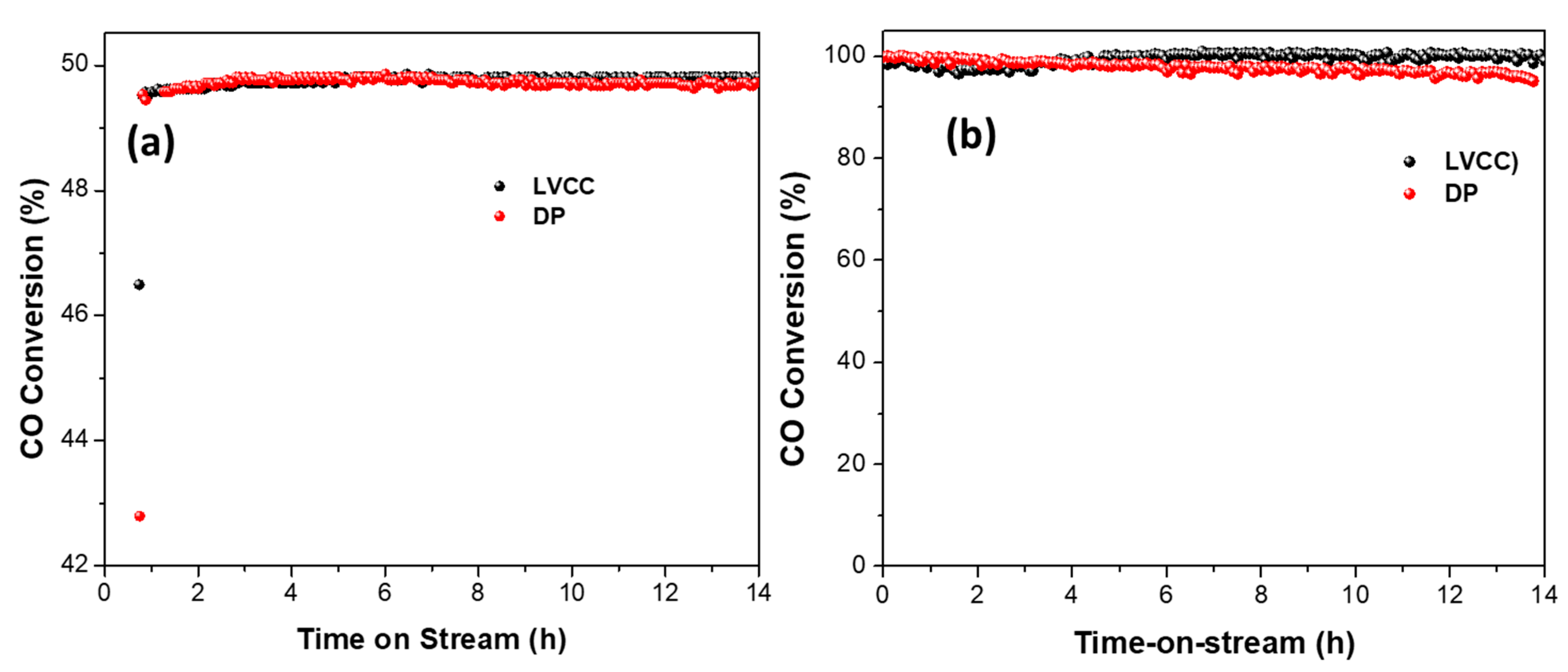
4. Long-Term Catalyst Stability of Au/CeO2 Catalysts Prepared by LVCC and DP Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- McCrea, K.R.; Parker, J.S.; Somorjai, G.A. The Role of Carbon Deposition from CO Dissociation on Platinum Crystal Surfaces during Catalytic CO Oxidation: Effects on Turnover Rate, Ignition Temperature, and Vibrational Spectra. J. Phys. Chem. B 2002, 106, 10854–10863. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Al Soubaihi, R.M.; Saoud, K.M.; Ye, F.; Zar Myint, M.T.; Saeed, S.; Dutta, J. Synthesis of hierarchically porous silica aerogel supported Palladium catalyst for low-temperature CO oxidation under ignition/extinction conditions. Microporous Mesoporous Mater. 2020, 292, 109758. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Catalysis by Gold; Catalysis Reviews; World Scientific: Singapore, 1999; Volume 41, pp. 319–388. [Google Scholar]
- Su, F.Z.; Liu, Y.M.; Wang, L.C.; Cao, Y.; He, H.Y.; Fan, K.N. Ga–Al Mixed-Oxide-Supported Gold Nanoparticles with Enhanced Activity for Aerobic Alcohol Oxidation. Angew. Chem. Int. Ed. 2008, 47, 334–337. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-Temperature Oxidation of CO over Gold Supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Abdelsayed, V.; Saoud, K.M.; El-Shall, M.S. Vapor phase synthesis and characterization of bimetallic alloy and supported nanoparticle catalysts. J. Nanoparticle Res. 2006, 8, 519–531. [Google Scholar] [CrossRef]
- Min, B.K.; Friend, C.M. Heterogeneous Gold-Based Catalysis for Green Chemistry: Low-Temperature CO Oxidation and Propene Oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef]
- Alhumaimess, M.; Aldosari, O.; Alshammari, H.; Kamel, M.M.; Betiha, M.A.; Hassan, H.M. Ionic liquid green synthesis of CeO2 nanorods and nano-cubes: Investigation of the shape dependent on catalytic performance. J. Mol. Liq. 2019, 279, 649–656. [Google Scholar] [CrossRef]
- Chen, T.; Xie, Z.; Jiang, W.; Jiang, W.; Zhang, X.; Liu, J. Synthesis of CeO2 nanosheets with a room temperature ionic liquid assisted method. J. Adv. Ceram. 2016, 5, 111–116. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.; Tian, M.; Wang, L.; Wang, Z. A Rational Design of the Sintering-Resistant Au-CeO2 Nanoparticles Catalysts for CO Oxidation: The Influence of H2 Pretreatments. Materials 2018, 11, 1952. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jang, M.G.; Shin, D.; Han, J.W. Design of Ceria Catalysts for Low-Temperature CO Oxidation. ChemCatChem 2020, 12, 11–26. [Google Scholar] [CrossRef]
- Reveles, J.U.; Saoud, K.M.; El-Shall, M.S. Water inhibits CO oxidation on gold cations in the gas phase. Structures and binding energies of the sequential addition of CO, H2O, O2, and N2 onto Au+. Phys. Chem. Chem. Phys. 2016, 18, 28606–28616. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Murayama, T.; Taketoshi, A.; Haruta, M. Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes. Chem. Rev. 2019, 120, 464–525. [Google Scholar] [CrossRef]
- Saavedra, J.; Doan, H.A.; Pursell, C.J.; Grabow, L.C.; Chandler, B.D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 1599–1602. [Google Scholar] [CrossRef] [PubMed]
- Tada, K.; Maeda, Y.; Koga, H.; Okumura, M. TiO2 Crystal Structure Dependence of Low-temperature CO Oxidation Catalyzed by Au/TiO2. Chem. Lett. 2018, 47, 200–203. [Google Scholar] [CrossRef]
- Duan, Z.; Henkelman, G. Calculations of CO Oxidation over a Au/TiO2 Catalyst: A Study of Active Sites, Catalyst Deactivation, and Moisture Effects. ACS Catal. 2018, 8, 1376–1383. [Google Scholar] [CrossRef]
- Bunluesin, T.; Cordatos, H.; Gorte, R.J. Study of CO Oxidation Kinetics on Rh/Ceria. J. Catal. 1995, 157, 222–226. [Google Scholar] [CrossRef]
- Bunluesin, T.; Putna, E.S.; Gorte, R.J. A comparison of CO oxidation on ceria-supported Pt, Pd, and Rh. Catal. Lett. 1996, 41, 1–5. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Wang, Q.; Zhou, X.; Li, J.; Song, S.; Zhang, H. CeO2 supported low-loading Au as an enhanced catalyst for low temperature oxidation of carbon monoxide. CrystEngComm 2019, 21, 7108–7113. [Google Scholar] [CrossRef]
- Al Soubaihi, R.; Saoud, K.; Dutta, J. Critical Review of Low-Temperature CO Oxidation and Hysteresis Phenomenon on Heterogeneous Catalysts. Catalysts 2018, 8, 660. [Google Scholar] [CrossRef]
- Huang, X.S.; Sun, H.; Wang, L.C.; Liu, Y.M.; Fan, K.N.; Cao, Y. Morphology effects of nanoscale ceria on the activity of Au/CeO2 catalysts for low-temperature CO oxidation. Appl. Catal. B Environ. 2009, 90, 224–232. [Google Scholar] [CrossRef]
- Si, R.; Flytzani-Stephanopoulos, M. Shape and Crystal-Plane Effects of Nanoscale Ceria on the Activity of Au-CeO2 Catalysts for the Water–Gas Shift Reaction. Angew. Chem. 2008, 120, 2926–2929. [Google Scholar] [CrossRef]
- Carrettin, S.; Concepción, P.; Corma, A.; Lopez Nieto, J.M.; Puntes, V.F. Nanocrystalline CeO2 Increases the Activity of Au for CO Oxidation by Two Orders of Magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef]
- Zhang, X.; Duan, D.; Li, G.; Feng, W.; Yang, S.; Sun, Z. Monolithic Au/CeO2 nanorod framework catalyst prepared by dealloying for low-temperature CO oxidation. Nanotechnology 2018, 29, 095606. [Google Scholar] [CrossRef]
- Luo, J.; Liu, Y.; Niu, Y.; Jiang, Q.; Huang, R.; Zhang, B.; Su, D. Insight into the chemical adsorption properties of CO molecules supported on Au or Cu and hybridized Au-CuO nanoparticles. Nanoscale 2017, 9, 15033–15043. [Google Scholar] [CrossRef]
- Palomino, R.M.; Gutiérrez, R.A.; Liu, Z.; Tenney, S.; Grinter, D.C.; Crumlin, E.; Waluyo, I.; Ramírez, P.J.; Rodriguez, J.A.; Senanayake, S.D. Inverse Catalysts for CO Oxidation: Enhanced Oxide-Metal Interactions in MgO/Au(111), CeO2/Au(111), and TiO2/Au(111). ACS Sustain. Chem. Eng. 2017, 5, 10783–10791. [Google Scholar] [CrossRef]
- Yinga, F.; Wang, S.; Au, C.-T.; Lai, S.-Y. Effect of the oxidation state of gold on the complete oxidation of isobutane on Au/CeO2 catalysts. Gold Bull. 2010, 43, 241–251. [Google Scholar] [CrossRef]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au–CeO2 catalysts for low-temperature water–gas shift. Catal. Lett. 2001, 77, 1–3. [Google Scholar] [CrossRef]
- He, Y.; Du, S.; Li, J.; Zhang, R.; Liang, X.; Chen, B. Mesoporous Ceria-Supported Gold Catalysts Self-Assembled from Monodispersed Ceria Nanoparticles and Nanocubes: A Study of the Crystal Plane Effect for the Low-Temperature Water Gas Shift Reaction. Chemcatchem 2017, 9, 4070–4082. [Google Scholar] [CrossRef]
- Yu, H.; Jiao, Y.; Li, N.; Pang, J.; Li, W.; Zhang, X.; Li, X.; Li, C. Au-CeO2 Janus-like nanoparticles fabricated by block copolymer templates and their catalytic activity in the degradation of methyl orange. Appl. Surf. Sci. 2018, 427, 771–778. [Google Scholar] [CrossRef]
- Scire, S.; Minico, S.; Crisafulli, C.; Pistone, A. Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts. Appl. Catal. B Gen. 2003, 40, 43–49. [Google Scholar] [CrossRef]
- Luengnaruemitchai, A.; Osuwan, S.; Gulari, E. Comparative studies of low-temperature water–gas shift reaction over Pt/CeO2, Au/CeO2, and Au/Fe2O3 catalysts. Catal. Commun. 2003, 4, 215–221. [Google Scholar] [CrossRef]
- Delmon, B.; Grange, P.; Jacobs, P.A.; Poncelet, G. Foreword. In Preparation of Catalysts V-Scientific Bases for the Preparation of Heterogeneous Catalysts. In Proceedings of the Fifth International Symposium; Elsevier: Amsterdam, The Netherlands, 1991; pp. xi–xii. [Google Scholar]
- Wolf, A.; Schuth, F. A systematic study of the synthesis conditions for the preparation of highly active gold catalysts. Appl. Catal. A 2002, 226, 1–13. [Google Scholar] [CrossRef]
- Li, J.; Li, W. Effect of preparation method on the catalytic activity of Au/CeO2 for VOCs oxidation. J. Rare Earths 2010, 28, 547–551. [Google Scholar] [CrossRef]
- Glaspell, G.; Abdelsayed, V.; Saoud, K.M.; El-Shall, M.S. Vapor-phase synthesis of metallic and intermetallic nanoparticles and nanowires: Magnetic and catalytic properties. Pure Appl. Chem. 2006, 78, 1667–1689. [Google Scholar] [CrossRef]
- Yang, Y.; Saoud, K.M.; Abdelsayed, V.; Glaspell, G.; Deevi, S.; El-Shall, M.S. Vapor phase synthesis of supported Pd, Au, and unsupported bimetallic nanoparticle catalysts for CO oxidation. Catal. Commun. 2006, 7, 281–284. [Google Scholar] [CrossRef]
- Herzing, A.A.; Tang, Z.R.; Edwards, J.K.; Enache, D.I.; Bartley, J.K.; Taylor, S.H.; Carley, A.F.; Kiely, C.J.; Hutchings, G.J. Characterization of Au-based Catalysts Using Novel Cerium Oxide Supports. Microsc. Microanal. 2007, 13, 102–103. [Google Scholar] [CrossRef][Green Version]
- Venezia, A.M.; Pantaleo, G.; Longo, A.; Di Carlo, G.; Casaletto, M.P.; Liotta, F.L.; Deganello, G. Relationship between Structure and CO Oxidation Activity of Ceria-Supported Gold Catalysts. J. Phys. Chem. B 2005, 109, 2821–2827. [Google Scholar] [CrossRef]
- Bera, P.; Hegd, M.S. Characterization and catalytic properties of combustion synthesized Au/CeO2 catalyst. Catal. Lett. 2002, 79, 75. [Google Scholar] [CrossRef]
- Liu, W.; Flytzani-Stephanopoulos, M.; Sarofim, A.F. Selective Catalytic Reduction of Sulfur Dioxide to Elemental Sulfur; Final Report; Office of Scientific and Technical Information (OSTI): Oak Ridge, TN, USA, 1995.
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Yuan, Y.Z.; Asukura Wan, H.L.; Tsai, K.; Iwasawa, Y. Preparation of supported gold catalysts from gold complexes and their catalytic activities for CO oxidation. Catal. Lett. 1996, 42, 15–20. [Google Scholar] [CrossRef]
- Mafune, F.; Kohno, J.; Takeda, Y.; Kondow, T. Formation of gold nanonetworks and small gold nanoparticles by irradiation of intense pulsed laser onto gold nanoparticles. J. Phys. Chem. B 2003, 107, 12589. [Google Scholar] [CrossRef]
- Sims, C.M.; Maier, R.A.; Johnston-Peck, A.C.; Gorham, J.M.; Hackley, V.A.; Nelson, B.C. Approaches for the quantitative analysis of oxidation state in cerium oxide nanomaterials. Nanotechnology 2018, 30, 085703. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, A.E.C.; Wirde, M.; Gelius, U.; Muhammed, M. Surfaces of doped nanophase cerium oxide catalysts. Nanostruct. Mater. 1999, 11, 995–1007. [Google Scholar] [CrossRef]
- Bêche, E.; Charvin, P.; Perarnau, D.; Abanades, S.; Flamant, G. Ce 3d XPS investigation of cerium oxides and mixed cerium oxide (CexTiyOz). Surf. Interface Anal. 2008, 40, 264–267. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Andreeva, D.; Tabakova, T. FTIR study of the low-temperature water–gas shift reaction on Au/Fe2O3 and Au/TiO2 catalysts. J. Catal. 1999, 188, 176–185. [Google Scholar] [CrossRef]
- Swain, G.; Sultana, S.; Naik, B.; Parida, K. Coupling of Crumpled-Type Novel MoS(2) with CeO(2) Nanoparticles: A Noble-Metal-Free p-n Heterojunction Composite for Visible Light Photocatalytic H(2) Production. ACS Omega 2017, 2, 3745–3753. [Google Scholar] [CrossRef]
- Fu, Q.; Kudriavtseva, S.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Gold–ceria catalysts for low-temperature water-gas shift reaction. Chem. Eng. J. 2003, 93, 41–53. [Google Scholar] [CrossRef]
- Wagner, F.E.; Galvagno, S.; Milone, C.; Visco, A.M.; Stievano, L.; Calogero, S. Mössbauer characterisation of gold/iron oxide catalysts. J. Chem. Soc. Faraday Trans. 1997, 93, 3404–3409. [Google Scholar] [CrossRef]
- Khoudiakov, M.; Gupta, M.C.; Deevi, S. Au/Fe2O3 nanocatalysts for CO oxidation by a deposition-prcipitation technique. Nanotechnology 2004, 15, 987–990. [Google Scholar] [CrossRef]
- Vernieres, J.; Steinhauer, S.; Zhao, J.; Grammatikopoulos, P.; Ferrando, R.; Nordlund, K.; Djurabekova, F.; Sowwan, M. Site-Specific Wetting of Iron Nanocubes by Gold Atoms in Gas-Phase Synthesis. Adv. Sci. 2019, 6, 1900447. [Google Scholar] [CrossRef] [PubMed]
- Kruis, F.E.; Fissan, H.; Peled, A. Synthesis of nanoparticles in the gas phase for electronic, optical, and magnetic applications—A review. J. Aerosol Sci. 1998, 29, 511–535. [Google Scholar] [CrossRef]
- Pithawalla, Y.B.; El-Shall, M.S.; Deevi, S.C.; Ström, V.; Rao, K.V. Synthesis of Magnetic Intermetallic FeAl Nanoparticles from a Non-Magnetic Bulk Alloy. J. Phys. Chem. B 2001, 105, 2085–2090. [Google Scholar] [CrossRef]
- El-Shall, M.S.; Abdelsayed, V.; Pithawalla, Y.B.; Alsharaeh, E.; Deevi, S.C. Vapor Phase Growth and Assembly of Metallic, Intermetallic, Carbon, and Silicon Nanoparticle Filaments. J. Phys. Chem. B 2003, 107, 2882–2886. [Google Scholar] [CrossRef]
- Li, S.; Germanenko, I.N.; El-Shall, M.S. Nanoparticles from the vapor phase: Synthesis and characterization of Si, Ge, MoO3, and WO3 nanocrystals. J. Clust. Sci. 1999, 10, 533–547. [Google Scholar] [CrossRef]
- Iizuka, Y.; Tode, T.; Takao, T.; Yatsu, K.; Takeuchi, T.; Tsubota, S.; Haruta, M. A kinetic and adsorption study of CO oxidation over unsupported fine gold powder and over gold supported on titanium dioxide. J. Catal. 1999, 187, 50. [Google Scholar] [CrossRef]
- Liang, R.; Hu, A.; Persic, J.; Zhou, Y.N. Palladium Nanoparticles Loaded on Carbon Modified TiO2 Nanobelts for Enhanced Methanol Electrooxidation. Nano Micro Lett. 2013, 5, 202–212. [Google Scholar] [CrossRef]
- Akita, T.; Tanaka, K.; Tsubota, S.; Haruta, M. Analytical High-Resolution TEM Study on Au/TiO2 Catalysts. MRS Proc. 2011, 589, 253. [Google Scholar] [CrossRef]
- Tada, K.; Koga, H.; Hayashi, A.; Kondo, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M. Theoretical Clarification of the Coexistence of Cl Effects on Au/TiO2: The Interaction between Au Clusters and the TiO2 Surface, and the Aggregation of Au Clusters on the TiO2 Surface. Bull. Chem. Soc. Jpn. 2017, 90, 506–519. [Google Scholar] [CrossRef]
- Camellone, M.F.; Fabris, S. Reaction Mechanisms for the CO Oxidation on Au/CeO2 Catalysts: Activity of Substitutional Au3+/Au+ Cations and Deactivation of Supported Au+ Adatoms. J. Am. Chem. Soc. 2009, 131, 10473–10483. [Google Scholar] [CrossRef] [PubMed]
- Tana; Wang, F.; Li, H.; Shen, W. Influence of Au particle size on Au/CeO2 catalysts for CO oxidation. Catal. Today 2011, 175, 541–545. [Google Scholar] [CrossRef]
- Kim, H.Y.; Henkelman, G. CO Oxidation at the Interface between Doped CeO2 and Supported Au Nanoclusters. J. Phys. Chem. Lett. 2012, 3, 2194–2199. [Google Scholar] [CrossRef]
- Zhou, Z.; Kooi, S. The Role of the Interface in CO Oxidation on Au/CeO2 Multilayer Nanotowers. Adv. Funct. Mater. 2008, 18, 2801–2807. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAMPLE | Atom% Au | Atom% Au0 | Atom% Au1+ |
|---|---|---|---|
| Au/CeO2 (LVCC) | 0.3 | 0.282 | 0.018 |
| Au/CeO2 (DP) | 1.2 | 0.8 | 0.4 |
| Sample 5% Au/CeO2 | 3% Conversion (Light-Off) Temp. (°C) | 50% Conversion Temp. (°C) | Maximum Conversion (%) | |
|---|---|---|---|---|
| Temp. (°C) | Conversion | |||
| Physical (LVCC) | 50.2 | 118.2 | 233.2 | 99.5 |
| Chemical (DP) | 0.1 | 14.7 | 110.0 | 100.0 |
| Sample 5% Au/CeO2 | 3% Conversion (Light-Off) Temp. (°C) | 50% Conversion Temp. (°C) | Maximum Conversion (%) | |
|---|---|---|---|---|
| Temp. (°C) | Conversion | |||
| Physical (LVCC) | 27.5 | 89.95 | 210.9 | 99.4 |
| Chemical (DP) | −28.3 | 19.2 | 178.3 | 100.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saoud, K.M.; El-Shall, M.S. Physical and Chemical Synthesis of Au/CeO2 Nanoparticle Catalysts for Room Temperature CO Oxidation: A Comparative Study. Catalysts 2020, 10, 1351. https://doi.org/10.3390/catal10111351
Saoud KM, El-Shall MS. Physical and Chemical Synthesis of Au/CeO2 Nanoparticle Catalysts for Room Temperature CO Oxidation: A Comparative Study. Catalysts. 2020; 10(11):1351. https://doi.org/10.3390/catal10111351
Chicago/Turabian StyleSaoud, Khaled Mohammad, and Mohamed Samy El-Shall. 2020. "Physical and Chemical Synthesis of Au/CeO2 Nanoparticle Catalysts for Room Temperature CO Oxidation: A Comparative Study" Catalysts 10, no. 11: 1351. https://doi.org/10.3390/catal10111351
APA StyleSaoud, K. M., & El-Shall, M. S. (2020). Physical and Chemical Synthesis of Au/CeO2 Nanoparticle Catalysts for Room Temperature CO Oxidation: A Comparative Study. Catalysts, 10(11), 1351. https://doi.org/10.3390/catal10111351