Influence of Topology and Brønsted Acid Site Presence on Methanol Diffusion in Zeolites Beta and MFI


Abstract
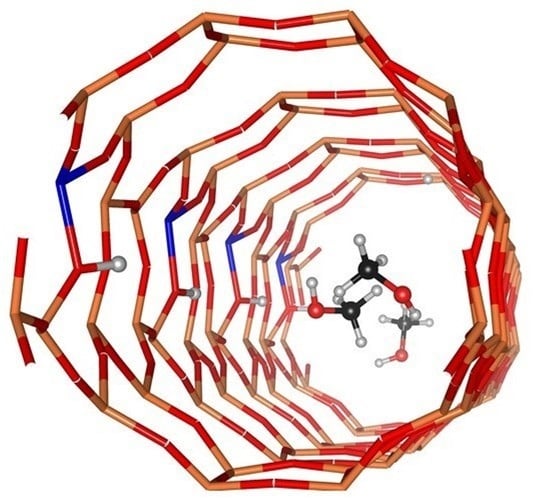
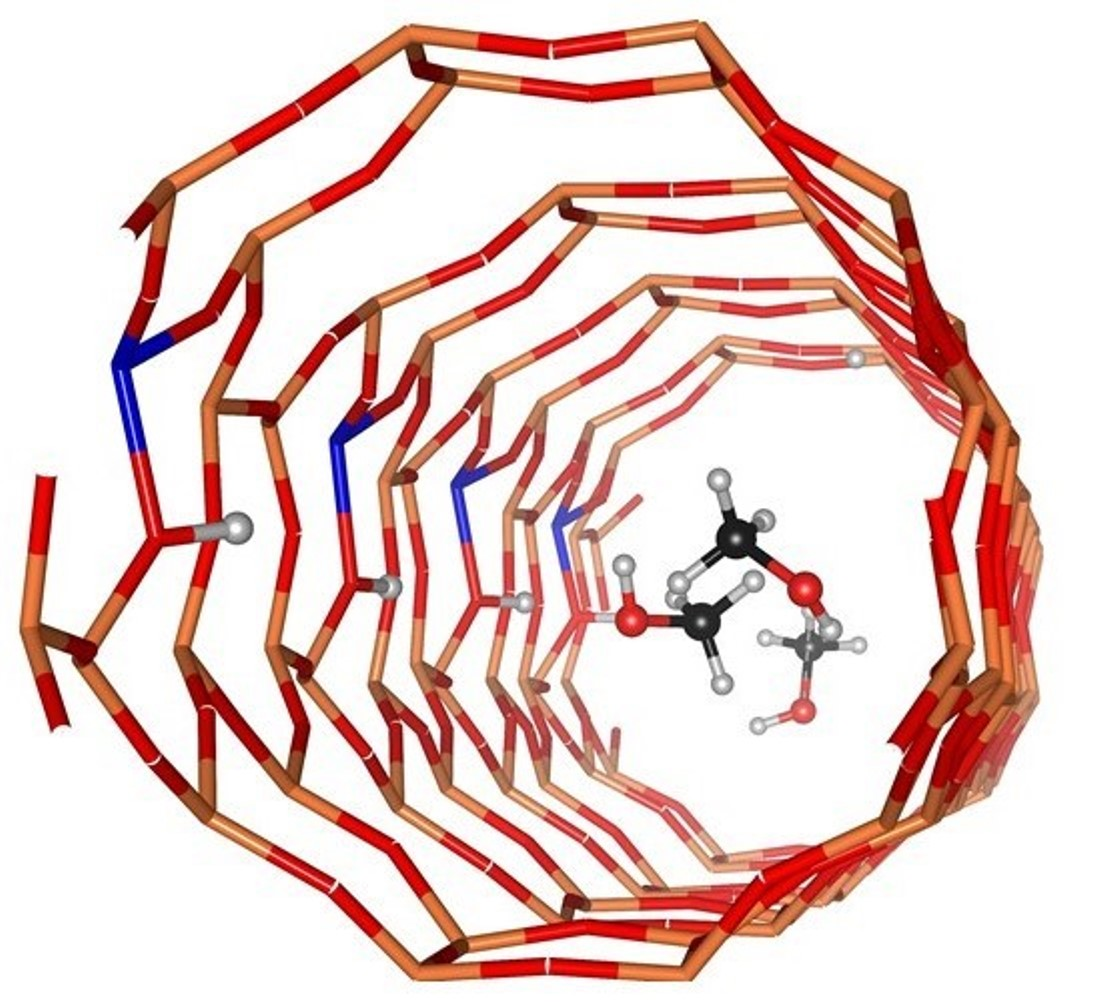
1. Introduction
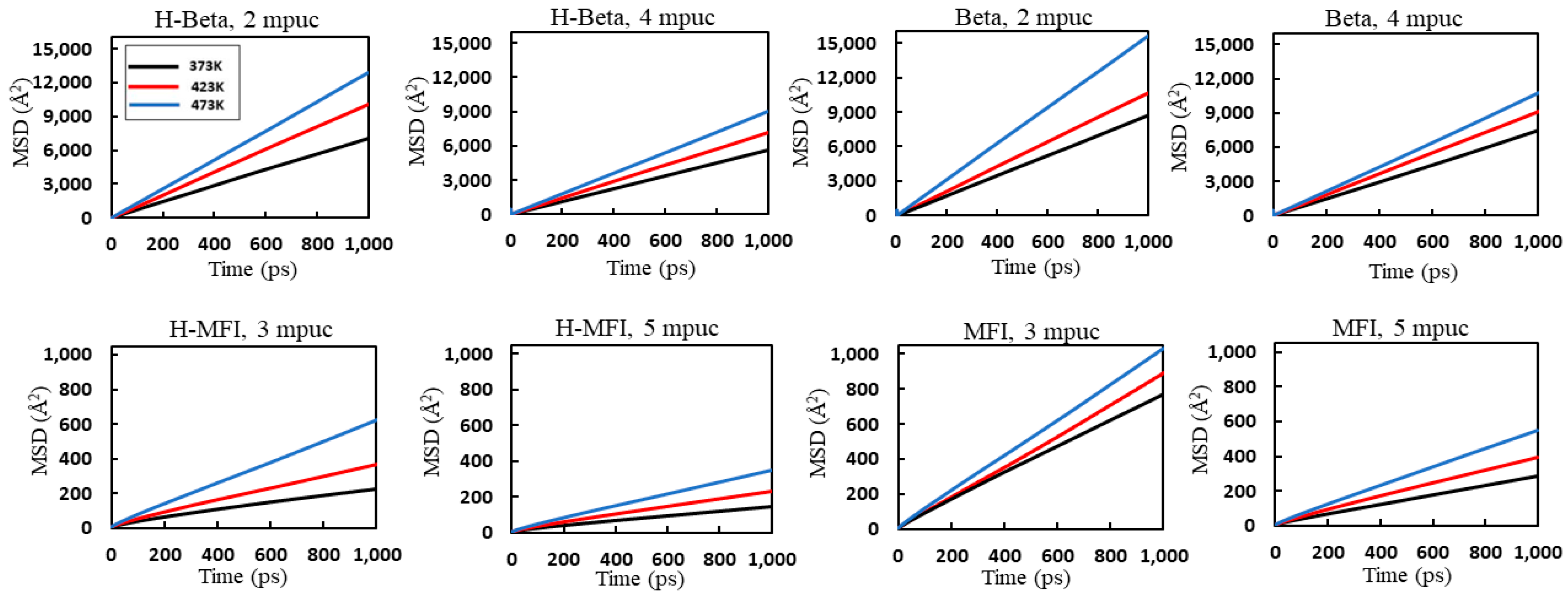
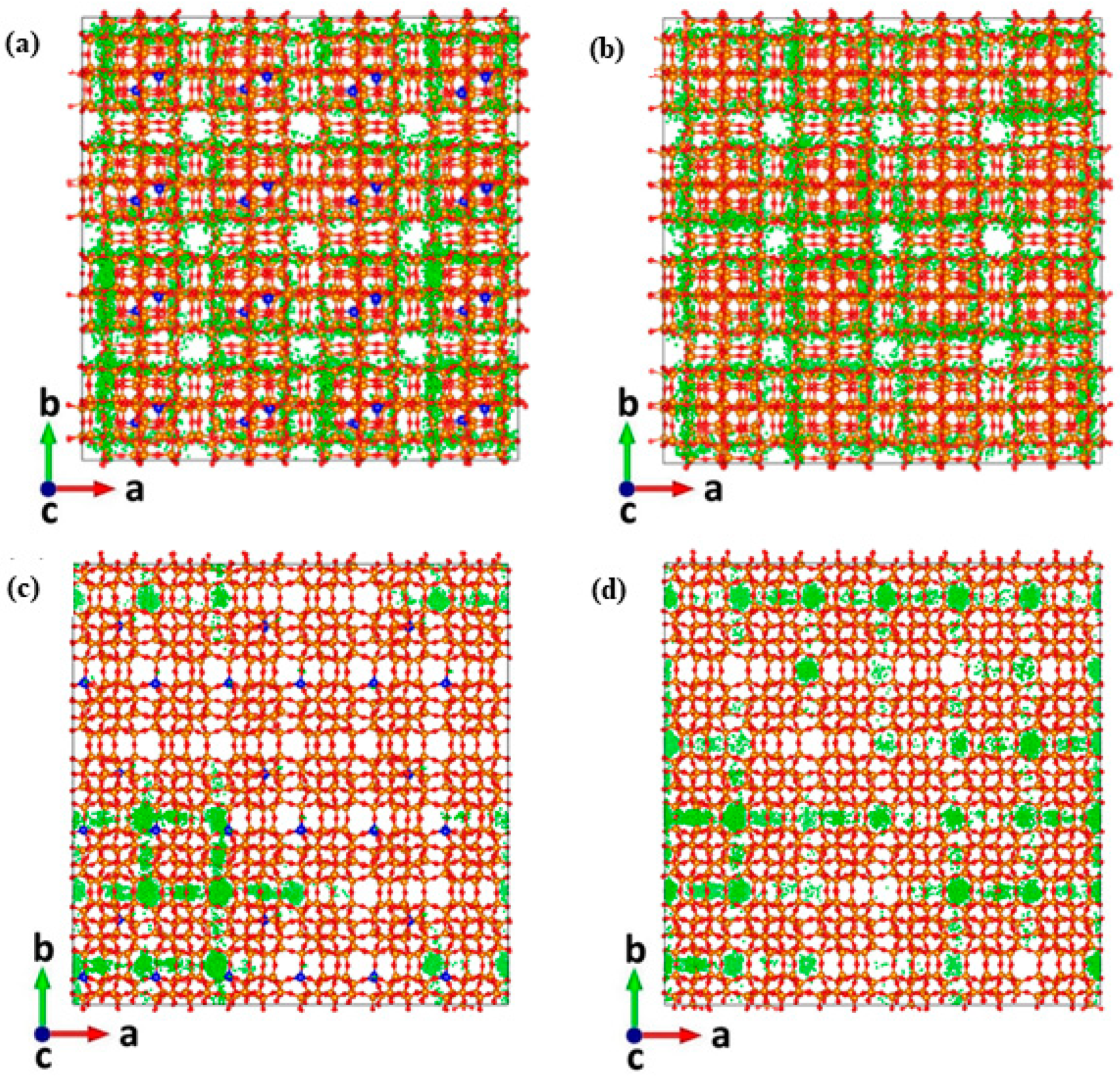
2. Results and Discussion
3. Method
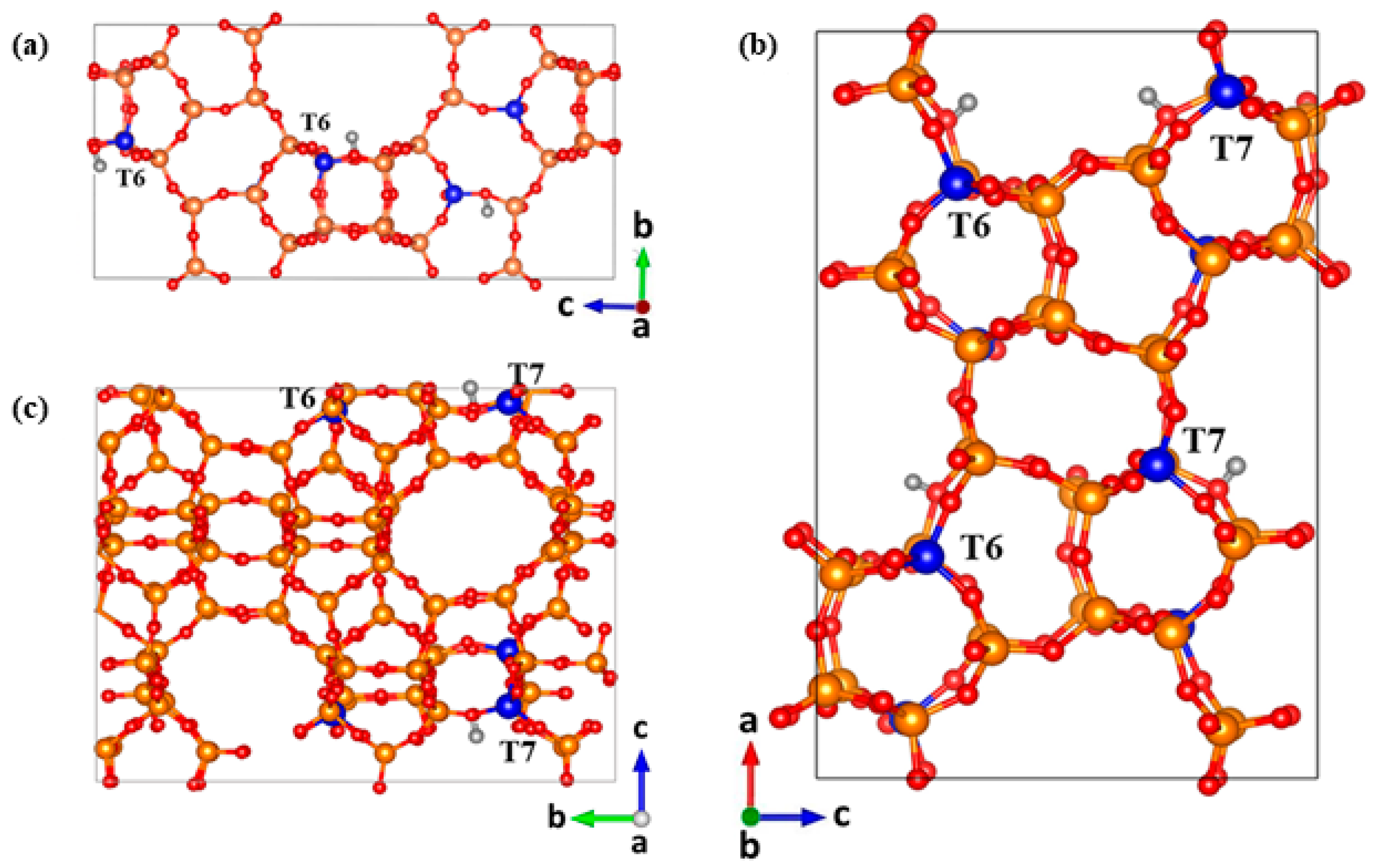
3.1. Models
3.2. Interatomic Potentials
3.3. Simulations
4. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arcoumanis, C.; Bae, C.; Crookes, R.; Kinoshita, E. The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel 2007, 87, 1014–1030. [Google Scholar] [CrossRef]
- Arcoya, A.; González, J.A.; Travieso, N.; Seoane, X.L. Physicochemical and Catalytic Properties of a Modified Natural Clinoptilolite. Clay Miner. 1994, 29, 123–131. [Google Scholar] [CrossRef]
- Yu, Y.; Li, X.; Su, L.; Zhang, Y.; Wang, Y.; Zhang, H. The role of shape selectivity in catalytic fast pyrolysis of lignin with zeolite catalysts. Appl. Catal. A Gen. 2012, 447–448, 115–123. [Google Scholar] [CrossRef]
- Corma, A.; Rey, F.; Valencia, S.; Jordá, J.L.; Rius, J. A zeolite with interconnected 8-, 10- and 12-ring pores and its unique catalytic selectivity. Nat. Mater. 2003, 2, 493–497. [Google Scholar] [CrossRef]
- Teketel, S.; Skistad, W.; Benard, S.; Olsbye, U.; Lillerud, K.P.; Beato, P.; Svelle, S. Shape selectivity in the conversion of methanol to hydrocarbons: The catalytic performance of one-dimensional 10-ring zeolites: ZSM-22, ZSM-23, ZSM-48, and EU-1. ACS Catal. 2011, 2, 26–37. [Google Scholar] [CrossRef]
- Müller, S.; Liu, Y.; Kirchberger, F.M.; Tonigold, M.; Sanchez-Sanchez, M.; Lercher, J.A. Hydrogen Transfer Pathways during Zeolite Catalyzed Methanol Conversion to Hydrocarbons. J. Am. Chem. Soc. 2016, 138, 15994–16003. [Google Scholar] [CrossRef]
- Teketel, S.; Olsbye, U.; Lillerud, K.P.; Beato, P.; Svelle, S. Selectivity control through fundamental mechanistic insight in the conversion of methanol to hydrocarbons over zeolites. Microporous Mesoporous Mater. 2010, 136, 33–41. [Google Scholar] [CrossRef]
- Dessau, R.M.; LaPierre, R.B. On the mechanism of methanol conversion to hydrocarbons over HZSM-5. J. Catal. 1982, 78, 136–141. [Google Scholar] [CrossRef]
- Chang, C.D.; Lang, W.H.; Smith, R.L. The Conversion of methanol and other O-compounds to hydrocarbons over zeolite catalysts. J. Catal. 1977, 47, 249–259. [Google Scholar] [CrossRef]
- Ono, Y.; Mori, T. Mechanism of methanol conversion into hydrocarbons over ZSM-5 zeolite. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1981, 77, 2209–2221. [Google Scholar] [CrossRef]
- Olsbye, U.; Svelle, S.; Lillerud, K.P.; Wei, Z.H.; Chen, Y.Y.; Li, J.F.; Wang, J.G.; Fan, W.B. The formation and degradation of active species during methanol conversion over protonated zeotype catalysts. Chem. Soc. Rev. 2015, 44, 7155–7176. [Google Scholar] [CrossRef] [PubMed]
- Teketel, S.; Erichsen, M.W.; Bleken, F.L.; Svelle, S.; Lillerud, K.P.; Olsbye, U. Shape selectivity in zeolite catalysis. The Methanol to Hydrocarbons (MTH) reaction. Catalysis 2014, 26, 179–217. [Google Scholar]
- Tang, Q.; Xu, H.; Zheng, Y.; Wang, J.; Li, H.; Zhang, J. Catalytic dehydration of methanol to dimethyl ether over micro-mesoporous ZSM-5/MCM-41 composite molecular sieves. Appl. Catal. A Gen. 2012, 413–414, 36–42. [Google Scholar] [CrossRef]
- Yang, J.Z.; Chen, Y.; Zhu, A.M.; Liu, Q.L.; Wu, J.Y. Analyzing diffusion behaviors of methanol/water through MFI membranes by molecular simulation. J. Membr. Sci. 2008, 318, 327–333. [Google Scholar] [CrossRef]
- Haw, J.F.; Song, W.; Marcus, D.M.; Nicholas, J.B. The mechanism of methanol to hydrocarbon catalysis. Acc. Chem. Res. 2003, 36, 317–326. [Google Scholar] [CrossRef]
- Yarulina, I.; Chowdhury, A.D.; Meirer, F.; Weckhuysen, B.M.; Gascon, J. Recent trends and fundamental insights in the methanol-to-hydrocarbons process. Nat. Catal. 2018, 1, 398–411. [Google Scholar] [CrossRef]
- Gogate, M.R. New insights into reaction mechanisms of the methanol-to-hydrocarbons (MTH) reactions: The formation of first C–C bond. Pet. Sci. Technol. 2019, 37, 28–37. [Google Scholar] [CrossRef]
- Omojola, T.; Silverwood, I.P.; O’Malley, A.J. Molecular behaviour of methanol and dimethyl ether in H-ZSM-5 catalysts as a function of Si/Al ratio: A quasielastic neutron scattering study. Catal. Sci. Technol. 2020, 10, 4305–4320. [Google Scholar] [CrossRef]
- Haase, F.; Sauer, J. Ab initio molecular dynamics simulation of methanol interacting with acidic zeolites of different framework structure. Microporous Mesoporous Mater. 2000, 35–36, 379–385. [Google Scholar] [CrossRef]
- O’Malley, A.J.; Parker, S.F.; Chutia, A.; Farrow, M.R.; Silverwood, I.P.; García-Sakai, V.; Catlow, C.R.A. Room temperature methoxylation in zeolites: Insight into a key step of the methanol-to-hydrocarbons process. Chem. Commun. 2016, 52, 2897–2900. [Google Scholar] [CrossRef]
- Forester, T.R.; Howe, R.F. In Situ FTIR Studies of Methanol and Dimethyl Ether in ZSM-5. J. Am. Chem. Soc. 1987, 109, 5076–5082. [Google Scholar] [CrossRef]
- Wang, W.; Buchholz, A.; Seiler, M.; Hunger, M. Evidence for an Initiation of the Methanol-to-Olefin Process by Reactive Surface Methoxy Groups on Acidic Zeolite Catalysts. J. Am. Chem. Soc. 2003, 125, 15260–15267. [Google Scholar] [CrossRef] [PubMed]
- Yarulina, I.; Kapteijn, F.; Gascon, J. The importance of heat effects in the methanol to hydrocarbons reaction over ZSM-5: On the role of mesoporosity on catalyst performance. Catal. Sci. Technol. 2016, 6, 5320–5325. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. Dimethyl ether synthesis via methanol dehydration: Effect of zeolite structure. Appl. Catal. A Gen. 2015, 502, 215–220. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, S.; Wei, Y.; Li, J.; Chen, J.; Wang, J.; Zhang, W.; Gao, S.; Li, X.; Wang, C.; et al. Methanol conversion on ZSM-22, ZSM-35 and ZSM-5 zeolites: Effects of 10-membered ring zeolite structures on methylcyclopentenyl cations and dual cycle mechanism. RSC Adv. 2016, 6, 95855–95864. [Google Scholar] [CrossRef]
- Rojo-Gama, D.; Etemadi, S.; Kirby, E.; Lillerud, K.P.; Beato, P.; Svelle, S.; Olsbye, U. Time- and space-resolved study of the methanol to hydrocarbons (MTH) reaction-influence of zeolite topology on axial deactivation patterns. Faraday Discuss. 2017, 197, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Le, T.T.; Fu, D.; Schmidt, J.E.; Filez, M.; Weckhuysen, B.M.; Rimer, J.D. Deconvoluting the Competing Effects of Zeolite Framework Topology and Diffusion Path Length on Methanol to Hydrocarbons Reaction. ACS Catal. 2018, 8, 11042–11053. [Google Scholar] [CrossRef]
- Park, J.W.; Kim, S.J.; Seo, M.; Kim, S.Y.; Sugi, Y.; Seo, G. Product selectivity and catalytic deactivation of MOR zeolites with different acid site densities in methanol-to-olefin (MTO) reactions. Appl. Catal. A Gen. 2008, 349, 76–85. [Google Scholar] [CrossRef]
- Derouane, E.G.; Nagy, J.B.; Fernandez, C.; Gabelica, Z.; Laurent, E.; Maljean, P. Diffusion of alkanes in molecular sieves. Evidence for Confinement Effects. Appl. Catal. 1988, 40, L1–L10. [Google Scholar] [CrossRef]
- Smit, B.; Maesen, T.L.M. Molecular simulations of zeolites: Adsorption, diffusion, and shape selectivity. Chem. Rev. 2008, 108, 4125–4184. [Google Scholar] [CrossRef] [PubMed]
- Jae, J.; Tompsett, G.A.; Foster, A.J.; Hammond, K.D.; Auerbach, S.M.; Lobo, R.F.; Huber, G.W. Investigation into the shape selectivity of zeolite catalysts for biomass conversion. J. Catal. 2011, 279, 257–268. [Google Scholar] [CrossRef]
- O’Malley, A.J.; García Sakai, V.; Silverwood, I.P.; Dimitratos, N.; Parker, S.F.; Catlow, C.R.A. Methanol diffusion in zeolite HY: A combined quasielastic neutron scattering and molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2016, 18, 17294–17302. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Tamargo, C.; O’Malley, A.; Silverwood, I.P.; De Leeuw, N.H. Molecular behaviour of phenol in zeolite Beta catalysts as a function of acid site presence: A quasielastic neutron scattering and molecular dynamics simulation study. Catal. Sci. Technol. 2019, 9, 6700–6713. [Google Scholar] [CrossRef]
- Omojola, T.; Cherkasov, N.; McNab, A.I.; Lukyanov, D.B.; Anderson, J.A.; Rebrov, E.V.; Van Veen, A.C. Mechanistic Insights into the Desorption of Methanol and Dimethyl Ether Over ZSM-5 Catalysts. Catal. Lett. 2018, 148, 474–488. [Google Scholar] [CrossRef]
- Caro, J.; Bülow, M.; Richter-Mendau, J.; Kärger, J.; Hunger, M.; Freude, D.; Rees, L.V.C. Nuclear magnetic resonance self-diffusion studies of methanol-water mixtures in pentasil-type zeolites. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1987, 83, 1843–1849. [Google Scholar] [CrossRef]
- Jobic, H.; Renouprez, A.; Bee, M.; Poinsignon, C. Quasi-elastic neutron scattering study of the molecular motions of methanol adsorbed on H-ZSM-5. J. Phys. Chem. 1986, 90, 1059–1065. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Nguyen, P.T.M.; Dang, L.X.; Mei, D.; Wick, C.D.; Do, D.D. A comparative study of the adsorption of water and methanol in zeolite BEA: A molecular simulation study. Mol. Simul. 2014, 40, 1113–1124. [Google Scholar] [CrossRef]
- Mikkelsen, O.; Kolboe, S. The conversion of methanol to hydrocarbons over zeolite H-beta. Microporous Mesoporous Mater. 1999, 9, 173–184. [Google Scholar] [CrossRef]
- Park, J.W.; Seo, G. IR study on methanol-to-olefin reaction over zeolites with different pore structures and acidities. Appl. Catal. A Gen. 2009, 356, 180–188. [Google Scholar] [CrossRef]
- Chua, Y.T.; Stair, P.C. An ultraviolet Raman spectroscopic study of coke formation in methanol to hydrocarbons conversion over zeolite H-MFI. J. Catal. 2003, 213, 39–46. [Google Scholar] [CrossRef]
- Rownaghi, A.A.; Rezaei, F.; Hedlund, J. Uniform mesoporous ZSM-5 single crystals catalyst with high resistance to coke formation for methanol deoxygenation. Microporous Mesoporous Mater. 2012, 151, 26–33. [Google Scholar] [CrossRef]
- Jiang, Y.; Hunger, M.; Wang, W. On the reactivity of surface methoxy species in acidic zeolites. J. Am. Chem. Soc. 2006, 128, 11679–11692. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Gale, J.D.; Payne, M.C. Methanol adsorption in zeolites—A first-principles study. J. Phys. Chem. 1996, 100, 11688–11697. [Google Scholar] [CrossRef]
- Blaszkowski, S.R.; Van Santen, R.A. Density functional theory calculations of the activation of methanol by a Brønsted zeolitic proton. J. Phys. Chem. 1995, 99, 11728–11738. [Google Scholar] [CrossRef]
- Nastase, S.A.F.; O’Malley, A.J.; Catlow, C.R.A.; Logsdail, A.J. Computational QM/MM investigation of the adsorption of MTH active species in H-Y and H-ZSM-5. Phys. Chem. Chem. Phys. 2019, 21, 2639–2650. [Google Scholar] [CrossRef]
- Mirth, G.; Lercher, J.A.; Anderson, M.W.; Klinowski, J. Adsorption complexes of methanol on zeolite ZSM-5. J. Chem. Soc. Faraday Trans. 1990, 86, 3039–3044. [Google Scholar] [CrossRef]
- Krishna, R.; Van Baten, J.M. Hydrogen bonding effects in adsorption of water-alcohol mixtures in zeolites and the consequences for the characteristics of the Maxwell-Stefan diffusivities. Langmuir 2010, 26, 10854–10867. [Google Scholar] [CrossRef]
- Newsam, J.M.; Treacy, M.M.J.; Koetsier, W.T.; De Gruyter, C.B. Structural characterization of zeolite beta. Proc. R. Soc. London, Ser. A Math. Phys. Sci. 1988, 420, 375–405. [Google Scholar]
- Olson, D.H.; Kokotailo, G.T.; Lawton, S.L.; Meler, W.M. Crystal Structure and Structure-Related Properties of ZSM-5. J. Phys. Chem. 1981, 85, 2238–2243. [Google Scholar] [CrossRef]
- Dempsey, E.; Kühl, G.H.; Olson, D.H. Variation of the lattice parameter with aluminum content in synthetic sodium faujasites. Evidence for ordering of the framework ions. J. Phys. Chem. 1969, 73, 387–390. [Google Scholar] [CrossRef]
- Born, M.; Huang, K.; Lax, M. Dynamical Theory of Crystal Lattices. Am. J. Phys. 1955, 23, 474. [Google Scholar] [CrossRef]
- Blanco, C.; Auerbach, S.M. Nonequilibrium molecular dynamics of microwave-driven zeolite-guest systems: Loading dependence of athermal effects. J. Phys. Chem. B 2003, 107, 2490–2499. [Google Scholar] [CrossRef]
- Plant, D.F.; Maurin, G.; Bell, R.G. Diffusion of methanol in zeolite NaY: A molecular dynamics study. J. Phys. Chem. B 2007, 111, 2836–2844. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Kubo, M. Structures of dimethyl ether and methyl alcohol. J. Chem. Phys. 1959, 30, 151–158. [Google Scholar] [CrossRef]
- Lees, R.M.; Baker, J.G. Torsion-vibration-rotation interactions in methanol. I. Millimeter wave spectrum. J. Chem. Phys. 1968, 48, 5299–5318. [Google Scholar] [CrossRef]
- Dauber-Osguthorpe, P.; Roberts, V.A.; Osguthorpe, D.J.; Monique, G.; Hagler, A.T. Adsorbate-induced lattice deformation in IRMOF-74 series. Porteins Struct. Funct. Genet. 1988, 4, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Rehfinger, A.; Hoffmann, U. Kinetics of methyl tertiary butyl ether liquid phase synthesis catalyzed by ion exchange resin-II. Macropore diffusion of methanol as rate-controlling step. Chem. Eng. Sci. 1990, 45, 1619–1626. [Google Scholar] [CrossRef]
- Sanders, M.J.; Leslie, M.; Catlow, C.R.A. Interatomic potentials for SiO2. J. Chem. Soc. Chem. Commun. 1984, 1271–1273. [Google Scholar] [CrossRef]
- Catlow, C.R.A.; Stoneham, A.M. Ionicity in solids. J. Phys. C Solid State Phys. 1983, 16, 4321–4338. [Google Scholar] [CrossRef]
- Shubin, A.A.; Catlow, C.R.A.; Thomas, J.M.; Zamaraev, K.I. A computational study of the adsorption of the isomers of butanol on silicalite and H-ZSM-5. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1994, 446, 411–427. [Google Scholar]
- Sastre, G.; Catlow, C.R.A.; Corma, A. Diffusion of benzene and propylene in MCM-22 zeolite. A molecular dynamics study. J. Phys. Chem. B 1999, 103, 5187–5196. [Google Scholar] [CrossRef]
- Raj, N.; Sastre, G.; Catlow, C.R.A. Diffusion of octane in silicalite: A molecular dynamics study. J. Phys. Chem. B 1999, 103, 11007–11015. [Google Scholar] [CrossRef]
- Schröder, K.P.; Sauer, J. Potential functions for silica and zeolite catalysts based on ab initio calculations. A shell model ion pair potential for silica and aluminosilicates. J. Phys. Chem. 1996, 100, 11043–11049. [Google Scholar] [CrossRef]
- Schröder, K.P.; Sauer, J.; Leslie, M.; Catlow, C.R.A.; Thomas, J.M. Bridging hydrodyl groups in zeolitic catalysts: A computer simulation of their structure, vibrational properties and acidity in protonated faujasites (HY zeolites). Chem. Phys. Lett. 1992, 188, 320–325. [Google Scholar] [CrossRef]
- Todorov, I.T.; Smith, W.; Trachenko, K.; Dove, M.T. DL_POLY_3: New dimensions in molecular dynamics simulations via massive parallelism. J. Mater. Chem. 2006, 16, 1911–1918. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Brandt, E.G.; Lyubartsev, A.P. Systematic Optimization of a Force Field for Classical Simulations of TiO2-Water Interfaces. J. Phys. Chem. C 2015, 119, 18110–18125. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
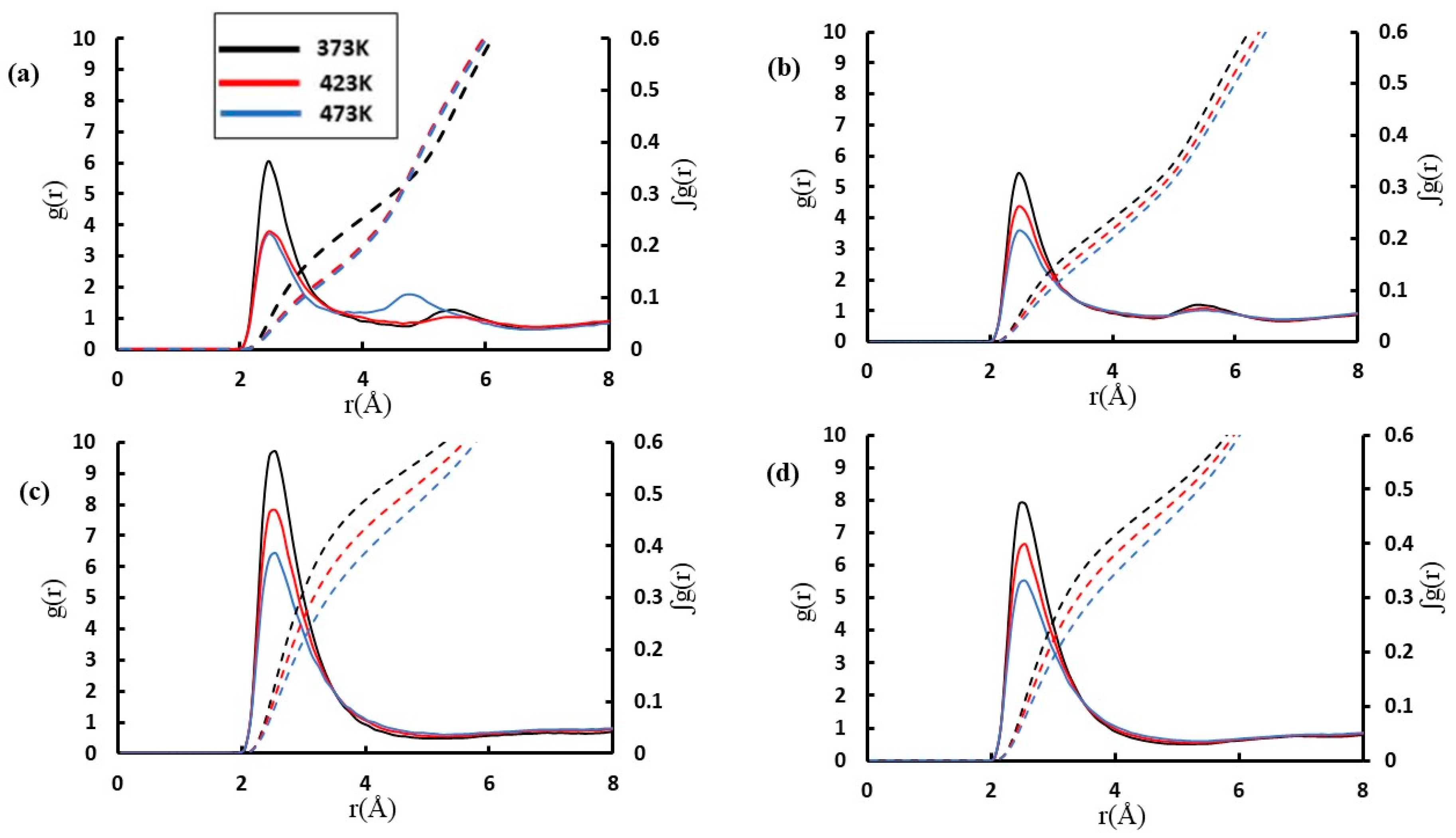{kind=link}
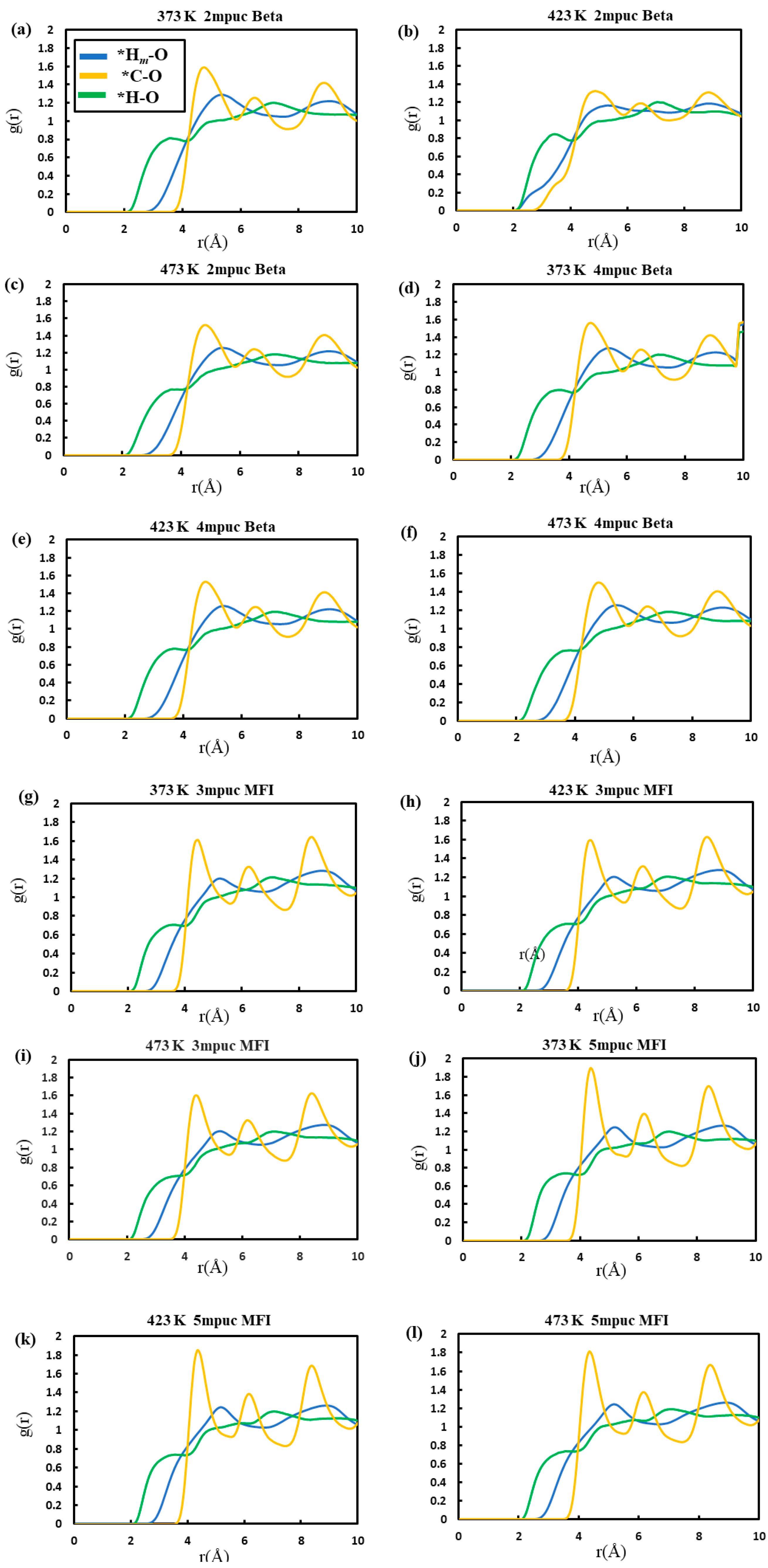{kind=link}
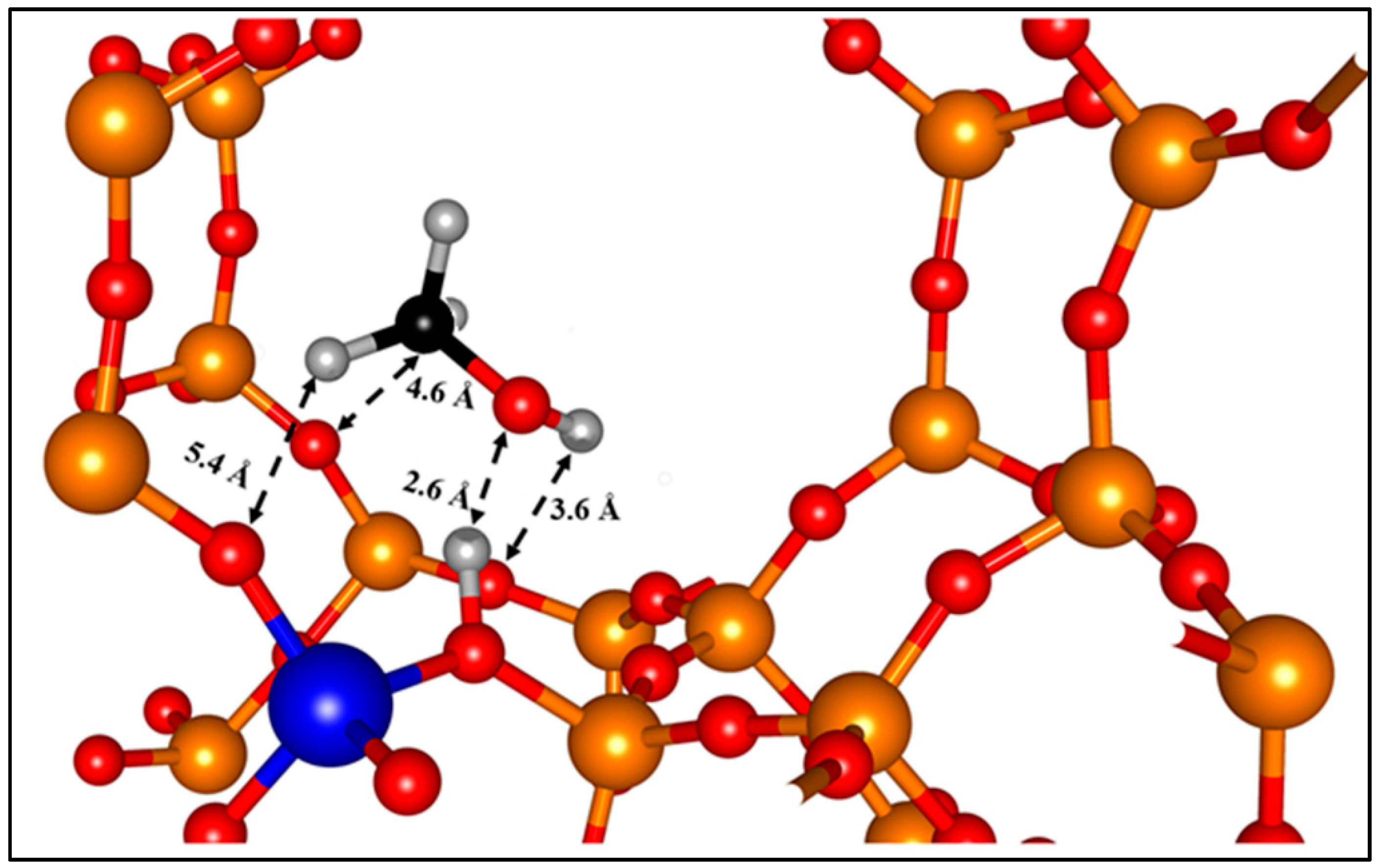{kind=link}
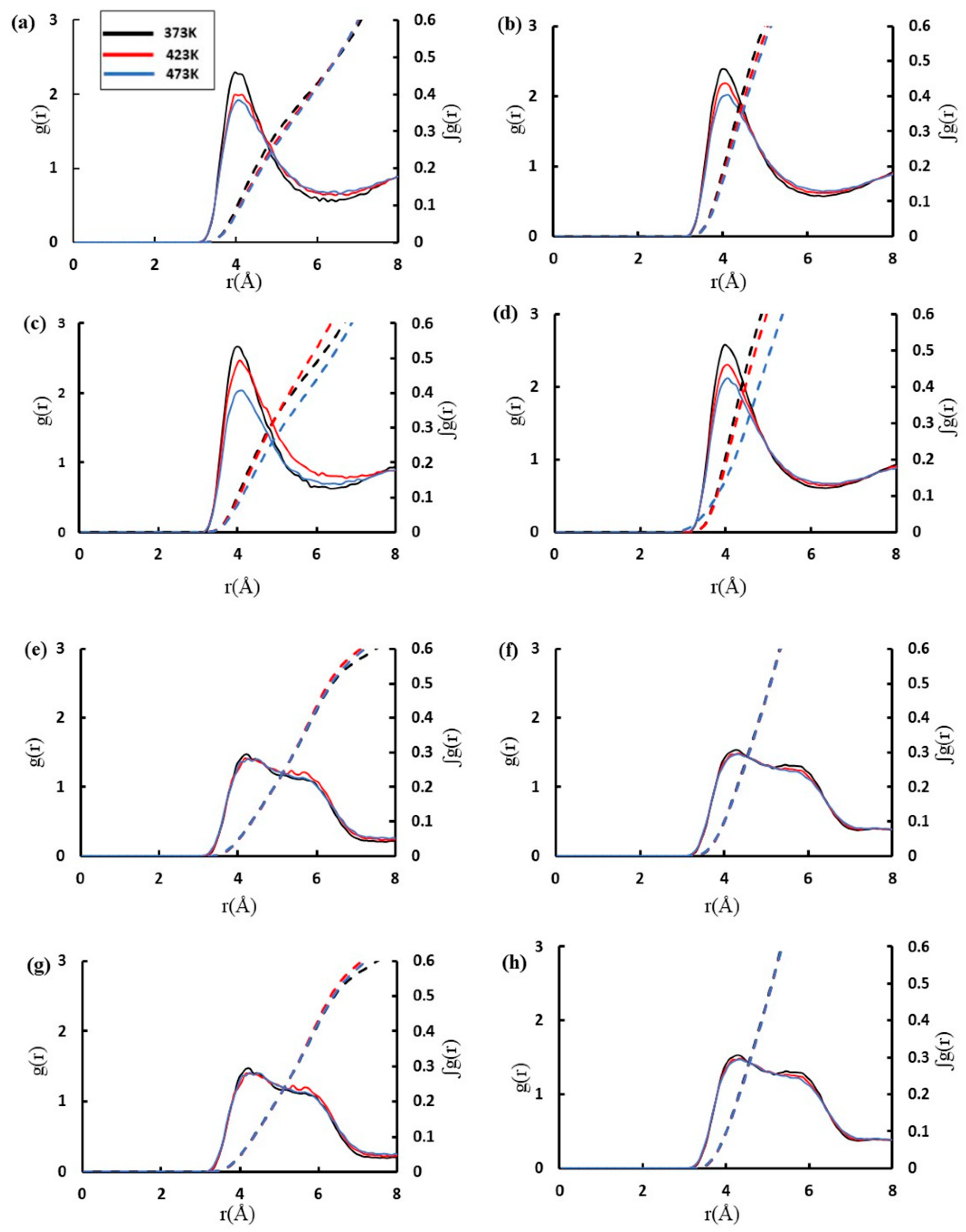{kind=link}
{kind=link}
| Acidic Beta | All-Silica Beta | |||
|---|---|---|---|---|
| Temp (K) | 2 mpuc | 4 mpuc | 2 mpuc | 4 mpuc |
| 373 | 112.4 | 89.9 | 139.1 | 1119.6 |
| 423 | 161.1 | 114.1 | 171.3 | 146.5 |
| 473 | 205.5 | 143.0 | 250.1 | 171.7 |
| Ea | 8.9 | 6.8 | 8.5 | 5.3 |
| Acidic MFI | All-Silica MFI | |||
| Temp (K) | 3 mpuc | 5 mpuc | 3 mpuc | 5 mpuc |
| 373 | 3.4 | 2.2 | 12.1 | 4.5 |
| 423 | 5.7 | 3.5 | 14.0 | 6.2 |
| 473 | 9.7 | 5.4 | 16.2 | 8.6 |
| Ea | 15.4 | 13.0 | 4.3 | 9.5 |
| Zeolite Structure and Loadings | 373 K | 423 K | 473 K |
|---|---|---|---|
| Beta 2 mpuc | 2.5 | 1.9 | 1.7 |
| Beta 4 mpuc | 2.3 | 1.6 | 1.5 |
| MFI 3 mpuc | 5.6 | 3.7 | 2.9 |
| MFI 5 mpuc | 5.1 | 3.6 | 2.7 |
| Atoms | Charges (a.u.) | ||
| C | −0.093 | ||
| H | 0.100 | ||
| Om | −0.432 | ||
| Hm | 0.225 | ||
| Harmonic bonds | |||
| Bonds | k (eV·Å−2) | r0 (Å) | |
| C-H | 29.56 | 1.105 | |
| C-Om | 33.33 | 1.420 | |
| Hm-Om | 46.97 | 0.945 | |
| Harmonic bond angles | |||
| Angles | k (eV·rad−2) | θ0 (˚) | |
| H-C-H | 4.4 | 108.38 | |
| H-C-Om | 5.5 | 106.90 | |
| C-Om-Hm | 5.6 | 108.32 | |
| Harmonic dihedral k | |||
| Dihedral | k (eV) | α (˚) | β |
| Hm-Om-C-H | 0.00762 | 1.0 | 3.0 |
| Lennard–Jones | |||
| Atomic pairs | |||
| C-C | 0.00694 | 3.475 | |
| C-H | 0.00338 | 2.920 | |
| C-Om | 0.00828 | 3.150 | |
| C-Hm | 0.00338 | 2.920 | |
| H-H | 0.00165 | 2.450 | |
| H-Om | 0.00404 | 2.650 | |
| H-Hm | 0.00165 | 2.450 | |
| Om-Om | 0.00988 | 2.860 | |
| Om-Hm | 0.004040 | 2.650 | |
| Hm-Hm | 0.001650 | 2.450 | |
| Buckingham Potentials | |||
| Atomic Pairs | A (eV) | ρ (Å) | C (eV·Å6) |
| O-O | 22764.0000 | 0.14900 | 27.88000 |
| O-Oa | 22764.0000 | 0.14900 | 27.88000 |
| O-Ha | 311.9700 | 0.25000 | 0.00000 |
| Si-O | 1283.9070 | 0.32052 | 10.66158 |
| Si-Oa | 983.5566 | 0.32052 | 10.66158 |
| Al-O | 1460.3000 | 0.29912 | 0.00000 |
| Al-Oa | 1142.6775 | 0.29912 | 0.00000 |
| Morse potential | |||
| Ionic Pair | D0 (eV) | k (Å−1) | r0 (Å) |
| Ha-Oa | 7.05250 | 2.19860 | 0.94850 |
| Three-body potentials | |||
| Angles | k (eV·rad−2) | θ0 (˚) | |
| O-T-O b | 2.09724 | 109.47 | |
| Lennard–Jones Potentials | ||
| Atomic Pairs | εij (eV) | σij (Å) |
| *C-O | 0.005910 | 4.310 |
| *C-Oa | 0.005910 | 4.310 |
| *C-Ha | 0.002991 | 2.806 |
| *H-O | 0.004987 | 2.557 |
| *H-Oa | 0.004987 | 2.557 |
| *H-Ha | 0.000851 | 1.785 |
| *Om-O | 0.010545 | 2.764 |
| *Om-Oa | 0.010545 | 2.764 |
| *Om-Ha | 0.004987 | 2.557 |
| *Hm-O | 0.004987 | 2.557 |
| *Hm-Oa | 0.004987 | 2.557 |
| *Hm-Ha | 0.000851 | 1.785 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botchway, C.H.; Tia, R.; Adei, E.; O’Malley, A.J.; Dzade, N.Y.; Hernandez-Tamargo, C.; de Leeuw, N.H. Influence of Topology and Brønsted Acid Site Presence on Methanol Diffusion in Zeolites Beta and MFI. Catalysts 2020, 10, 1342. https://doi.org/10.3390/catal10111342
Botchway CH, Tia R, Adei E, O’Malley AJ, Dzade NY, Hernandez-Tamargo C, de Leeuw NH. Influence of Topology and Brønsted Acid Site Presence on Methanol Diffusion in Zeolites Beta and MFI. Catalysts. 2020; 10(11):1342. https://doi.org/10.3390/catal10111342
Chicago/Turabian StyleBotchway, Cecil H., Richard Tia, Evans Adei, Alexander J. O’Malley, Nelson Y. Dzade, Carlos Hernandez-Tamargo, and Nora H. de Leeuw. 2020. "Influence of Topology and Brønsted Acid Site Presence on Methanol Diffusion in Zeolites Beta and MFI" Catalysts 10, no. 11: 1342. https://doi.org/10.3390/catal10111342
APA StyleBotchway, C. H., Tia, R., Adei, E., O’Malley, A. J., Dzade, N. Y., Hernandez-Tamargo, C., & de Leeuw, N. H. (2020). Influence of Topology and Brønsted Acid Site Presence on Methanol Diffusion in Zeolites Beta and MFI. Catalysts, 10(11), 1342. https://doi.org/10.3390/catal10111342