Resolving the Acid Site Distribution in Zn-Exchanged ZSM-5 with Stimulated Raman Scattering Microscopy



Abstract

1. Introduction
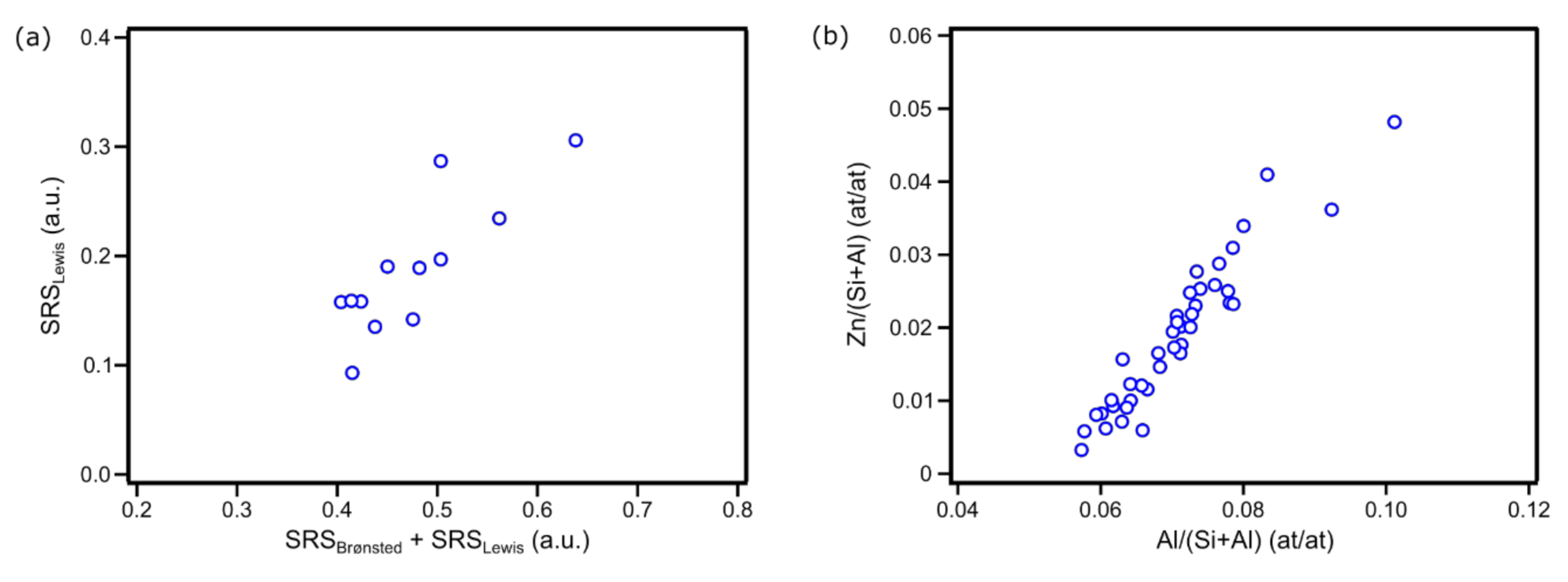
2. Results
3. Conclusions
4. Materials and Methods
4.1. Material Preparation
4.2. Bulk-Scale Characterization
4.3. Spontaneous Raman Microspectroscopy and SRS Imaging—Sample Preparation
4.4. Spontaneous Raman Microspectroscopy
4.5. Stimulated Raman Scattering Microscopy
4.6. SEM—Sample Preparation
4.7. Scanning Electron Microscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 978-7-80471-457-9. [Google Scholar]
- Corma, A.; García, H. Lewis acids: From conventional homogeneous to green homogeneous and heterogeneous catalysis. Chem. Rev. 2003, 103, 4307–4366. [Google Scholar] [CrossRef] [PubMed]
- Dapsens, P.Y.; Mondelli, C.; Pérez-Ramírez, J. Design of Lewis-acid centres in zeolitic matrices for the conversion of renewables. Chem. Soc. Rev. 2015, 44, 7025–7043. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.Y.; Lewis, J.D.; Román-Leshkov, Y. Lewis acid zeolites for biomass conversion: Perspectives and challenges on reactivity, synthesis, and stability. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Pidko, E.A. The nature and catalytic function of cation sites in zeolites: A computational perspective. ChemCatChem 2019, 11, 134–156. [Google Scholar] [CrossRef]
- Mole, T.; Anderson, J.R.; Creer, G. The reaction of propane over ZSM-5-H and ZSM-5-Zn zeolite catalysts. Appl. Catal. 1985, 17, 141–154. [Google Scholar] [CrossRef]
- Ono, Y.; Adachi, H.; Senoda, Y. Selective conversion of methanol into aromatic hydrocarbons over zinc-exchanged ZSM-5 zeolites. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 1091–1099. [Google Scholar] [CrossRef]
- Ono, Y.; Kanae, K. Transformation of butanes over ZSM-5 zeolites. Part 2.—Formation of aromatic hydrocarbons over Zn-ZSM-5 and Ga-ZSM-5. J. Chem. Soc. Faraday Trans. 1991, 87, 669–675. [Google Scholar] [CrossRef]
- Niu, X.; Gao, J.; Miao, Q.; Dong, M.; Wang, G.; Fan, W.; Qin, Z.; Wang, J. Influence of preparation method on the performance of Zn-containing HZSM-5 catalysts in methanol-to-aromatics. Microporous Mesoporous Mater. 2014, 197, 252–261. [Google Scholar] [CrossRef]
- Biscardi, J.A.; Iglesia, E. Structure and function of metal cations in light alkane reactions catalyzed by modified H-ZSM5. Catal. Today 1996, 31, 207–231. [Google Scholar] [CrossRef]
- Biscardi, J.A.; Meitzner, G.D.; Iglesia, E. Structure and density of active Zn species in Zn/H-ZSM5 Propane aromatization catalysts. J. Catal. 1998, 179, 192–202. [Google Scholar] [CrossRef]
- Tabacchi, G. Supramolecular organization in confined nanospaces. ChemPhysChem 2018, 19, 1249–1297. [Google Scholar] [CrossRef] [PubMed]
- Bryce, D.L. NMR crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Morra, E.; Berlier, G.; Borfecchia, E.; Bordiga, S.; Beato, P.; Chiesa, M. Electronic and geometrical structure of Zn+ ions stabilized in the Porous structure of Zn-loaded Zeolite H-ZSM-5: A multifrequency CW and Pulse EPR study. J. Phys. Chem. C 2017, 121, 14238–14245. [Google Scholar] [CrossRef]
- van Bokhoven, J.A.; Lamberti, C. Structure of aluminum, iron, and other heteroatoms in zeolites by X-ray absorption spectroscopy. Coord. Chem. Rev. 2014, 277–278, 275–290. [Google Scholar] [CrossRef]
- Hagen, A.; Hallmeier, K.-H.; Hennig, C.; Szargan, R.; Inui, T.; Roessner, F. State of Zinc in MFI Type Zeolites Characterized by XANES and EXAFS. In Studies in Surface Science and Catalysis; Beyer, H.K., Karge, H.G., Kiricsi, I., Nagy, J.B., Eds.; Catalysis by Microporous Materials; Elsevier: Amsterdam, The Netherlands, 1995; Volume 94, pp. 195–202. [Google Scholar] [CrossRef]
- Weckhuysen, B.M. Chemical imaging of spatial heterogeneities in catalytic solids at different length and time scales. Angew. Chem. Int. Ed. 2009, 48, 4910–4943. [Google Scholar] [CrossRef] [PubMed]
- Buurmans, I.L.C.; Weckhuysen, B.M. Heterogeneities of individual catalyst particles in space and time as monitored by spectroscopy. Nat. Chem. 2012, 4, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Meirer, F.; Weckhuysen, B.M. Spatial and temporal exploration of heterogeneous catalysts with synchrotron radiation. Nat. Rev. Mater. 2018, 3, 324–340. [Google Scholar] [CrossRef]
- Janssen, K.P.F.; Cremer, G.D.; Neely, R.K.; Kubarev, A.V.; Loon, J.V.; Martens, J.A.; Vos, D.E.D.; Roeffaers, M.B.J.; Hofkens, J. Single molecule methods for the study of catalysis: From enzymes to heterogeneous catalysts. Chem. Soc. Rev. 2014, 43, 990–1006. [Google Scholar] [CrossRef]
- Stavitski, E.; Weckhuysen, B.M. Infrared and Raman imaging of heterogeneous catalysts. Chem. Soc. Rev. 2010, 39, 4615–4625. [Google Scholar] [CrossRef]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent developments in the synthesis of supported catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Plessers, E.; Stassen, I.; Sree, S.P.; Janssen, K.P.F.; Yuan, H.; Martens, J.; Hofkens, J.; De Vos, D.; Roeffaers, M.B.J. Resolving interparticle heterogeneities in composition and hydrogenation performance between individual supported silver on silica catalysts. ACS Catal. 2015, 5, 6690–6695. [Google Scholar] [CrossRef] [PubMed]
- Plessers, E.; van den Reijen, J.E.; de Jongh, P.E.; de Jong, K.P.; Roeffaers, M.B.J. Origin and abatement of heterogeneity at the support granule scale of silver on silica catalysts. ChemCatChem 2017, 9, 4562–4569. [Google Scholar] [CrossRef]
- van der Bij, H.E.; Aramburo, L.R.; Arstad, B.; Dynes, J.J.; Wang, J.; Weckhuysen, B.M. Phosphatation of Zeolite H-ZSM-5: A combined microscopy and spectroscopy study. ChemPhysChem 2014, 15, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Zečević, J.; van der Eerden, A.M.J.; Friedrich, H.; de Jongh, P.E.; de Jong, K.P. Heterogeneities of the Nanostructure of Platinum/Zeolite Y catalysts revealed by electron tomography. ACS Nano 2013, 7, 3698–3705. [Google Scholar] [CrossRef]
- Mehdad, A.; Lobo, R.F. Ethane and ethylene aromatization on zinc-containing zeolites. Catal. Sci. Technol. 2017, 7, 3562–3572. [Google Scholar] [CrossRef]
- Roeffaers, M.B.J.; Sels, B.F.; Uji-i, H.; Blanpain, B.; L’hoëst, P.; Jacobs, P.A.; De Schryver, F.C.; Hofkens, J.; De Vos, D.E. Space- and time-resolved visualization of acid catalysis in ZSM-5 crystals by fluorescence microscopy. Angew. Chem. 2007, 119, 1736–1739. [Google Scholar] [CrossRef]
- Kox, M.H.F.; Stavitski, E.; Groen, J.C.; Pérez-Ramírez, J.; Kapteijn, F.; Weckhuysen, B.M. Visualizing the crystal structure and locating the catalytic activity of micro- and mesoporous ZSM-5 Zeolite Crystals by using in situ optical and fluorescence microscopy. Chem. Eur. J. 2008, 14, 1718–1725. [Google Scholar] [CrossRef]
- Kennes, K.; Demaret, C.; Van Loon, J.; Kubarev, A.V.; Fleury, G.; Sliwa, M.; Delpoux, O.; Maury, S.; Harbuzaru, B.; Roeffaers, M.B.J. Assessing Inter and Intra-Particle Heterogeneity in Alumina-Poor H-ZSM-5 Zeolites. ChemCatChem 2017, 9, 3440–3445. [Google Scholar] [CrossRef]
- Kerssens, M.M.; Sprung, C.; Whiting, G.T.; Weckhuysen, B.M. Selective staining of zeolite acidity: Recent progress and future perspectives on fluorescence microscopy. Microporous Mesoporous Mater. 2014, 189, 136–143. [Google Scholar] [CrossRef]
- Bordiga, S.; Lamberti, C.; Bonino, F.; Travert, A.; Thibault-Starzyk, F. Probing zeolites by vibrational spectroscopies. Chem. Soc. Rev. 2015, 44, 7262–7341. [Google Scholar] [CrossRef]
- Aramburo, L.R.; Karwacki, L.; Cubillas, P.; Asahina, S.; de Winter, D.A.M.; Drury, M.R.; Buurmans, I.L.C.; Stavitski, E.; Mores, D.; Daturi, M.; et al. The porosity, acidity, and reactivity of dealuminated Zeolite ZSM-5 at the single particle level: The influence of the Zeolite architecture. Chem. Eur. J. 2011, 17, 13773–13781. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.-L.; Kubarev, A.V.; Van Loon, J.; Uji-i, H.; De Vos, D.E.; Hofkens, J.; Roeffaers, M.B.J. Rationalizing inter- and intracrystal heterogeneities in dealuminated acid mordenite zeolites by stimulated raman scattering microscopy correlated with super-resolution fluorescence microscopy. ACS Nano 2014, 8, 12650–12659. [Google Scholar] [CrossRef] [PubMed]
- Berndt, H.; Lietz, G.; Völter, J. Zinc promoted H-ZSM-5 catalysts for conversion of propane to aromatics II. Nature of the active sites and their activation. Appl. Catal. Gen. 1996, 146, 365–379. [Google Scholar] [CrossRef]
- Almutairi, S.M.T.; Mezari, B.; Magusin, P.C.M.M.; Pidko, E.A.; Hensen, E.J.M. Structure and reactivity of Zn-modified ZSM-5 Zeolites: The importance of clustered cationic Zn complexes. ACS Catal. 2012, 2, 71–83. [Google Scholar] [CrossRef]
- Bi, Y.; Wang, Y.; Chen, X.; Yu, Z.; Xu, L. Methanol aromatization over HZSM-5 catalysts modified with different zinc salts. Chin. J. Catal. 2014, 35, 1740–1751. [Google Scholar] [CrossRef]
- Wei, Z.; Chen, L.; Cao, Q.; Wen, Z.; Zhou, Z.; Xu, Y.; Zhu, X. Steamed Zn/ZSM-5 catalysts for improved methanol aromatization with high stability. Fuel Process. Technol. 2017, 162, 66–77. [Google Scholar] [CrossRef]
- Pinilla-Herrero, I.; Borfecchia, E.; Holzinger, J.; Mentzel, U.V.; Joensen, F.; Lomachenko, K.A.; Bordiga, S.; Lamberti, C.; Berlier, G.; Olsbye, U.; et al. High Zn/Al ratios enhance dehydrogenation vs hydrogen transfer reactions of Zn-ZSM-5 catalytic systems in methanol conversion to aromatics. J. Catal. 2018, 362, 146–163. [Google Scholar] [CrossRef]
- Valecillos, J.; Epelde, E.; Albo, J.; Aguayo, A.T.; Bilbao, J.; Castaño, P. Slowing down the deactivation of H-ZSM-5 zeolite catalyst in the methanol-to-olefin (MTO) reaction by P or Zn modifications. Catal. Today 2020, 348, 243–256. [Google Scholar] [CrossRef]
- Egerton, T.A.; Hardin, A.H.; Sheppard, N. Raman spectra of pyridine adsorbed on a series of ion-exchanged forms of zeolite Y. Can. J. Chem. 1976, 54, 586–598. [Google Scholar] [CrossRef]
- Yamada, H.; Yamamoto, Y. Ultraviolet excitation of Raman spectra of pyridines adsorbed on oxides. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1979, 75, 1215–1221. [Google Scholar] [CrossRef]
- Ferwerda, R.; van der Maas, J.H.; Hendra, P.J. Pyridine adsorbed on Na-faujasite: A FT-Raman spectroscopic study. J. Phys. Chem. 1993, 97, 7331–7336. [Google Scholar] [CrossRef]
- Ferwerda, R.; van der Maas, J.H.; Hendra, P.J. Fourier transform Raman spectroscopy of pyridine adsorbed on faujasites. Vib. Spectrosc. 1994, 7, 37–47. [Google Scholar] [CrossRef]
- Ferwerda, R.; van der Maas, J.H.; van Duijneveldt, F.B. Pyridine adsorption onto metal oxides: An ab initio study of model systems. J. Mol. Catal. Chem. 1996, 104, 319–328. [Google Scholar] [CrossRef]
- Kassab, E.; Castellà-Ventura, M. Theoretical study of Pyridine and 4,4′-Bipyridine adsorption on the lewis acid sites of alumina surfaces based on Ab Initio and density functional cluster calculations. J. Phys. Chem. B 2005, 109, 13716–13728. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, L.I.; de Jong, K.P.; Zečević, J. The origin of metal loading heterogeneities in Pt/Zeolite Y bifunctional catalysts. ChemCatChem 2019, 11, 4081–4088. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zn Content (wt. %) | CBronsted (μmol/g) | CLewis (μmol/g) | |
|---|---|---|---|
| Zn000 | 0 | 306 | 21 |
| Zn030 | 1.04 | 209 | 165 |
| Zn600 | 1.65 | 158 | 245 |
| Mean SRS Intensity (a.u.) | |||
|---|---|---|---|
| Zn000 | Zn030 | Zn600 | |
| Brønsted acid sites | 0.50 ± 0.09 | 0.38 ± 0.08 | 0.28 ± 0.04 |
| Lewis acid sites | 0.02 ± 0.02 | 0.09 ± 0.03 | 0.19 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fleury, G.; Roeffaers, M.B.J. Resolving the Acid Site Distribution in Zn-Exchanged ZSM-5 with Stimulated Raman Scattering Microscopy. Catalysts 2020, 10, 1331. https://doi.org/10.3390/catal10111331
Fleury G, Roeffaers MBJ. Resolving the Acid Site Distribution in Zn-Exchanged ZSM-5 with Stimulated Raman Scattering Microscopy. Catalysts. 2020; 10(11):1331. https://doi.org/10.3390/catal10111331
Chicago/Turabian StyleFleury, Guillaume, and Maarten B. J. Roeffaers. 2020. "Resolving the Acid Site Distribution in Zn-Exchanged ZSM-5 with Stimulated Raman Scattering Microscopy" Catalysts 10, no. 11: 1331. https://doi.org/10.3390/catal10111331
APA StyleFleury, G., & Roeffaers, M. B. J. (2020). Resolving the Acid Site Distribution in Zn-Exchanged ZSM-5 with Stimulated Raman Scattering Microscopy. Catalysts, 10(11), 1331. https://doi.org/10.3390/catal10111331