CO2 and H2O Coadsorption and Reaction on the Low-Index Surfaces of Tantalum Nitride: A First-Principles DFT-D3 Investigation


Abstract

1. Introduction
2. Results and Discussion
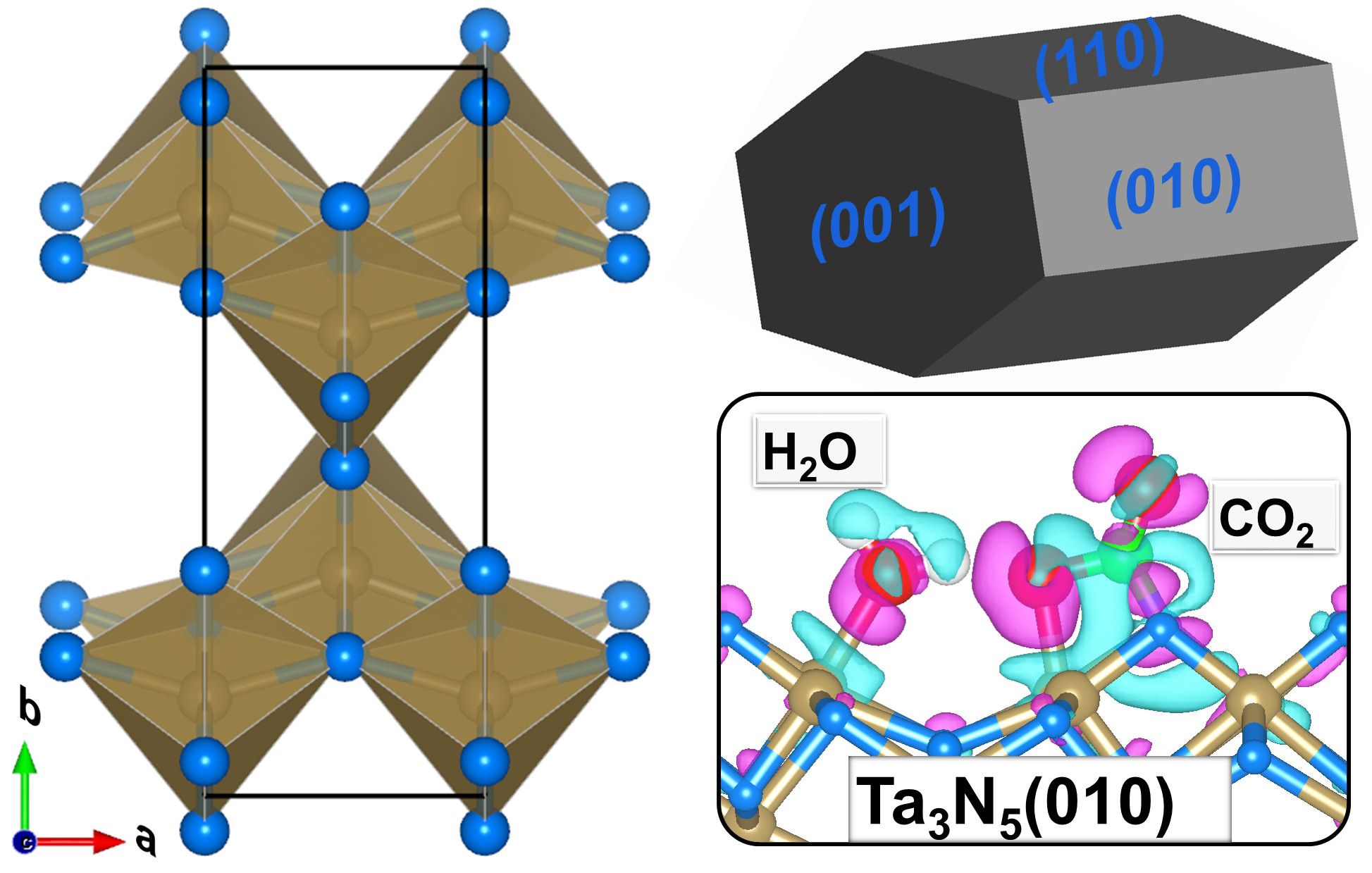
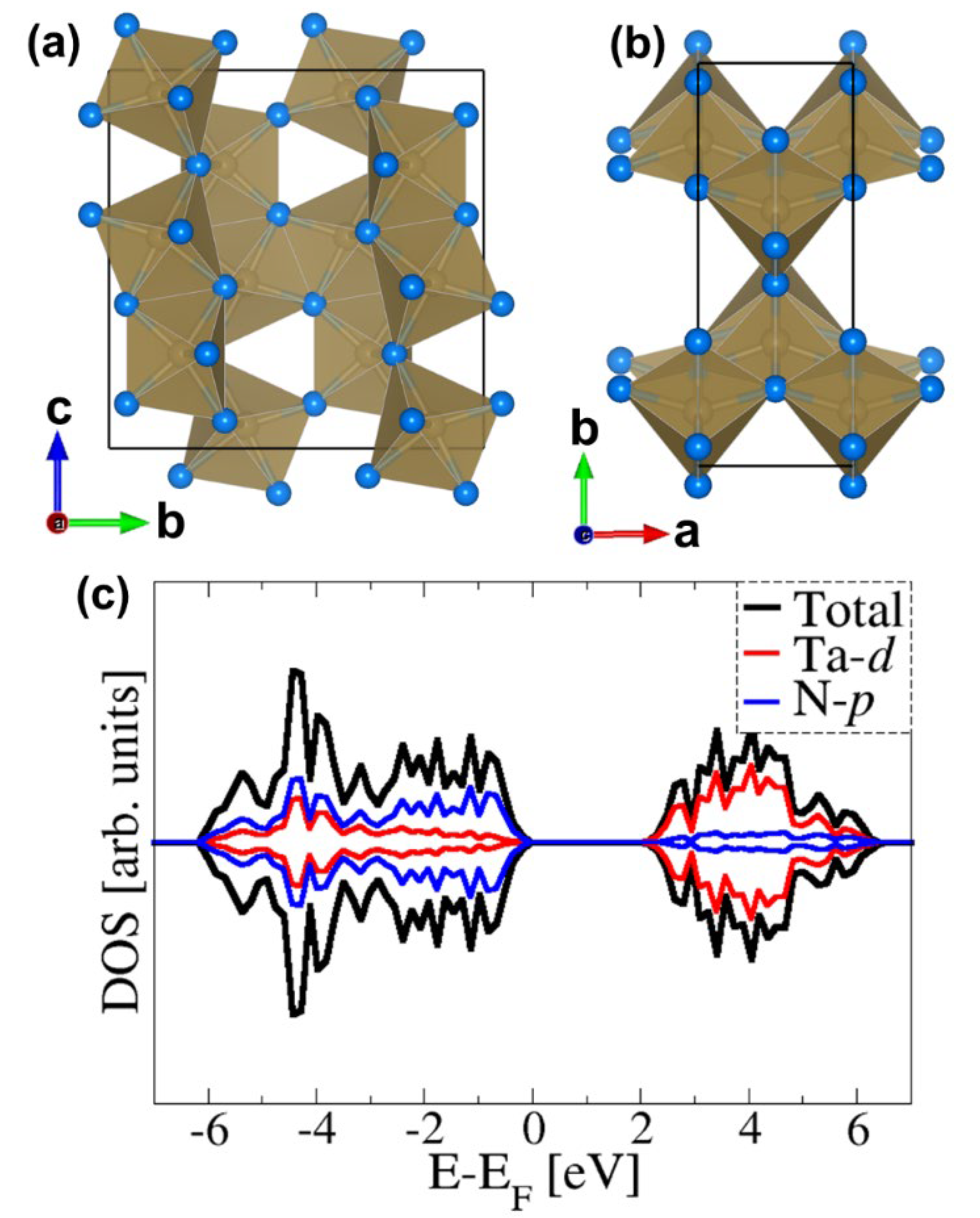
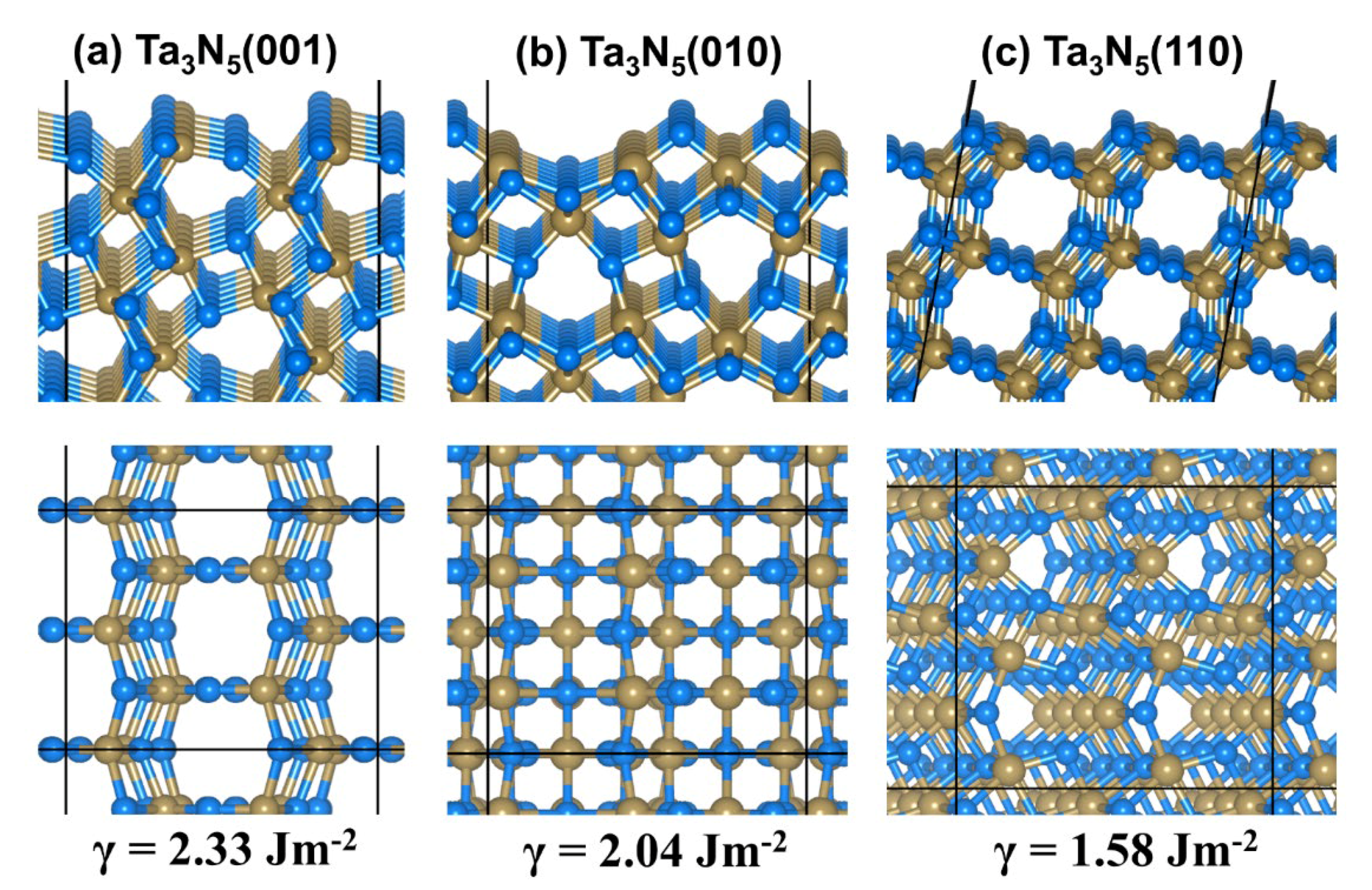
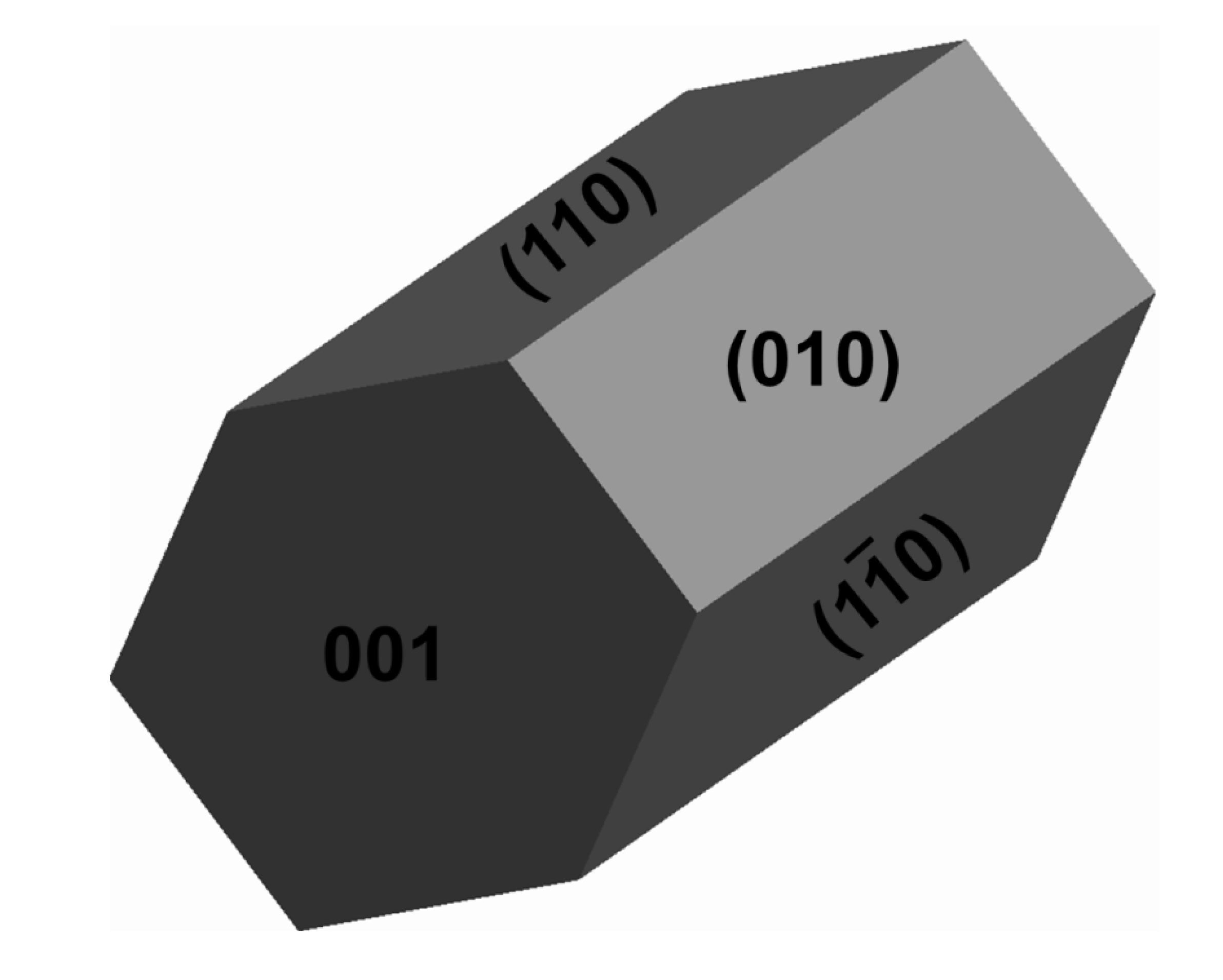
2.1. Bulk and Surface Properties
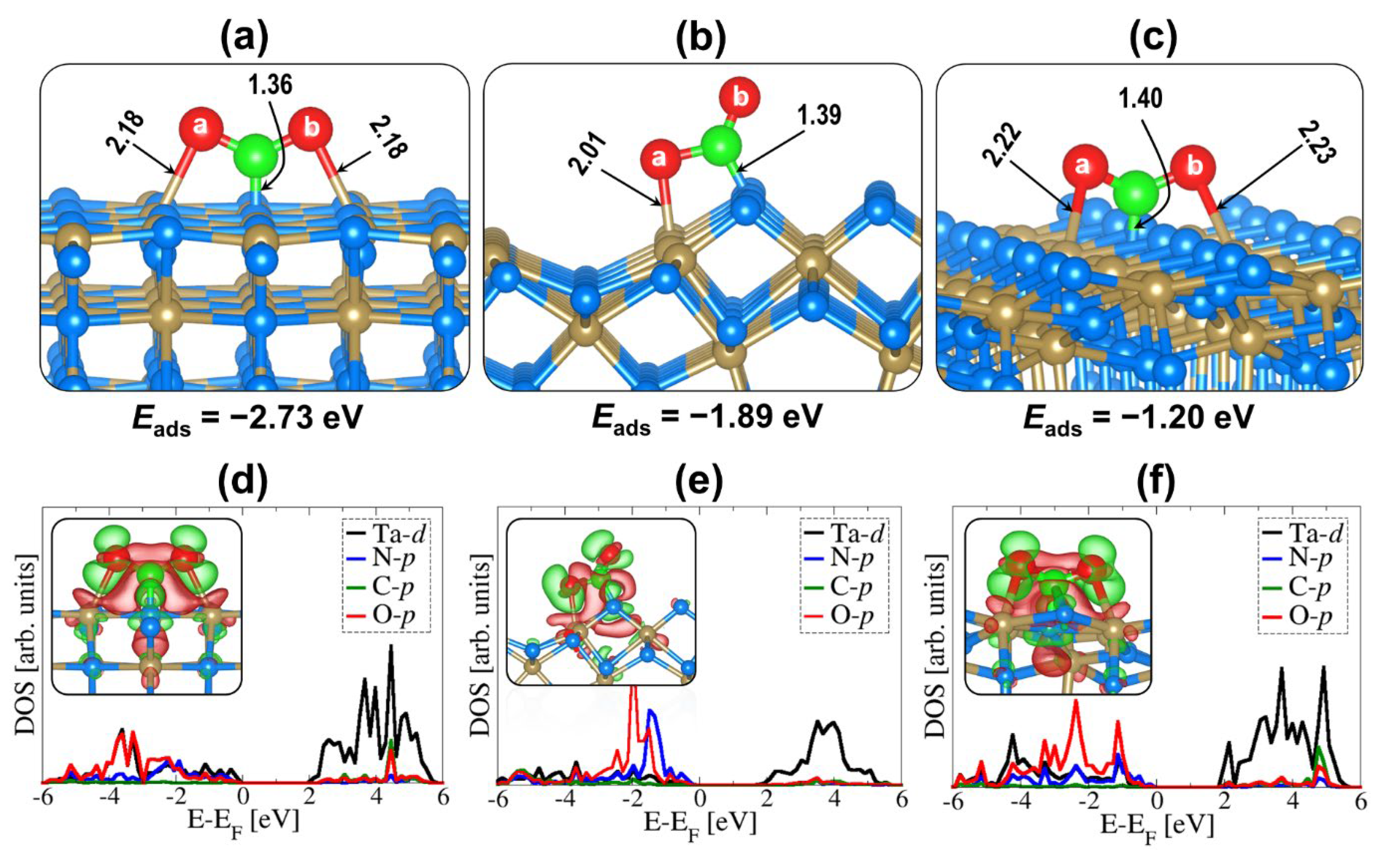
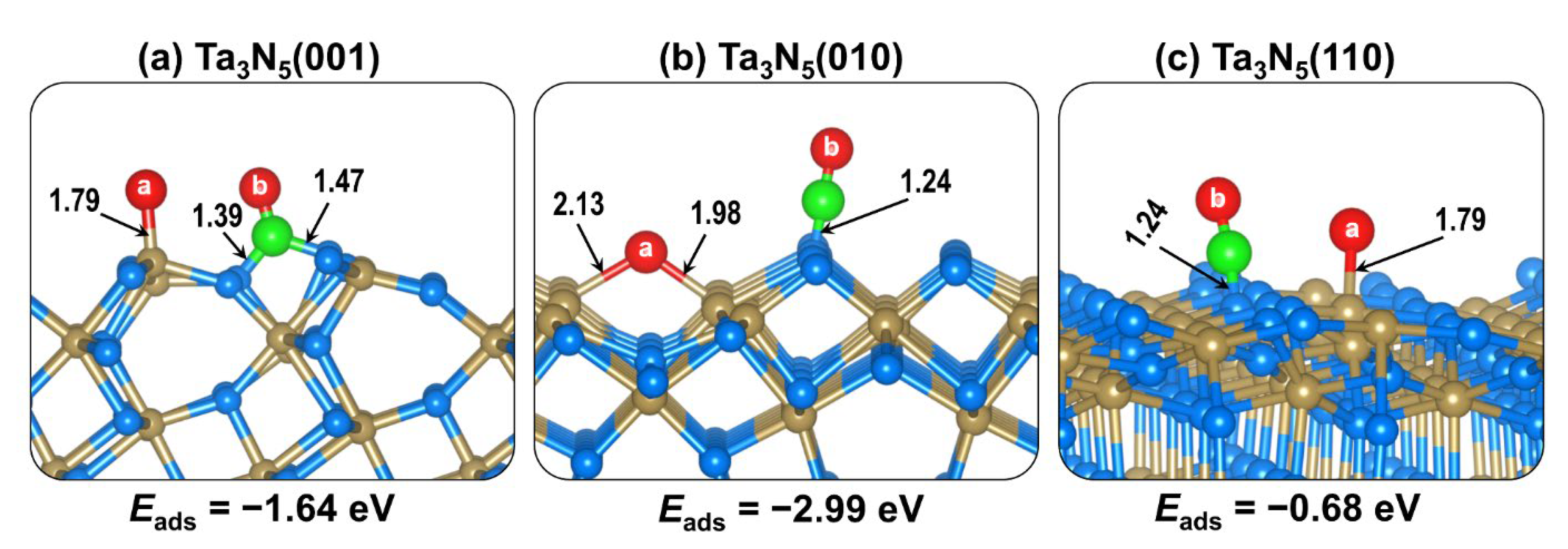
2.2. CO2 Adsorption on (001), (010), and (110) Ta3N5 Surfaces
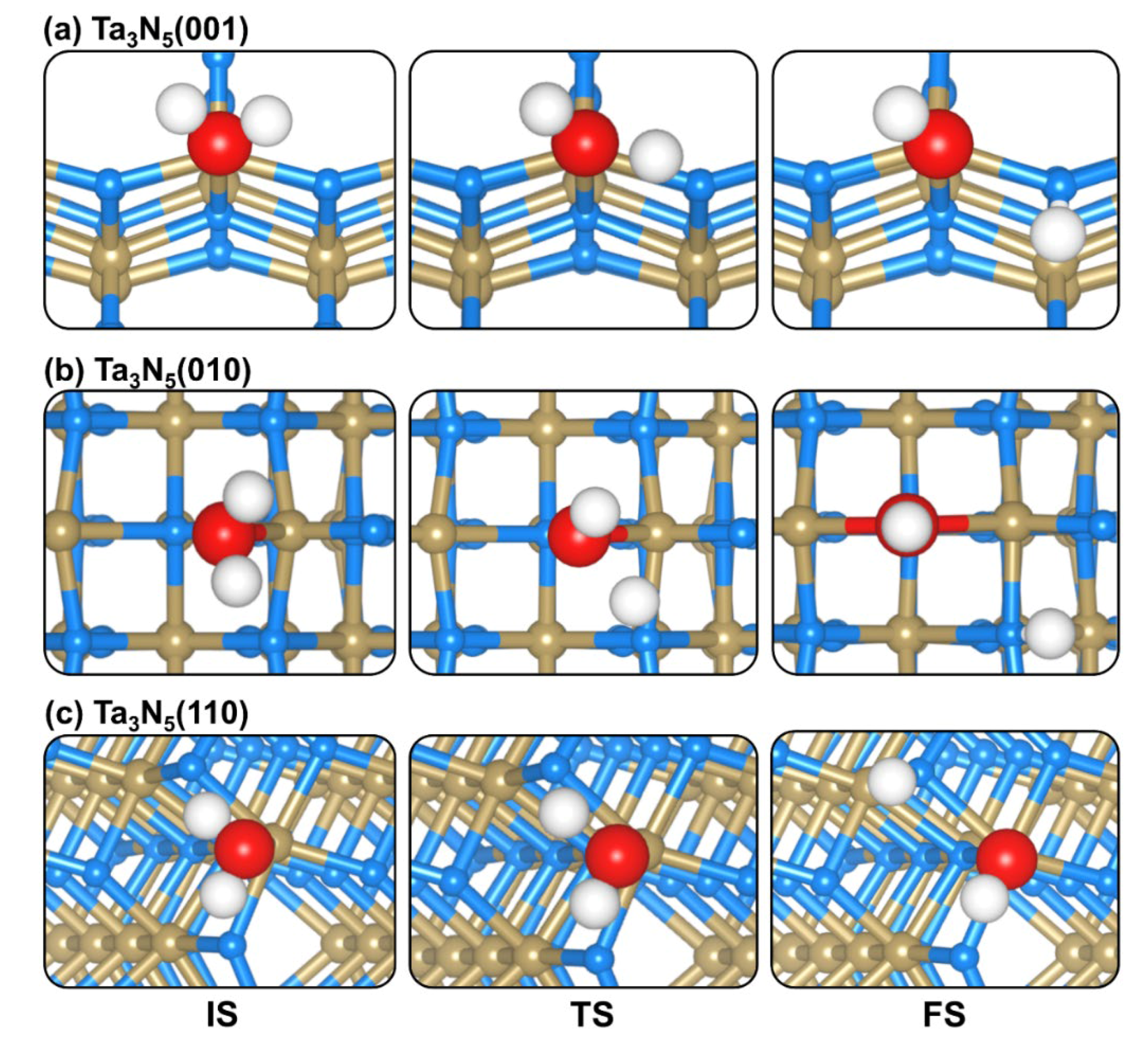
2.3. CO2 Dissociation on (001), (010), and (110) Ta3N5 Surfaces
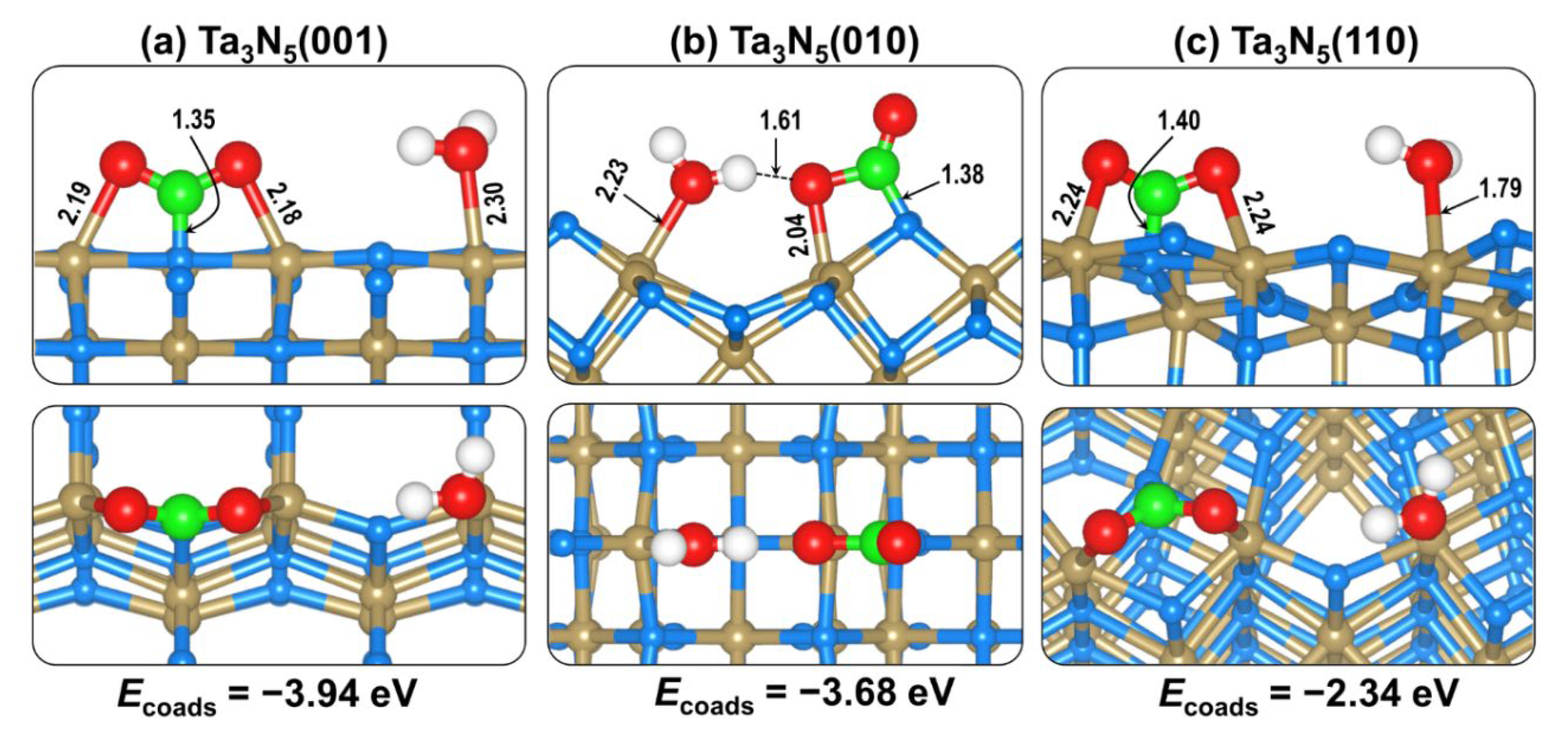
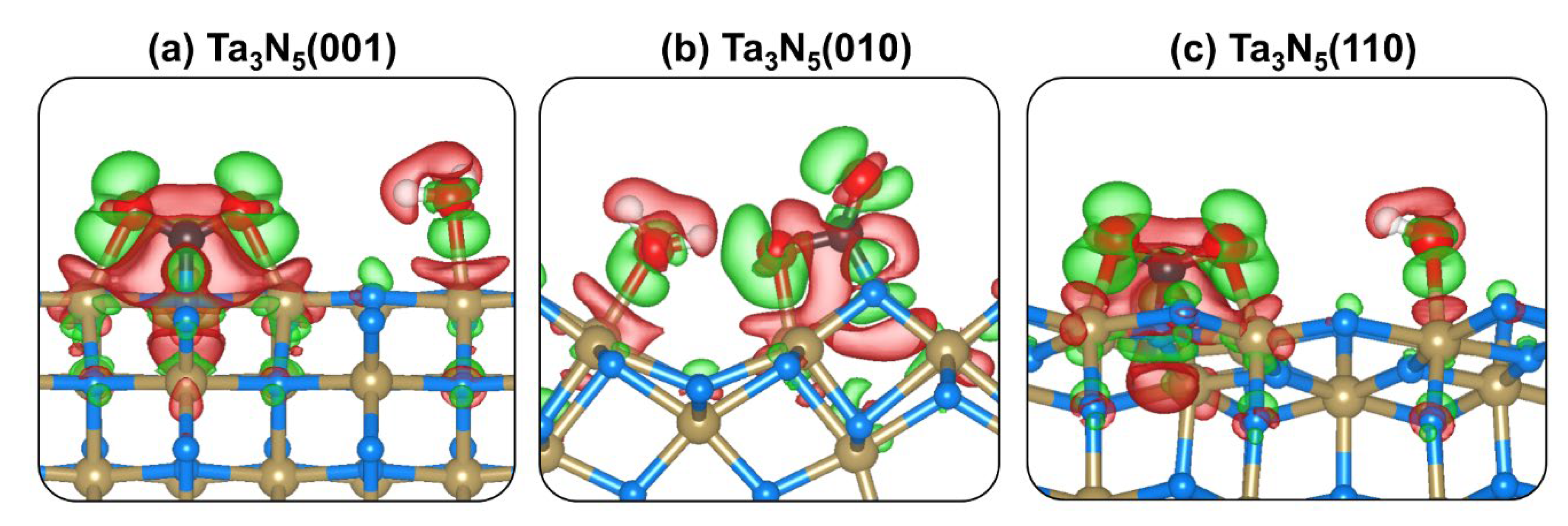
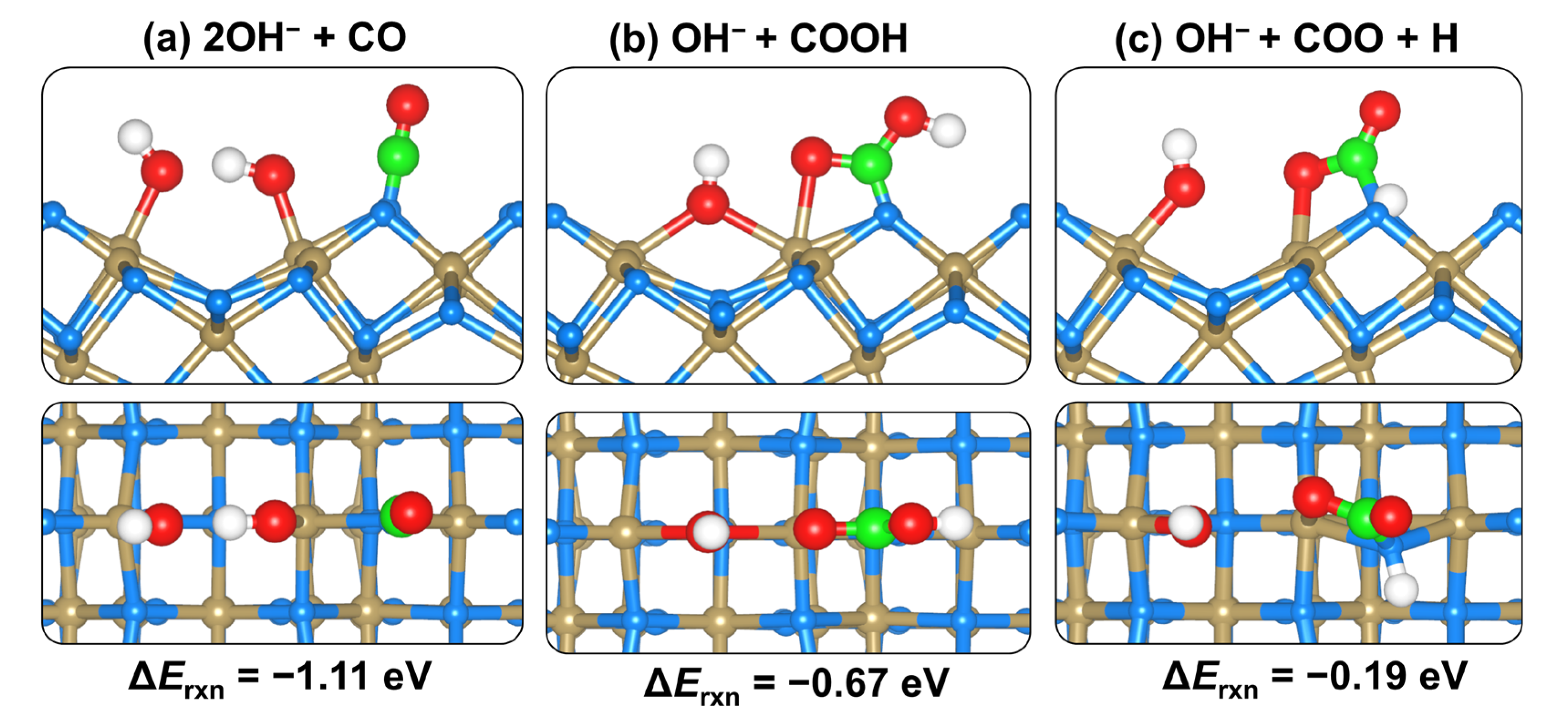
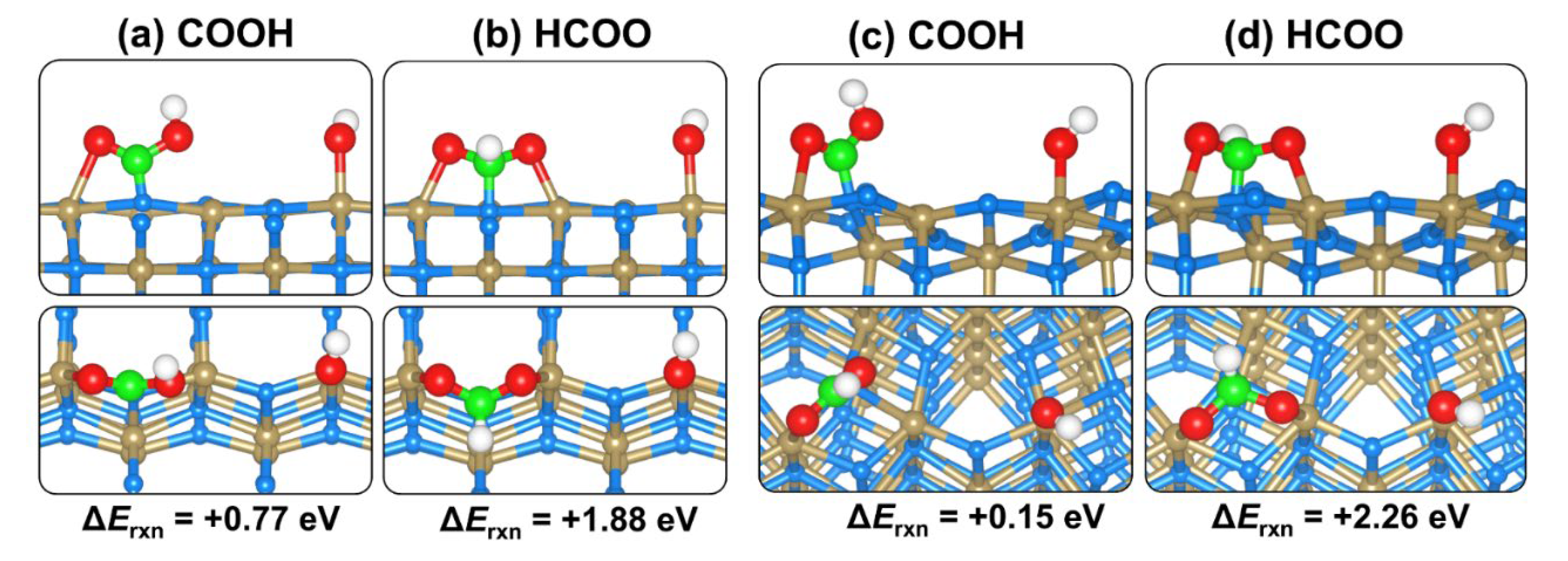
2.4. CO2 and H2O Coadsorption and Reactions
3. Summary and Conclusions
4. Computational Details
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Li, K.; Peng, B.; Peng, T. Recent Advances in Heterogeneous Photocatalytic CO2 Conversion to Solar Fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Chang, X.; Wang, T.; Gong, J. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef] [PubMed]
- Lingampalli, S.R.; Ayyub, M.M.; Rao, C.N.R. Recent Progress in the Photocatalytic Reduction of Carbon Dioxide. ACS Omega 2017, 2, 2740–2748. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic Reduction of Carbon Dioxide in Aqueous Suspensions of Semiconductor Powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Morris, A.J.; Meyer, G.J.; Fujita, E. Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels. Acc. Chem. Res. 2009, 42, 983–1994. [Google Scholar] [CrossRef]
- Varghese, O.K.; Paulose, M.; LaTempa, T.J.; Grimes, C.A. High-Rate Solar Photocatalytic Conversion of CO2 and Water Vapor to Hydrocarbon Fuels. Nano Lett. 2009, 9, 731–737. [Google Scholar] [CrossRef]
- Roy, S.C.; Varghese, O.K.; Paulose, M.; Grimes, C.A. Toward Solar Fuels: Photocatalytic Conversion of Carbon Dioxide to Hydrocarbons. ACS Nano 2010, 4, 1259–1278. [Google Scholar] [CrossRef]
- Li, Y.; Chan, S.H.; Sun, Q. Heterogeneous Catalytic Conversion of CO2: A Comprehensive Theoretical Review. Nanoscale 2015, 7, 8663–8683. [Google Scholar] [CrossRef]
- Todorova, T.K.; Schreiber, M.W.; Fontecave, M. Mechanistic Understanding of CO2 Reduction Reaction (CO2RR) Toward Multicarbon Products by Heterogeneous Copper-Based Catalysts. ACS Catal. 2020, 10, 1754–1768. [Google Scholar] [CrossRef]
- Paulino, P.N.; Salim, V.M.M.; Resende, N.S. Zn-Cu promoted TiO2 photocatalyst for CO2 reduction with H2O under UV light. Appl. Catal. B Environ. 2016, 185, 362–370. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, H.; Andino, J.M.; Li, Y. Photocatalytic CO2 Reduction with H2O on TiO2 Nanocrystals: Comparison of Anatase, Rutile, and Brookite Polymorphs and Exploration of Surface Chemistry. ACS Catal. 2012, 2, 1817–1828. [Google Scholar] [CrossRef]
- Mino, L.; Spoto, G.; Ferrari, A.M. CO2 Capture by TiO2 Anatase Surfaces: A Combined DFT and FTIR Study. J. Phys. Chem. C 2014, 118, 25016–25026. [Google Scholar] [CrossRef]
- Ye, J.Y.; Liu, C.-J.; Ge, Q.F. DFT Study of CO2 Adsorption and Hydrogenation on the In2O3 Surface. J. Phys. Chem. C 2012, 116, 7817–7825. [Google Scholar] [CrossRef]
- Ye, J.Y.; Liu, C.-J.; Mei, D.H.; Ge, Q.F. Active Oxygen Vacancy Site for Methanol Synthesis from CO2 Hydrogenation on In2O3(110): A DFT Study. ACS Catal. 2013, 3, 1296–1306. [Google Scholar] [CrossRef]
- Pa, Y.X.; Mei, D.; Liu, C.J.; Ge, Q. Adsorption on Ga2O3 Surface: A Combined Experimental and Computational Study. J. Phys. Chem. C 2011, 115, 10140–10146. [Google Scholar]
- Pan, Y.X.; Liu, C.-J.; Mei, D.H.; Ge, Q.F. Effects of Hydration and Oxygen Vacancy on CO2 Adsorption and Activation on β-Ga2O3(100). Langmuir 2010, 26, 5551–5558. [Google Scholar] [CrossRef]
- Pan, Y.X.; Liu, C.-J.; Ge, Q.F. Adsorption and Protonation of CO2 on Partially Hydroxylated γ-Al2O3 Surfaces: A Density Functional Theory Study. Langmuir 2008, 24, 12410–12419. [Google Scholar] [CrossRef]
- Pan, Y.X.; Liu, C.-J.; Ge, Q.F. Effect of surface hydroxyls on selective CO2 hydrogenation over Ni4/γ-Al2O3: A density functional theory study. J. Catal. 2010, 272, 227–234. [Google Scholar] [CrossRef]
- Liu, X.; Ye, L.; Liu, S.; Li, Y.; Ji, X. Photocatalytic Reduction of CO2 by ZnO Micro/nanomaterials with Different Morphologies and Ratios of {0001} Facets. Sci. Rep. 2016, 6, 38474. [Google Scholar] [CrossRef]
- Tang, Q.-L.; Luo, Q.-H. Adsorption of CO2 at ZnO: A Surface Structure Effect from DFT+U Calculations. J. Phys. Chem. C 2013, 117, 22954–22966. [Google Scholar] [CrossRef]
- Cheng, Z.; Sherman, B.J.; Lo, C.S. Carbon dioxide activation and dissociation on ceria (110): A density functional theory study. J. Chem. Phys. 2013, 138, 014702. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Haider, M.A.; Agarwal, M.; Sinha, N.; Basu, S. Role of Reduced CeO2(110) Surface for CO2 Reduction to CO and Methanol. J. Phys. Chem. C 2016, 120, 16626–16635. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.; Kou, J.; Chen, X.; Tian, Z.; Gao, J.; Yan, S.; Zou, Z. High-Yield Synthesis of Ultralong and Ultrathin Zn2GeO4 Nanoribbons Toward Improved Photocatalytic Reduction of CO2 into Renewable Hydrocarbon Fuel. J. Am. Chem. Soc. 2010, 132, 14385–14387. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Peng, T.; Zhang, X.; Li, K.; Zan, L. Selective methanol production from photocatalytic reduction of CO2 on BiVO4 under visible light irradiation. Catal. Commun. 2012, 28, 38–41. [Google Scholar] [CrossRef]
- Sommers, J.M.; Alderman, N.P.; Viasus, C.J.; Gambarotta, S. Revisiting the behaviour of BiVO4 as a carbon dioxide reduction photo-catalyst. Dalton Trans. 2017, 46, 6404–6408. [Google Scholar] [CrossRef] [PubMed]
- Harb, M.; Cavallo, L.; Basset, J.-M. Designing an active Ta3N5 photocatalyst for H2 and O2 evolution reactions by specific exposed facet engineering: A first-principles study. Phys. Chem. Chem. Phys. 2020, 22, 10295–10304. [Google Scholar] [CrossRef]
- Li, Y.; Takata, T.; Cha, D.; Takanabe, K.; Minegishi, T.; Kubota, J.; Domen, K. Vertically aligned Ta3N5 nanorod arrays for solar-driven photoelectrochemical water splitting. Adv. Mater. 2012, 25, 125–131. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, M.H.; Kim, J.-H.; Kim, C.W.; Youn, D.H. Facile nanocrystalline Ta3N5 synthesis for photocatalytic dye degradation under visible light. Chem. Phys. Lett. 2020, 738, 136900. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, J.; Gong, J. Tantalum-based semiconductors for solar water splitting. Chem. Soc. Rev. 2014, 43, 4395–4422. [Google Scholar] [CrossRef]
- He, Y.; Ma, P.; Zhu, S.; Liu, M.; Dong, Q.; Espano, J.; Yao, X.; Wang, D. Photo-Induced Performance Enhancement of Tantalum Nitride for Solar Water Oxidation. Joule 2017, 1, 831–842. [Google Scholar] [CrossRef]
- Zhang, Q.; Gao, L. Ta3N5 nanoparticles with enhanced photocatalytic efficiency under visible light irradiation. Langmuir 2004, 20, 9821–9827. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.S.K.; Hisatomi, T.; Maeda, K.; Moriya, Y.; Domen, K. Enhanced water oxidation on Ta3N5 Photocatalysts by modification with alkaline metal salts. J. Am. Chem. Soc. 2012, 134, 19993–19996. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R.; Gomez, V.; Bear, J.C.; Rome, B.; Mazzali, F.; McGettrick, J.; Lewis, A.R.; Margadonna, S.; Al-Masry, W.A.; Dunnill, C.W. Active removal of waste dye pollutants using Ta3N5/W18O49 nanocomposite fibres. Sci. Rep. 2017, 7, 4090. [Google Scholar] [CrossRef]
- Jiang, Y.; Jing, X.; Zhu, K.; Peng, Z.Y.; Zhang, J.; Liu, Y.; Zhang, W.; Nia, L.; Liub, Z. Ta3N5 nanoparticles/TiO2 hollow sphere (0D/3D) heterojunction: Facile synthesis and enhanced photocatalytic activities of levofloxacin degradation and H2 evolution. Dalton Trans. 2018, 47, 13113–13125. [Google Scholar] [CrossRef]
- Nguyen, T.D.C.; Nguyen, T.P.L.C.; Mai, H.T.T.; Dao, V.D.; Nguyen, M.P.; Nguyen, V.N. Novel photocatalytic conversion of CO2 by vanadium-doped tantalum nitride for valuable solar fuel production. J. Catal. 2017, 352, 67–74. [Google Scholar] [CrossRef]
- Lu, L.; Wang, S.; Zhou, C.; Shi, Z.; Zhu, H.; Xin, Z.; Wang, X.; Yan, S.; Zoua, Z. Surface chemistry imposes selective reduction of CO2 to CO over Ta3N5/LaTiO2N photocatalyst. J. Mater. Chem. A 2018, 6, 14838–14846. [Google Scholar] [CrossRef]
- Dzade, N.Y.; Roldan, A.; de Leeuw, N.H. Activation and dissociation of CO2 on the (001), (011), and (111) surfaces of mackinawite (FeS): A dispersion-corrected DFT study. J. Chem. Phys. 2015, 143, 094703. [Google Scholar] [CrossRef]
- Yin, W.J.; Krack, M.; Wen, B.; Ma, S.Y.; Liu, L.M. CO2 Capture and Conversion on Rutile TiO2(110) in the Water Environment: Insight by First-Principles Calculations. J. Phys. Chem. Lett. 2015, 6, 2538–2545. [Google Scholar] [CrossRef] [PubMed]
- Santos-Carballal, D.; Roldan, A.; Dzade, N.Y.; de Leeuw, N.H. Reactivity of CO2 on the surfaces of magnetite (Fe3O4), greigite (Fe3S4) and mackinawite (FeS). Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2018, 376, 20170065. [Google Scholar]
- Meng, X.G.; Ouyang, S.X.; Kako, T.; Li, P.; Yu, Q.; Wang, T.; Ye, J.H. Photocatalytic CO2 conversion over alkali modified TiO2 without loading noble metal cocatalyst. Chem. Commun. 2014, 50, 11517–11519. [Google Scholar] [CrossRef] [PubMed]
- Toda, Y.; Hirayama, H.; Kuganathan, N.; Torrisi, A.; Sushko, P.V.; Hosono, H. Activation and splitting of carbon dioxide on the surface of an inorganic electride material. Nat. Commun. 2013, 4, 2378. [Google Scholar] [CrossRef] [PubMed]
- Brese, N.E.; O’Keeffe, M.; Rauch, P.; DiSalvo, F.J. Structure of Ta3N5 at 16 K by time-of-flight neutron diffraction. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1991, 47, 2291. [Google Scholar] [CrossRef]
- Morbec, J.M.; Narkeviciute, I.; Jaramillo, T.F.; Galli, G. Optoelectronic properties of Ta3N5: A joint theoretical and experimental study. Phys. Rev. B 2014, 90, 155204. [Google Scholar] [CrossRef]
- Wang, J.; Luo, W.; Feng, J.; Zhang, L.; Li, Z.; Zou, Z. Theoretical study of water adsorption and dissociation on Ta3N5(100) surfaces. Phys. Chem. Chem. Phys. 2013, 15, 16054–16064. [Google Scholar] [CrossRef]
- Fan, G.; Fang, T.; Wang, X.; Zhu, Y.; Fu, H.; Feng, J.; Li, Z.; Zou, Z. Interfacial Effects on the Band Edges of Ta3N5 Photoanodes in an Aqueous Environment: A Theoretical View. iScience 2019, 13, 432–439. [Google Scholar] [CrossRef]
- Zeinalipour-Yazdi, C.D.; Hargreaves, J.S.J.; Laassiri, S.; Catlow, C.R.A. DFT-D3 study of H2 and N2 chemisorption over cobalt promoted Ta3N5-(100), (010) and (001) surfaces. Phys. Chem. Chem. Phys. 2017, 19, 11968–11974. [Google Scholar] [CrossRef]
- Chun, W.-J.; Ishikawa, A.; Fujisawa, H.; Takata, T.; Kondo, J.N.; Hara, M.; Kawai, M.; Matsumoto, Y.; Domen, K. Conduction and Valence Band Positions of Ta2O5, TaON, and Ta3N5 by UPS and Electrochemical Methods. J. Phys. Chem. B 2003, 107, 1798–1803. [Google Scholar] [CrossRef]
- Pinaud, B.A.; Vailionis, A.; Jaramillo, T.F. Controlling the Structural and Optical Properties of Ta3N5 Films through Nitridation Temperature and the Nature of the Ta Metal. Chem. Mater. 2014, 26, 1576–1582. [Google Scholar] [CrossRef]
- Wang, J.; Feng, J.; Zhang, L.; Li, Z.; Zou, Z. Role of oxygen impurity on the mechanical stability and atomic cohesion of Ta3N5 semiconductor photocatalyst. Phys. Chem. Chem. Phys. 2014, 16, 15375–15380. [Google Scholar] [CrossRef]
- Harb, M.; Basset, J.-M. Insights into the Most Suitable TiO2 Surfaces for Photocatalytic O2 and H2 Evolution Reactions from DFT Calculations. J. Phys. Chem. C 2020, 124, 2472–2480. [Google Scholar] [CrossRef]
- Suzuki, S.; Wagata, H.; Komatsus, M.; Minegishi, T.; Domen, K.; Oishia, S.; Teshima, K. A novel flux coating method for the fabrication of layers of visible-light-responsive Ta3N5 crystals on tantalum substrates. Mater. Chem. A 2015, 3, 13946–13952. [Google Scholar] [CrossRef]
- Freund, H.-J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazáabal, G.O.; Pérez-Ramírez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef]
- Wang, J.; Ma, A.; Li, Z.; Jiang, J.; Feng, J.; Zou, Z. Theoretical study on the surface stabilities, electronic structures and water adsorption behavior of the Ta3N5(110) surface. Phys. Chem. Chem. Phys. 2016, 18, 7938–7945. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Dzade, N.Y.; de Leeuw, N.H. Periodic DFT+U investigation of the bulk and surface properties of marcasite (FeS2). Phys. Chem. Chem. Phys. 2017, 19, 27478–27488. [Google Scholar] [CrossRef] [PubMed]
- Himmetoglu, B.; Floris, A.; de Gironcoli, S.; Cococcioni, M. Hubbard-corrected DFT energy functionals: The LDA+U description of correlated systems. Int. J. Quantum Chem. 2014, 114, 14–49. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Aryasetiawan, F.; Lichtenstein, A.I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: The LDA+ U method. J. Phys. Condens. Matter 1997, 9, 767–808. [Google Scholar] [CrossRef]
- Wang, J.; Fang, T.; Zhang, L.; Feng, J.; Li, Z.; Zou, Z. Effects of oxygen doping on optical band gap and band edge positions of Ta3N5 photocatalyst: A GGA plus U calculation. J. Catal. 2014, 309, 291–299. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B Solid State 1976, 13, 5188. [Google Scholar] [CrossRef]
- Watson, G.W.; Kelsey, E.T.; de Leeuw, N.H.; Harris, D.J.; Parker, S.C. Atomistic simulation of dislocations, surfaces and interfaces in MgO. J. Chem. Soc. Faraday Trans. 1996, 92, 433. [Google Scholar] [CrossRef]
- Tasker, P.W. The stability of ionic crystal surfaces. J. Phys. C Solid State Phys. 1979, 12, 4977. [Google Scholar] [CrossRef]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B Condens. Matter Mater. Phys. 1995, 51, 4014. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 84204. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.; Jónsson, H.; Schenter, G.K. Reversible work transition state theory: Application to dissociative adsorption of hydrogen. Surf. Sci. 1995, 324, 305–337. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Free CO2 | Ta3N5(001) | Ta3N5(010) | Ta3N5(110) |
|---|---|---|---|---|
| Eads (eV) | - | −2.73 | −1.89 | −1.20 |
| Δq(CO2) (|e|) | 0.0 | 0.40 | 0.34 | 0.31 |
| d(C−Oa) (Å) | 1.176 | 1.302 | 1.422 | 1.293 |
| d(C−Ob) (Å) | 1.176 | 1.301 | 1.202 | 1.290 |
| OaCOb (o) | 180.0 | 130.0 | 123.9 | 125.5 |
| d(C−N) (Å) | - | 1.361 | 1.396 | 1.400 |
| d(Oa−Ta) (Å) | - | 2.176 | 2.012 | 2.217 |
| d(Ob−Ta) (Å) | - | 2.180 | - | 2.230 |
| υas (cm−1) | 2373 | 1465 | 1804 | 1718 |
| υs (cm−1) | 1323 | 1265 | 911 | 881 |
| υb (cm−1) | 631 | 819 | 737 | 691 |
| Parameter | Ta3N5(001) | Ta3N5(010) | Ta3N5(110) |
|---|---|---|---|
| Ecoads (eV) | −1.69 | −2.99 | −0.68 |
| Erxn (eV) | 1.04 | −1.09 | 0.52 |
| Eact (eV) | 1.34 | 1.15 | 1.56 |
| d(C−N) (Å) | 1.467/1.388 | 1.237 | 1.242 |
| d(Oa−Ta) (Å) | 1.788 | 2.126/1.979 | 1.788 |
| d(C−Ob) (Å) | 1.227 | 1.173 | 1.170 |
| Parameter | Ta3N5(001) | Ta3N5(010) | Ta3N5(110) | |||
|---|---|---|---|---|---|---|
| State | Molecular | Dissociative | Molecular | Dissociative | Molecular | Dissociative |
| Eads (eV) | −1.42 | −2.16 | −1.07 | −1.35 | −1.08 | −2.12 |
| Erxn (eV) | - | −0.74 | - | −0.28 | - | −1.04 |
| Eact (eV) | 0.18 | - | 0.34 | - | 0.27 | - |
| d(O−Ta) (Å) | 2.307 | 1.987 | 2.395 | 2.297 | 2.295 | 1.978 |
| d(H−N) (Å) | - | 1.031 | - | 1.056 | - | 1.022 |
| d(O−H) (Å) | 0.977/0.977 | 0.969 | 0.980/0.979 | 0.976 | 0.986/1.006 | 0.970 |
| Parameter | Ta3N5(001) | Ta3N5(010) | Ta3N5(110) |
|---|---|---|---|
| Ecoads (eV) | −3.94 | −3.68 | −2.34 |
| Δq(CO2+H2O) (|e|) | 0.40 | 0.35 | 0.31 |
| Δq(H2O) (|e|) | 0.03 | 0.03 | 0.02 |
| d(C−Oa) (Å) | 1.305 | 1.460 | 1.292 |
| d(C−Ob) (Å) | 1.307 | 1.202 | 1.292 |
| OaCOb (o) | 125.6 | 122.1 | 125.5 |
| d(C−N) (Å) | 1.354 | 1.378 | 1.399 |
| d(Oa−Ta) (Å) | 2.187 | 2.045 | 2.231 |
| d(Ob−Ta) (Å) | 2.188 | - | 2.244 |
| d(Ow−H) (Å) | 0.981 | 1.022 | 0.978 |
| d(Ow−Ta) (Å) | 2.299 | 2.226 | 2.315 |
| d( O–H) (Å) | 3.619 | 1.606 | 3.454 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzade, N.Y. CO2 and H2O Coadsorption and Reaction on the Low-Index Surfaces of Tantalum Nitride: A First-Principles DFT-D3 Investigation. Catalysts 2020, 10, 1217. https://doi.org/10.3390/catal10101217
Dzade NY. CO2 and H2O Coadsorption and Reaction on the Low-Index Surfaces of Tantalum Nitride: A First-Principles DFT-D3 Investigation. Catalysts. 2020; 10(10):1217. https://doi.org/10.3390/catal10101217
Chicago/Turabian StyleDzade, Nelson Y. 2020. "CO2 and H2O Coadsorption and Reaction on the Low-Index Surfaces of Tantalum Nitride: A First-Principles DFT-D3 Investigation" Catalysts 10, no. 10: 1217. https://doi.org/10.3390/catal10101217
APA StyleDzade, N. Y. (2020). CO2 and H2O Coadsorption and Reaction on the Low-Index Surfaces of Tantalum Nitride: A First-Principles DFT-D3 Investigation. Catalysts, 10(10), 1217. https://doi.org/10.3390/catal10101217