1. Introduction
The catalytic potential of gold in its various oxidation states has clearly emerged in the course of the past decades, and it is nowadays evident that this metal, long considered useless for catalytic purposes, can provide indeed unsurpassed catalytic performances in several chemical reactions of technological interest [
1]. Whereas gold metal nanoparticles are mostly employed in oxidation processes [
2], gold(I) and, to a lesser extent, gold(III) species have a clear attitude to activate unsaturated organic substrates, such as alkenes, alkynes, allenes, arenes, and heteroarenes, for reactions that mostly involve (cyclo)additions or the introduction of functional groups in the molecule [
3,
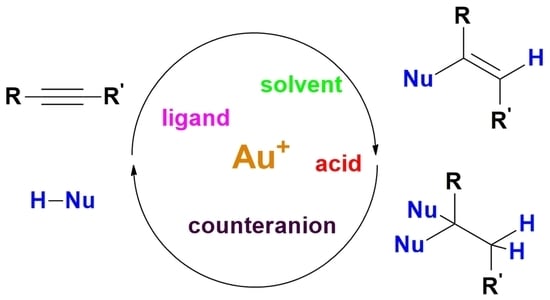
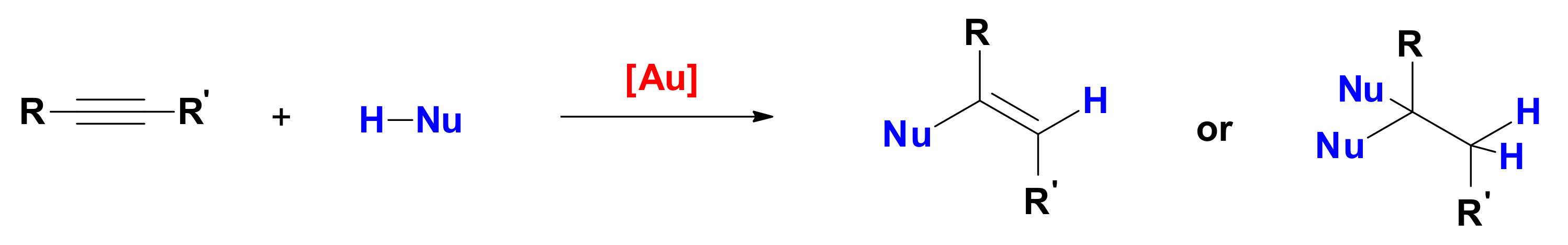
4]. Particularly well investigated are alkyne hydrofunctionalization reactions, in which a nucleophile HNu attacks an alkyne and produces a functionalized alkene (
Scheme 1) [
5,
6,
7,
8]; the resulting product can be stable under reaction conditions, it can undergo subsequent tautomerization (not shown), or it can react with a second molecule of HNu to produce saturated, difunctionalized molecules.
Reactions of this kind can also produce more complex products if the gold-vinyl intermediates that form in the course of the reaction (see below) are intercepted or undergo further evolution before the actual hydrofunctionalization product is liberated [
8,
9,
10]. This interesting possibility is however available mostly in the case of more complex substrates, in which these evolution reactions generally take place by involving other functional group present in the same intermediate (i.e., in an intramolecular fashion); alternatively, such a possibility is provided also by the use of unprotonated nucleophiles with oxidizing character [
9,
10]. Since this review article is specifically focused on the factors affecting the mechanism and rates of intermolecular alkyne hydrofuncionalizations, further reactions of the type outlined above will not be specifically covered herein.
The disclosure of gold-catalyzed alkyne hydrofunctionalization reactions dates back to the end of the 1990s, and in the course of the last 20 years the field has evolved considerably: novel potentially useful, gold-based catalytic systems are continuously being proposed in the literature, new synthetic applications are being devised and, most notably, several mechanistic studies have been performed to highlight the essential features of these processes. However, a unifying view on this reaction class is still lacking, as the mechanistic features of these reaction, which as will be highlighted below are heavily dependent on the kind of ligand linked to gold, on its counteranion and on the solvent, are also obviously a function of the nature of the substrates and particularly of the incoming nucleophile HNu. Consequently, we deem it useful to provide a comprehensive picture of the state-of-the-art knowledge of the mechanistic features of these reaction, which could provide advice to the interested reader in the choice of a peculiar catalytic system and conditions for a given reaction.
We will concern ourselves only with intermolecular alkyne hydrofunctionalization reactions, since they are more challenging and general compared to intramolecular ones. Reference to selected intramolecular processes will be given when appropriate. Furthermore, we do not aim at providing a comprehensive coverage of the gold-based catalytic systems proposed to date for these reactions, but rather to focus on the contributions that shed light on the mechanistic features of these reactions. For a more general coverage of hydrofunctionalization reactions, catalyzed by gold but also by other metals, the interested reader is referred to several other dedicated reviews that have recently appeared in the literature [
5,
6,
7,
8,
9,
10,
11].
2. General Reaction Mechanism
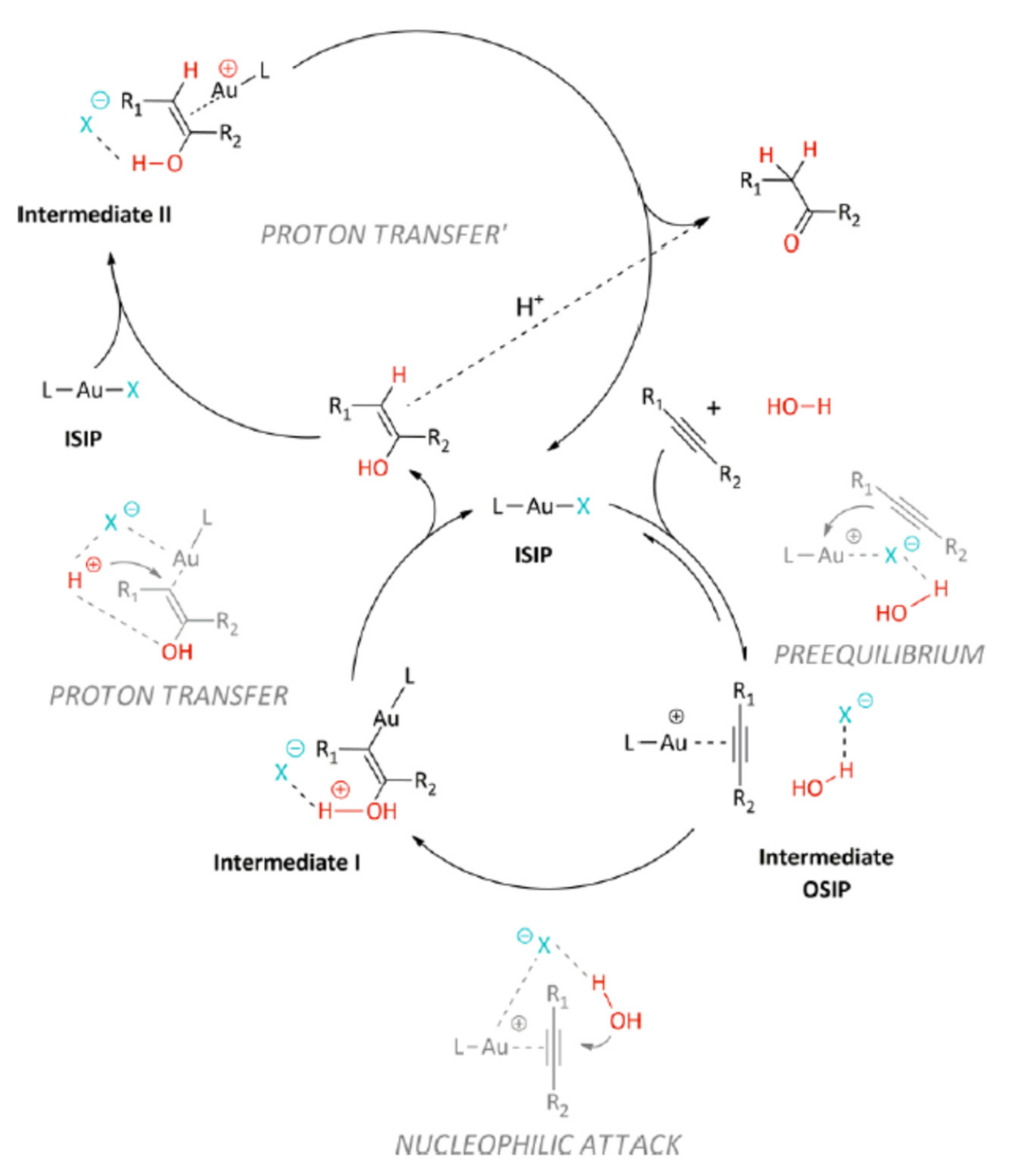
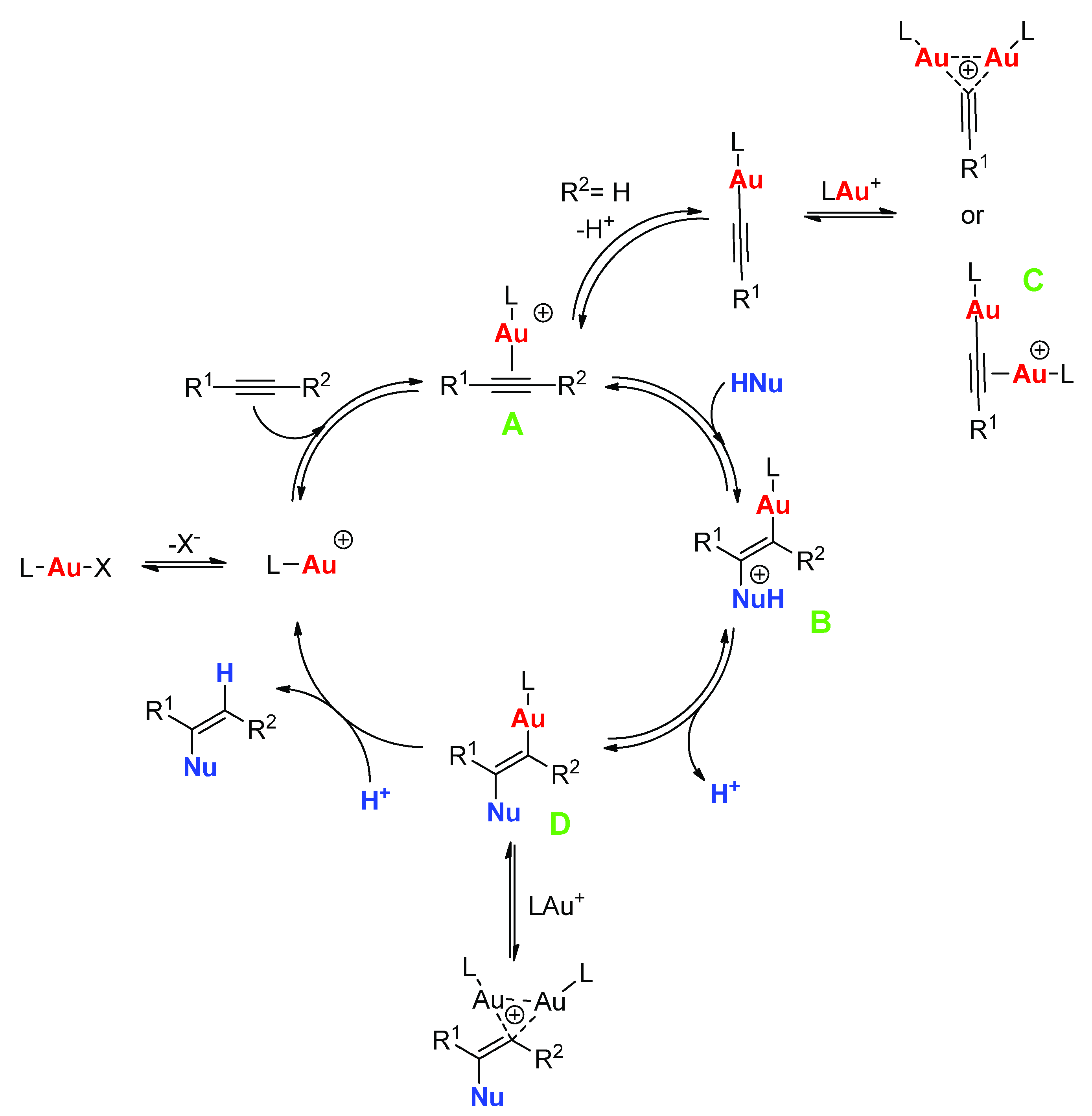
By now there is a quite generally accepted reaction mechanism for gold-catalyzed alkyne hydrofunctionalization reactions. In its essential features, the mechanism involves a cationic gold center (e.g., LAu
+ for a gold(I) catalyst) stabilized by a weakly coordinated inner sphere anion as catalytically competent species, and it follows a sequence based on alkyne activation bycoordination to gold, external nucleophilic attack of HNu providing a gold-vinyl intermediate, proton transfer from HNu to the vinyl carbon linked to gold with consequent protonolysis and liberation of the product of formal
anti-addition of HNu to the alkyne (
Scheme 2). An internal nucleophilic attack of a gold-precoordinated HNu has been sometimes proposed as the main alternative to this mechanism (see also below). Very recent computational analyses support this alternative mechanism in the case of the hydration of internal alkynes [
12]; however, to the best of our knowledge this proposal still awaits experimental verification.
This apparently rather simple mechanism involves however several peculiar features that need to be highlighted:
- (1)
Formation of the-complex A between the cationic gold center and the alkyne is in competition with the interaction of the gold center with its counteranion, and to some extent also with the solvent and any other potentially present Lewis basic additive. This limitation can be obviously overcome by using poorly coordinating solvents and counteranions; however, this often hampers the solubility and especially the stability of the gold complex: under these conditions, gold is reduced with relative ease to the zerovalent state, and the ligand L that is liberated coordinates to the remaining cationic gold complex forming catalytically inactive L2Au+ species and ultimately resulting in complete catalyst deactivation.
- (2)
Once that the complex
A is formed, it can transform into a coordinated alkynyl complex with concomitant liberation of a proton, if the employed alkyne is terminal. The ease of formation of such species, which is a quite common reaction, particularly with group 10 and 11 metal centers [
13], depends on the acidity of the alkyne substrate and on the presence of suitable proton acceptors in the solution. Although expectedly reversible, such a reaction obviously subtracts catalytically active species. Further to this, it is by now well-known that the resulting gold alkynyl can associate with another cationic gold center to produce a dinuclear, complex
C, which is sometimes alternatively viewed as a (2e, 3c)-
gem-dicoordinated alkynyl stabilized by an aurophilic interaction between the two gold centers [
14,
15]. These dinuclear complexes have been found to be much more reluctant than mononuclear gold alkynyls to undergo protonolysis [
15]; nevertheless, there has been considerable debate in the course of the last years on their possible involvement as catalytically competent species in hydrofunctionalizations and other gold-catalyzed reactions [
16,
17]. While their involvement has been disproved in some cases [
18], in which it has been demonstrated that these complexes rather act as catalyst reservoirs for the true catalytically competent species (the cationic mononuclear complex), the question remains open in some others [
19,
20]. Formation of these dinuclear complexes has been also considered to ultimately favor catalyst decomposition with the formation of Au metal [
21]; this can apparently be avoided by using bulky ligands at gold that stabilize the dinuclear complexes or even prevent their formation.
- (3)
After nucleophilic attack, generally taking place at the more substituted carbon atom of the triple bond, a proton needs to be removed from the original nucleophile in intermediate
B and transferred to the gold-bound carbon of the resulting vinyl group in intermediate
D, determining protonolysis of the Au-C bond and liberation of the product. This proton transfer is generally believed to be not concerted, and consequently it has been represented as two separate steps in
Scheme 2. Proton transfer can be facilitated by the solvent, by the counteranion, or by other species with basic character present in solution; all these possible contributions need to be taken into account. Furthermore, the Au-vinyl intermediate
D can evolve differently: for example, it can coordinate an additional cationic gold center forming a
gem-dicoordinated vinyl with variable electron distribution and reactivity [
22].
- (4)
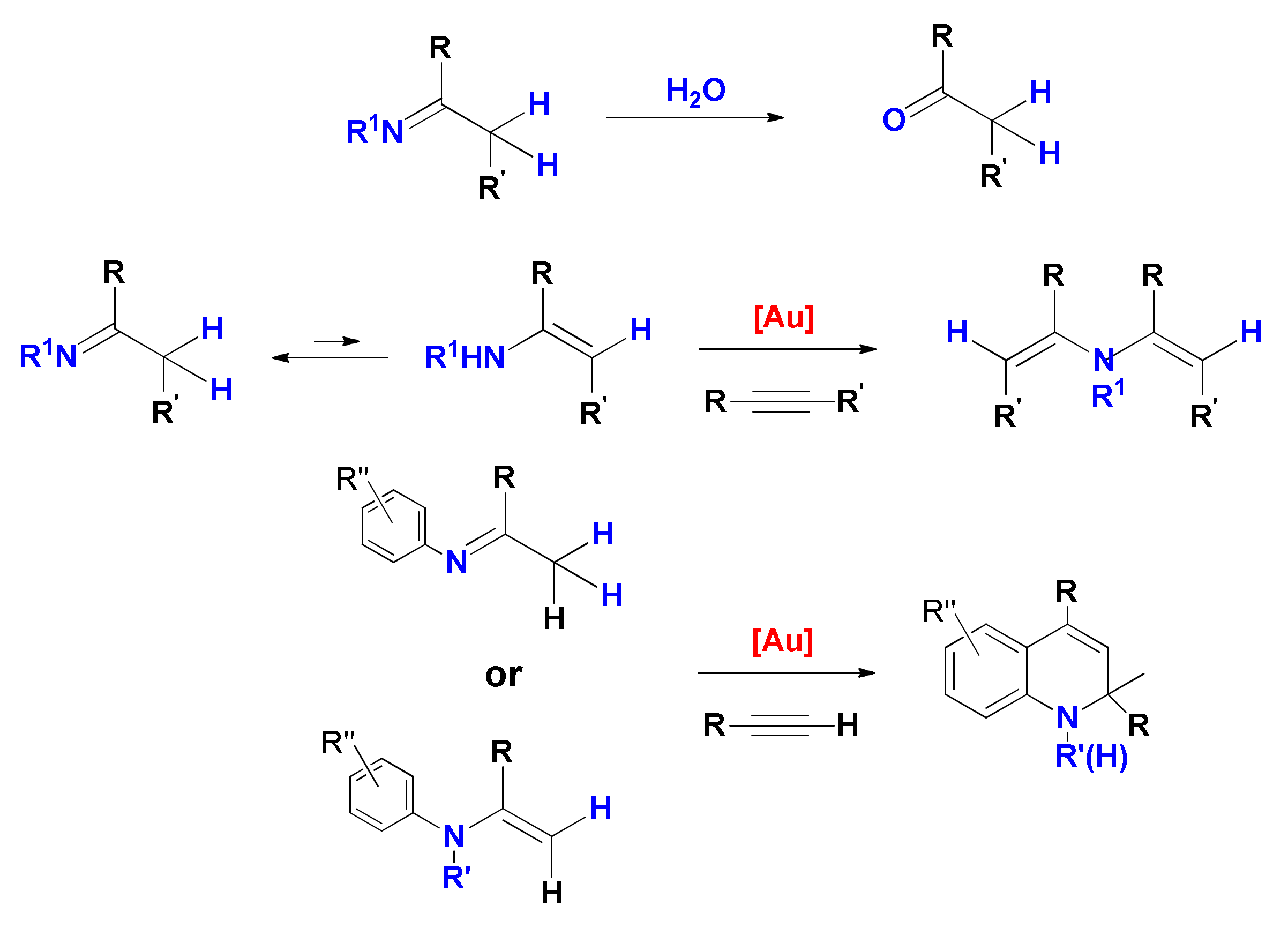
Finally, it must be borne in mind that the reaction product can further evolve after it has been formed within the catalytic cycle, and such evolution may or may not involve the gold catalyst. For example, as it will be discussed in more detail in the subsequent sections, when products of double hydrofunctionalization are formed (
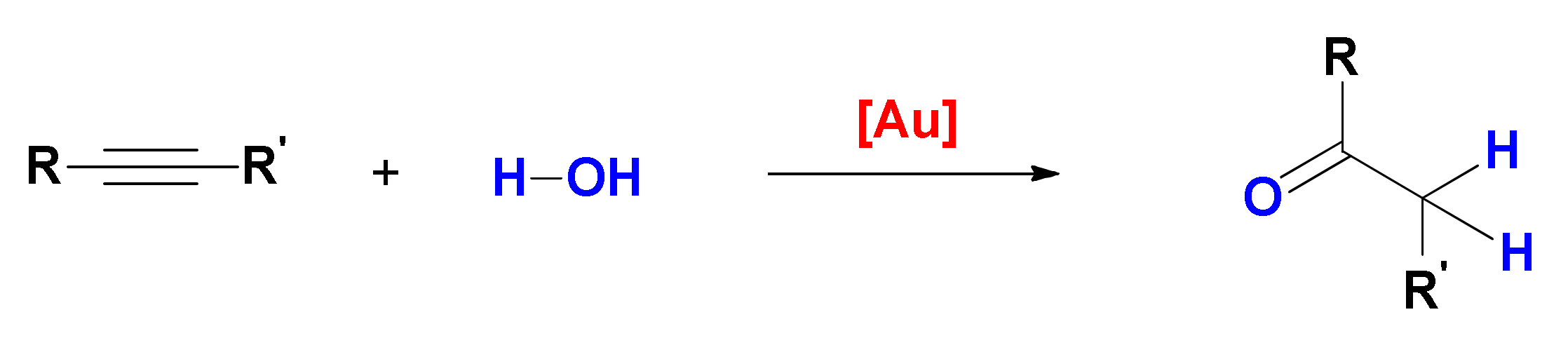
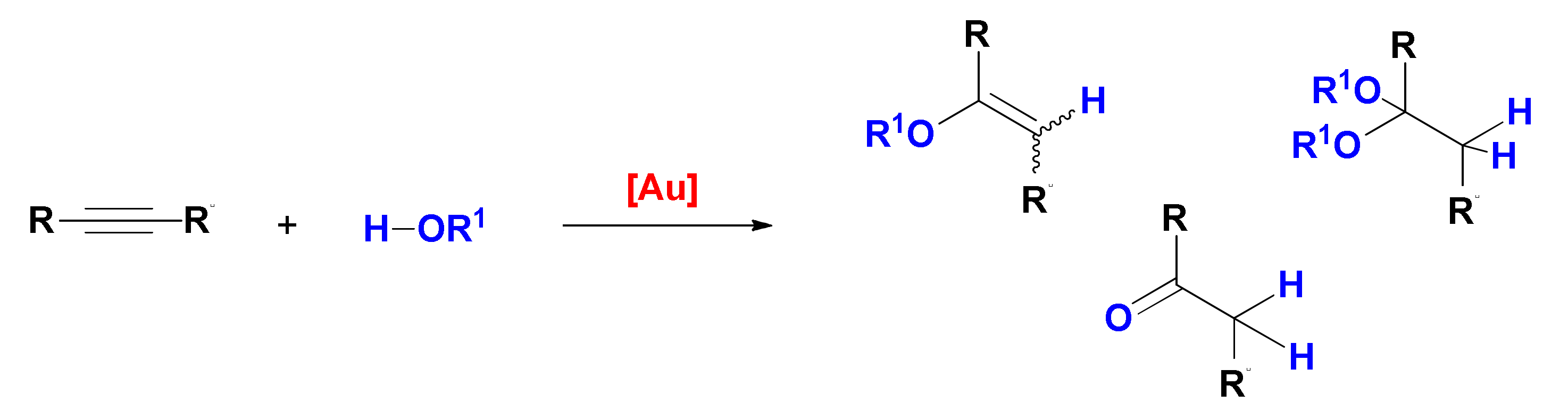
Scheme 1) the two hydrofunctionalization reactions are consecutive and the second one may be promoted by traces of acid present in the system instead of by the gold catalyst. Furthermore, the imine product of hydroamination or the enolether/ketal product of single/double hydroalkoxylation can undergo prompt hydrolysis by traces of water, leading to the corresponding carbonyl compound, i.e., the product of formal hydration of the triple bond.
In the subsequent sections, we will discuss the main classes of hydrofunctionalization reactions according to the kind of employed nucleophile, and we will focus our attention on the current degree of understanding of the mechanistic features of each reaction, basing on the general mechanism listed above. Some general trends regarding the effect of the various reaction components and conditions can be however anticipated here.
Effect of the nucleophile. The hydrofunctionalization reaction can be potentially run with a wide range of nucleophiles. However, it should be borne in mind that the nucleophile is a Lewis base and can compete with the alkyne for coordination to gold, causing catalyst deactivation. Consequently, nucleophiles of intermediate to low Lewis basicity are conventionally employed. The nucleophilic character of this reagent obviously influences the rate of step A, which consequently has a greater chance of being the rate-determining step of the reaction, the less nucleophilic is the employed reagent.
Effect of the alkyne. The nature of the alkyne has also a dramatic importance for the overall efficiency of the hydrofunctionalizaion process. First of all, as already outlined above, π-coordinated terminal alkynes can transform into coordinated alkynyls, thereupon contributing to catalyst deactivation. However, π-coordinated terminal alkynes also undergo nucleophilic attack more quickly and form more stable vinyl intermediates [
12]. Sterically more encumbered alkynes can experience difficulties in coordinating to gold, with consequent slower, less effective reaction. Most important are however electronic effects provided by the alkyne substituents. Electron-poor alkynes are activated more easily toward external nucleophilic attack by coordination to the gold center, whereas electron-rich ones are less easily attacked. Electronic effects also govern the regioselectivity of the gold-catalyzed hydrofunctionalization process: the nucleophile attacks at the coordinated carbon atom which stabilizes best the cationic charge on intermediate
A. As it will be shown below, the presence of directing functional groups on the alkyne substituents can however determine alternative regioselectivities.
Effect of the ligand. Both steric and electronic factors come into play when considering the effect of the neutral ligand L at gold. Sterically encumbered ligands are generally employed, which impart stabilization to the gold complex toward decomposition and also prevent the formation of dinuclear gold complexes, which as outlined above often decrease the reactivity of the catalytic system. The effect of ligand electronic factors is instead ambivalent. As already pointed out by Xu and Hammond [
23], they influence in an opposite way the nucleophilic attack step and the protonolysis step: the former is facilitated by electron-withdrawing ligands whereas the latter by electron-donating ligands. While in several cases protonolysis is the rate-determining step of the reaction, in some instances, in which weak nucleophiles or internal/electron-rich alkynes are employed, nucleophilic attack can take over as the rate-determining step. Consequently, the effect of a change in electronic properties of the ligand might be different, and it can even result in a switch of the rate-determining step of the reaction.
Effect of the counteranion. Counteranion effects in gold catalysis have been very intensively investigated in recent years [
24]. In the context of alkyne hydrofunctionalization reactions, the Lewis basicity of the inner sphere anion is obviously an important factor, since more coordinating anions tend to stick to the gold center and to deactivate the catalyst; on the other hand, a very poorly coordinating anion leads to an unstable gold complex, which will tend to decompose in solution unless reaction conditions are provided to slow down decomposition. The Brønsted basicity of the anionic ligand may also become important, since it can aid the proton transfer leading to the final product after nucleophilic attack; an anion that is a too strong Brønsted base will however sequester the proton and block the protonolysis step [
24,
25]. Finally, the hydrogen bond acceptor character of the anion may also come into play, since the anion can activate HNu for nucleophilic attack to the coordinated alkyne by establishing a hydrogen bond with it; the effectiveness of such an activating effect is however strongly dependent on the mutual position and orientation of the counteranion with respect to the cationic gold-alkyne intermediate complex
A [
25].
Effect of gold oxidation state. Although most of the research work on gold-catalyzed alkyne hydrofunctionalization reactions has been carried out on gold(I) complexes, some studies have been performed with gold(III) compounds as well. Care should be taken, though, in evaluating the results of these studies, since occasionally gold(III) complexes have been employed that can easily transform in gold(I) compounds by reductive elimination under reaction conditions [
17]; consequently, in these cases there is the legitimate doubt that the gold(III) complex merely acts as a precatalyst for a catalytically competent gold(I) species. In cases in which the gold(III) complex is sufficiently stable, the gold center is clearly expected to be more electrophilic than gold(I); consequently, activation of the alkyne towards nucleophilic attack upon coordination to gold should be enhanced and result in a more efficient reaction in cases in which this is the rate-determining step. On the other hand, the higher electronegativity of gold(III) also renders the gold-vinyl bond in intermediate
D less polarized towards the carbon atom and consequently less prone to undergo protonolysis; this reaction step becomes therefore more easily rate-determining and often needs to be aided by using acids as additives.
Finally, it needs to be anticipated at this point that beside gold(I) and gold(III) complexes, also gold(0) in the form of (supported) gold clusters or nanoparticles has been employed as catalyst and displays activity, albeit at high temperature and generally in the presence of acid cocatalysts, in some hydrofunctionalization reactions (hydration, hydroamination, hydrochlorination) [
26,
27,
28,
29,
30]. Only in one case, though, use of gold metal as a catalyst has been paralleled by a detailed mechanistic study demonstrating a heterogeneous reaction catalyzed by the gold surface [
30]; in the remaining cases, it remains unclear whether the reaction is truly catalyzed by the metal surface or by some form of oxidized gold species, leached into solution or anchored at the external surface of the gold nanoparticle, which operates through a more traditional organometallic mechanism.
4. Hydroalkoxylation
First reported in the late nineties [
49], gold-catalyzed alkyne hydroalkoxylation is a 100% atom-economic reaction, that can be used to prepare vinyl ethers (addition of one alcohol molecule) and acetals/ketals (addition of two alcohol molecules,
Scheme 5); whereas the former are stable compounds that can be obtained stereoselectively, the latter undergo rapid hydrolysis in the presence of water, yielding the ketone product of formal hydration of the alkyne [
50]. It is however important to remark that whereas the process that affords the vinyl ether is a gold-catalyzed reaction, the following addition of a second alcohol and the obtainment of the ketal or of its hydrolysis product are classical proton-catalyzed processes. As outlined in the preceding section, intentional addition of water to this reaction system is also a viable strategy to form products of formal hydration of the triple bond.
The chemoselectivity of the reaction is driven primarily by thermodynamics, namely by the relative stability of the mono-hydroalkoxylation product, the vinyl ether, with respect to that of the acetal/ketal formed after the addition of a second alcohol molecule. In this regard, some general trends can be found in the literature. For example, by increasing the steric bulk of the alcohol (e.g., using phenol instead of methanol) the selectivity is shifted toward the one-to-one addition and the obtainment of the vinyl ethers [
51]. The same can be stated for the alkyne substrate: bulky substituents disfavor the addition of a second alcohol molecule [
51]. In the reaction between diphenylacetylene and phenol, usually only the vinyl ether is observed [
52]; conversely, in the reaction between methanol and 3-hexyne the isolation of the vinyl ether is not possible, and formation of the ketal or, in the presence of water, of the hydration product is always observed [
48].
Reaction conditions are also crucial in determining the selectivity of the system, and by playing with them it is possible to drive the reaction toward the desired product: for example, use of the alcohol as solvent (neat conditions) pushes the reaction to full alkyne conversion, but inevitably favors the formation of the ketal and of the related hydration product. The reaction time is also very important: the ratio between the vinyl ether and the ketal products obviously decreases by prolonging the reaction time, in particular if the alcohol is used in excess [
51].
The gold-catalyzed intermolecular hydroalkoxylation of alkynes is very challenging from the regioselectivity point of view [
32]. High selectivity is usually obtained only with terminal alkynes. In that case, the addition of the OR fragment is usually observed at the internal carbon atom (Markovnikov regioselectivity) [
49]. There are however exceptions. For example, ethyl propiolate reacts affording mainly (but not fully) functionalization in the terminal position showing the typical reactivity of α-β unsaturated carbonyl compounds [
51]. On the other hand, controlling the regiochemistry in the case of internal alkynes is more difficult, although several examples of regioselective reaction have been reported so far. Again, steric effects play an important role: when reacting an internal alkyne bearing alkyl substituents of different steric bulkiness, as for example a methyl and an isopropyl substituent, the nucleophilic attack occurs preferentially at the less encumbered position (the methyl substituted one) [
53]. Electronic effects are fundamental as well: if the internal alkyne presents an alkyl and an aromatic substituent the selectivity will be toward the nucleophilic attack at the position bearing the aromatic substituent in most cases [
53]. Finally, the employed catalyst can also influence the regiochemistry of the reaction. There is in fact at least one example in which by changing the catalyst it is possible to reverse the regioselectivity: the
p-nitrophenoxylation of 1-phenyl-1-propyne. Soho et al. reported that this reaction, when catalyzed by in situ formed gold(III) complex [AuCl
3L] (L = phosphine), affords selectively the product in which the nucleophilic attack occurs at the phenyl bearing position [
53]. On the contrary, Nolan et al. reported that by using as catalyst the dinuclear complex [{Au(IPr)}
2(
μ-OH)][BF
4] the selectivity is reversed [
54]. The authors however did not further rationalize this opposite selectivity.
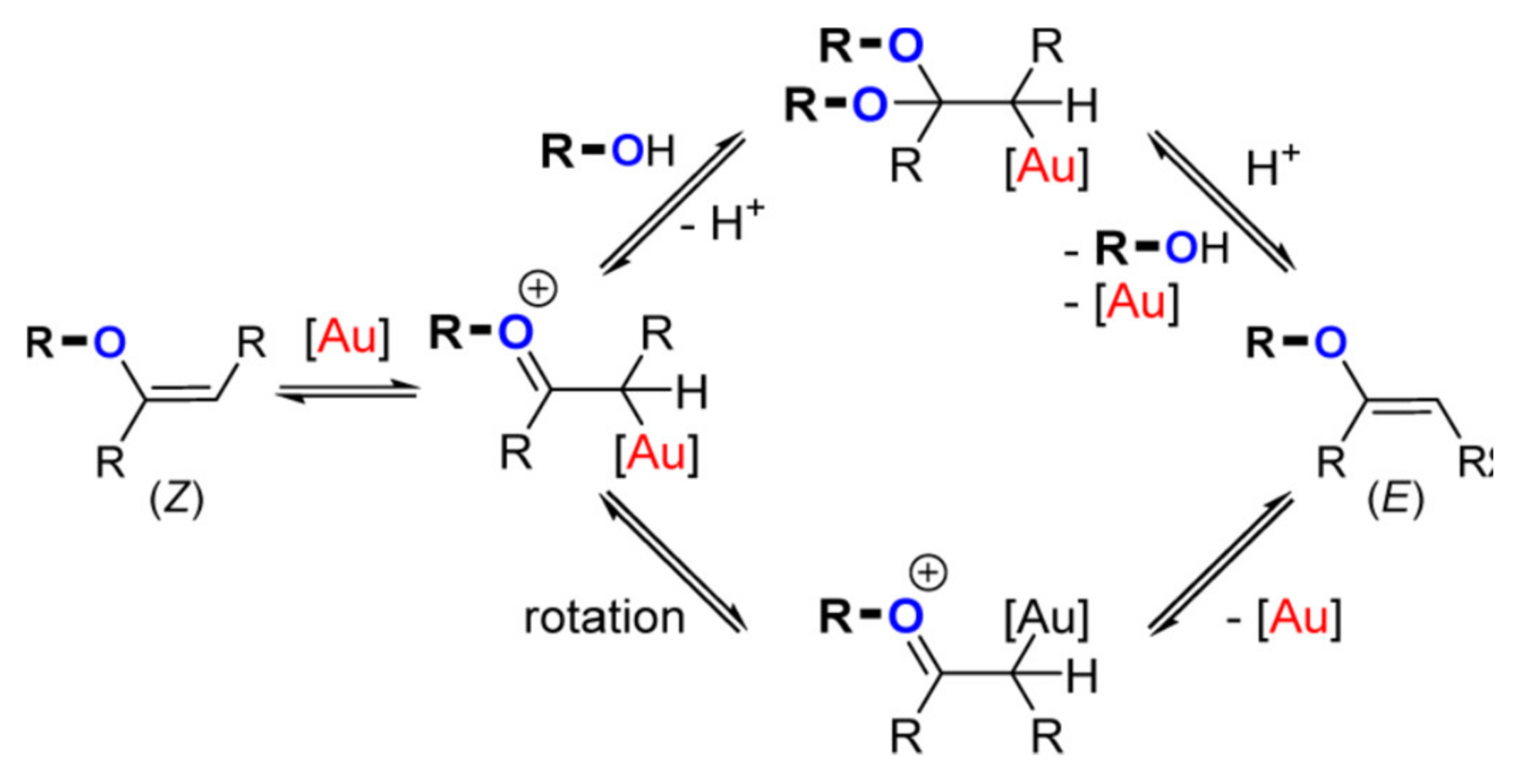
The gold-catalyzed hydroalkoxylation of alkynes usually proceed with good stereoselectivity [
55,
56]. A seminal work in this field was reported by Corma and coworkers [
55]: in particular, they showed that for this gold-catalyzed process the
Z isomer is formed first via a
trans-hydroalkoxylation mechanism. Successively, the
E isomer can form through a gold-catalyzed isomerization process that can proceed alternatively through the formation of an acetal or an oxonium intermediate (
Scheme 6). More recently, Nolan et al. demonstrated that the isomerization predominantly proceeds through the oxonium route [
56]. Stereoselectivity has thus been reported to be substrate dependent, because different substrates may have a different tendency to undergo the isomerization process, e.g., methyl vinyl ethers isomerize more readily than benzyl vinyl ethers [
56]. Moreover, also the choice of the catalyst is crucial, because different complexes may have a different impact on the isomerization process. In this frame, as reported by Nolan, the mononuclear complexes [IPrAu(CH
3CN)][BF
4] tends to deliver in general a lower stereoselectivity compared to the dinuclear complex [{Au(IPr)}
2(
μ-OH)][BF
4] under the same reaction conditions [
56].
As in the case of the hydration reaction discussed in the previous section, the most common active form of the catalyst is a cationic gold(I) complex of general formula [AuL]
+, in which L is an ancillary ligand capable of stabilizing the gold(I) center, usually a phosphine or an NHC donor. The active catalyst is usually prepared in situ, using the same strategies outlined in the preceding section. The reaction mechanism for the gold-catalyzed hydroalkoxylation reaction has been elucidated by Zhdanko and Mayer in a seminal work [
57], and conforms to the general mechanism reported in
Scheme 2, with the same peculiarities and pitfalls described in
Section 2 and
Section 3. Indeed, the dependence of the catalytic performance of a gold(I) catalyst on the nature of the ligand is substantially the same as previously illustrated for the hydration reaction. On the other hand, the effect of the counteranion has been elucidated in greater detail and is slightly different compared to hydration, as it will be apparent in the following.
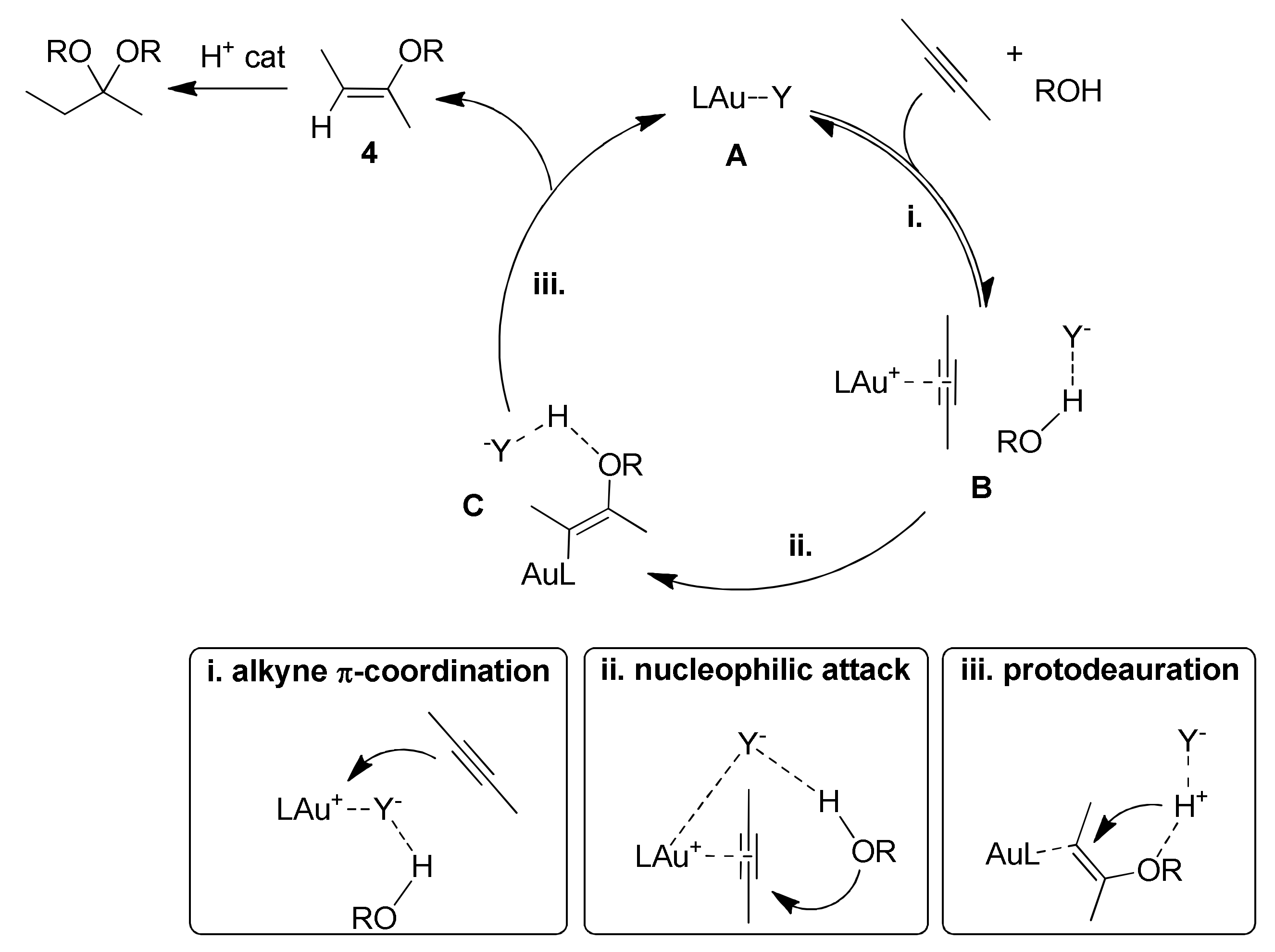
4.1. Effect of the Counteranion
The anion effect in the hydroalkoxylation reaction has been extensively studied by Zuccaccia and coworkers [
58,
59]. As in the case of the alkyne hydration reaction, the anion present in the system plays an important role at different stages of the catalytic cycle (
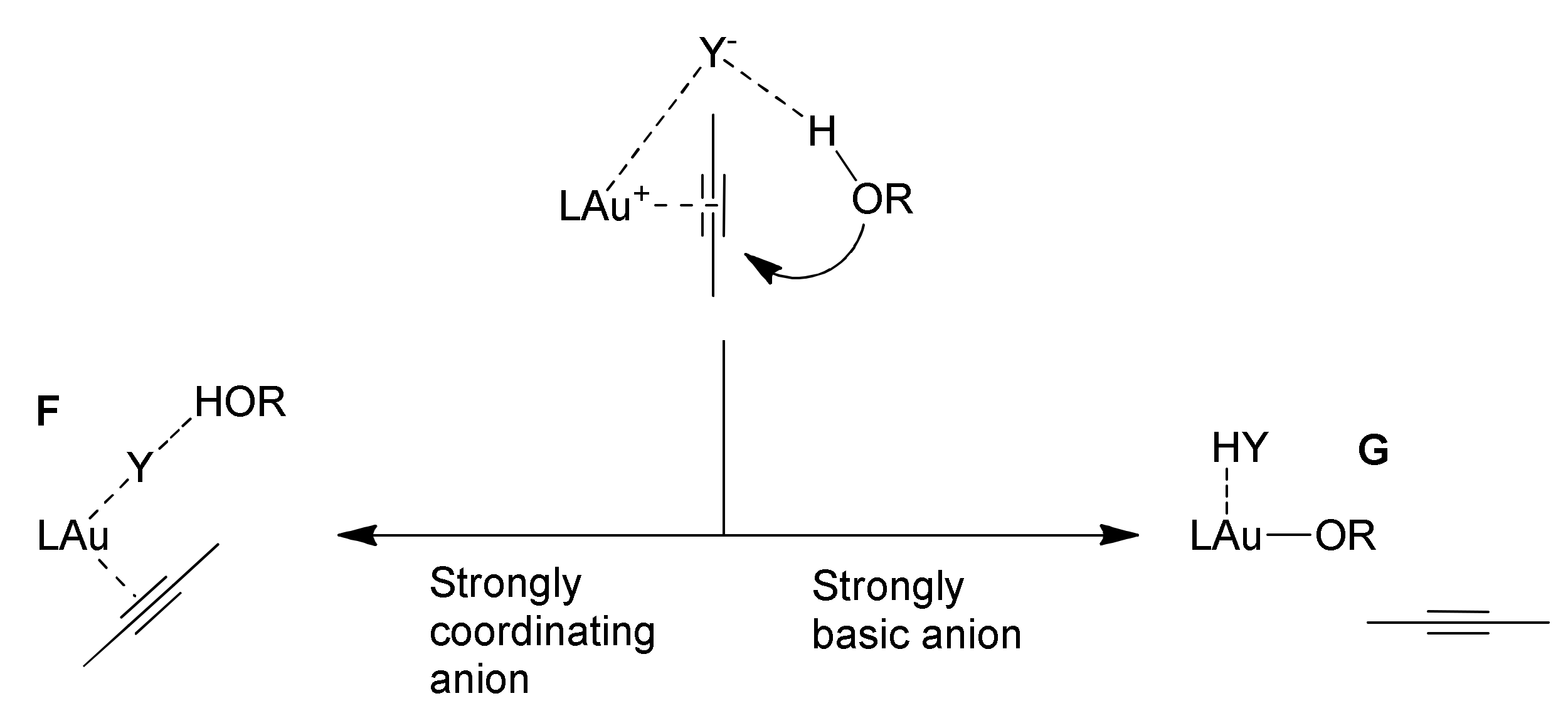
Scheme 7): in the formation of the π-coordinated gold(I) complex upon alkyne coordination (i), in the alcohol nucleophilic attack (ii) and finally in the protodeauration step (iii). In particular, the anion properties that can affect the catalysis are its basicity and its coordinating ability.
By means of experimental and theoretical studies the role of the anion in the three mechanistic steps depicted in
Scheme 7 has been well described. In the alkyne π-coordination pre-equilibrium step (i), the anion can be detrimental to the process in two ways, if it has strong coordinating or basic properties (
Scheme 8) [
58].
A strongly coordinating anion can form the tricoordinated species F, that prevents the proper alkyne π-coordination and activation. Otherwise, a strongly basic anion can generate the alkoxide species G upon deprotonation of a coordinated alcohol. The resulting coordinated alkoxide inhibits again alkyne π-coordination and activation.
The alcohol nucleophilic attack step (ii) is anion-assisted through the formation of a hydrogen bond with the nucleophile. Here the anion acts as a templating agent, by keeping the alcohol in the proper spatial orientation, and also as a hydrogen bonding acceptor, enhancing alcohol nucleophilicity. Finally, in the protodeauration step (iii) the anion works as proton shuttle for the proton transfer. In this step however it has to be remarked that the ligand present in the system also plays a crucial role. In fact, strongly electron donating ligands favor the Au-C bond breaking in intermediate
C (
Scheme 7) [
60].
Zuccaccia et al. have studied in detail the mechanism reported in
Scheme 7, by focusing on the methoxylation of 3-hexyne at 30 °C in CDCl
3. Conversion versus time obtained by using [LAu]
+ catalyst (L = 1,3-dimethylimidazol-2-ylidene) and different anions (
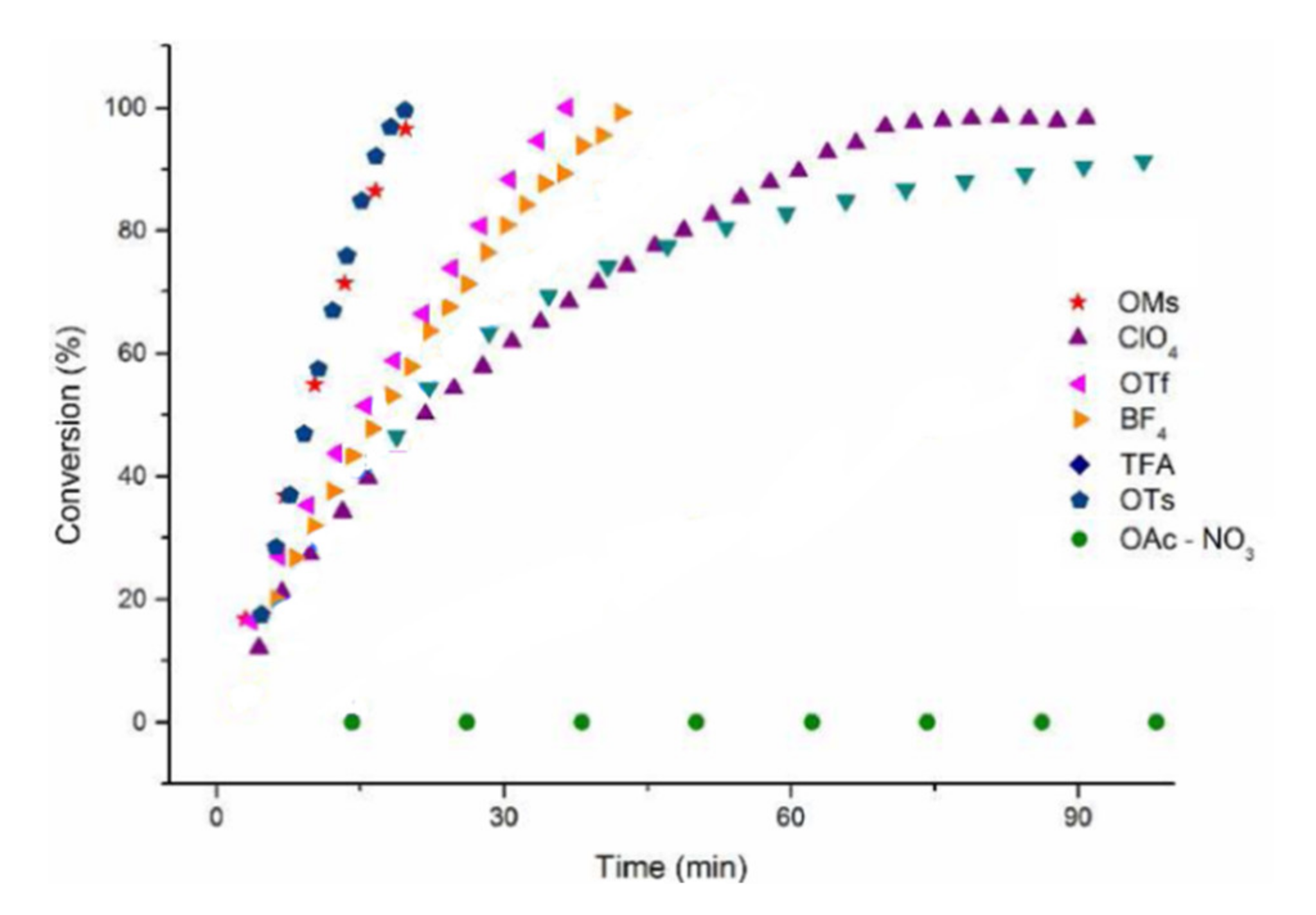
Scheme 9) are reported in
Figure 2 [
58].
Some of the data obtained by Zuccaccia in the catalytic evaluation of the anions are reported in
Table 1 [
58]; the bond dissociation energy (BDE) for the reaction LAuX = [LAu]
+ + [X]
− is reported as well (L = 1,3-dimethylimidazol-2-ylidene). Anion coordinating ability is estimated by the mentioned BDE and increases in the sequence BF
4− < ClO
4− < OTf
− < OTs
− < OMs
− < NO
3− < TFA
− < OAc
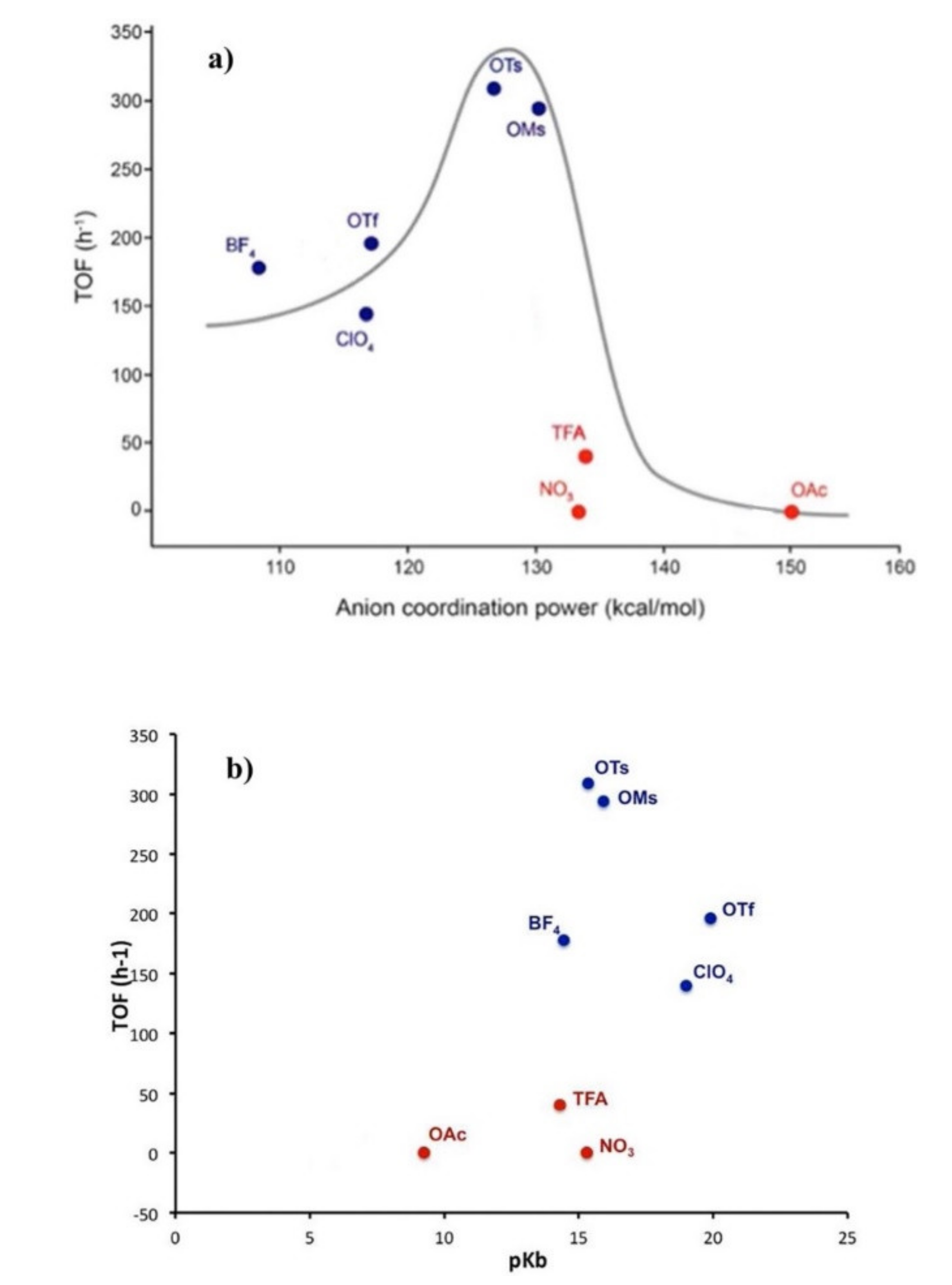
−. An interesting picture emerges when TOF values are plotted against anion BDE and pK
B (
Figure 3): clearly, the anions delivering the best performances are OTs
− and OMs
−, and the authors attribute this to their balanced coordinating and hydrogen bonding accepting ability.
The BF
4− anion has a weak coordinating ability, but having intermediate basic properties, affords anyway moderate catalytic results. OTf
− and ClO
4− are the anions with the lowest pK
B value in the series, but having a comparatively lower BDE value afford quite good catalysts, particularly in the case of OTf
−. NO
3− and TFA
− have a much lower activity compared to OTs
− even though they have a similar BDE value; even considering their pK
B value it is not possible to account for their poor activity, considering that they also have an intermediate pK
B value between the best anions in the series (OTs
− and OMs
−). In this case the authors invoked the different molecular shape. In fact, the most active anions present a spherical shape (OTs
− and OMs
−), whereas those having a planar shape (NO
3− and TFA
−) present lower catalytic activity. A different shape of the counteranion may result in a different location and orientation of the counteranion relative to the cationic gold complex in the ion pair responsible for catalysis, and consequently in a different capability of the counteranion to actively promote the process [
61]. The total inactivity of OAc
− is explained by its high coordination ability toward the gold(I) species [LAu]
+, consequently preventing alkyne coordination and activation; moreover, this anion is also the one with the lowest pK
B value.
It is quite remarkable that the results of the studies by Zuccaccia et al. point at OTs
− and OMs
− as the anions yielding the most efficient catalytic systems, whereas in the mechanistically strictly related hydration reaction discussed in the preceding section less coordinating anions such as OTf
− appear optimum. To add even more complexity to this mechanistic puzzle, it has been demonstrated that the anion effect is ligand dependent [
62]. In a study carried out by Zuccaccia et al. on the methoxylation of 3-hexyne at 30 °C in CDCl
3, the anion effect has been compared to different gold(I) catalysts bearing different ancillary ligands: triphenylphosphine (PPh
3), tritertbutylphosphine (P(t-Bu)
3), tri(3,5-bis(trifluoromethyl)phenyl)-phosphine (PArF) and 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr). It is interesting to note that the catalytic efficiency of the system depends on the ligand-anion combination. Complexes with phosphine-based ligands show the same trend by changing the anion: OTf
− anion turned out to be the best in this case. Differently, in the case of the NHC ligand IPr, the OTs
− anion shows the best activity (
Figure 4). Here again, effects related to the location and orientation of the counteranion with varying ancillary ligand can come into play and justify the different observed dependence. Finally, the anion effect is also solvent dependent: as demonstrated by Zhdanko and Mayer, by increasing the polarity of the solvent, the impact of the anion effect on the catalysis is reduced [
63].
4.2. Dual Gold Catalysis
An interesting feature of homogeneous gold catalysis is the possibility of having a double substrate activation. The involvement of two gold centers in a single catalytic cycle is known as dual gold catalysis [
64,
65,
66,
67].
As illustrated in
Section 2, formation of
gem-diaurated species or of a dinuclear σ,π-alkynyl complex in the course of gold-catalyzed reactions with alkynes is quite common (
Scheme 2). In particular,
gem-diaurated vinyls have been identified as relevant gold-containing species in hydroalkoxylation reaction mixtures [
68]. Their accumulation is however considered detrimental in the case of alkyne hydroalkoxylation reactions, since these species are not involved in the main catalytic cycle and undergo slow protonolysis to liberate the reaction product [
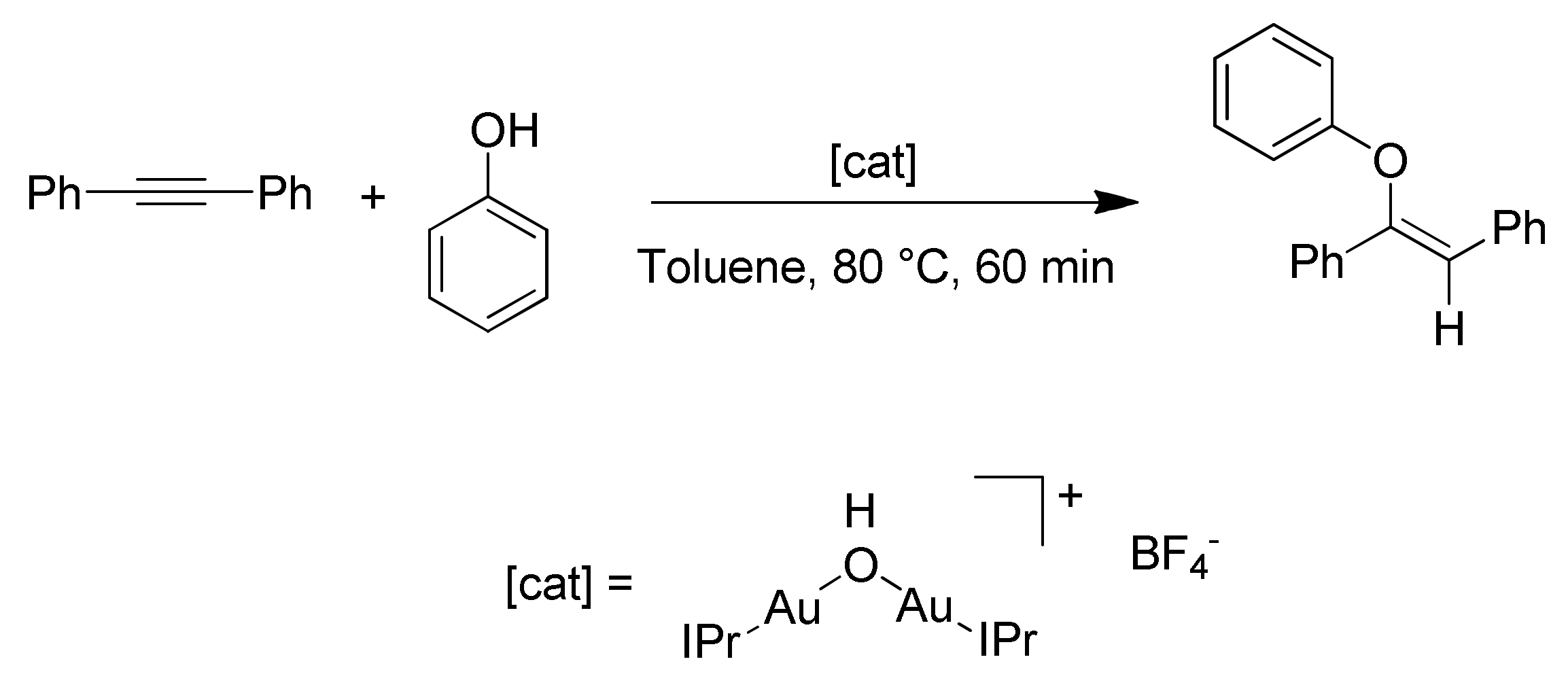
41]. There is however at least one exception to this general trend, reported by Nolan and coworkers [
52,
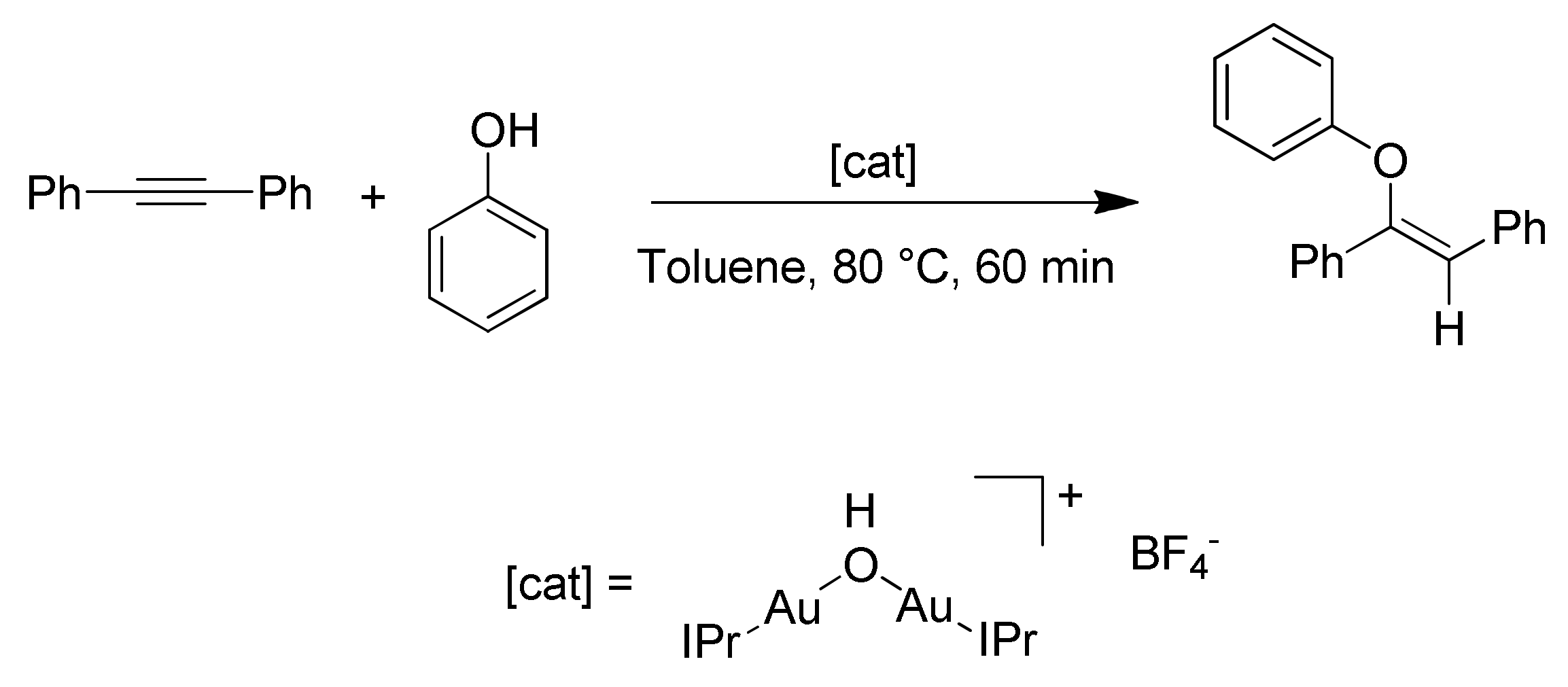
54]. In their studies, the authors analyzed the hydrophenoxylation of diphenylacetylene using the dinuclear gold(I) complex [{Au(IPr)}
2(
μ-OH)][BF
4] as catalyst (
Scheme 10).
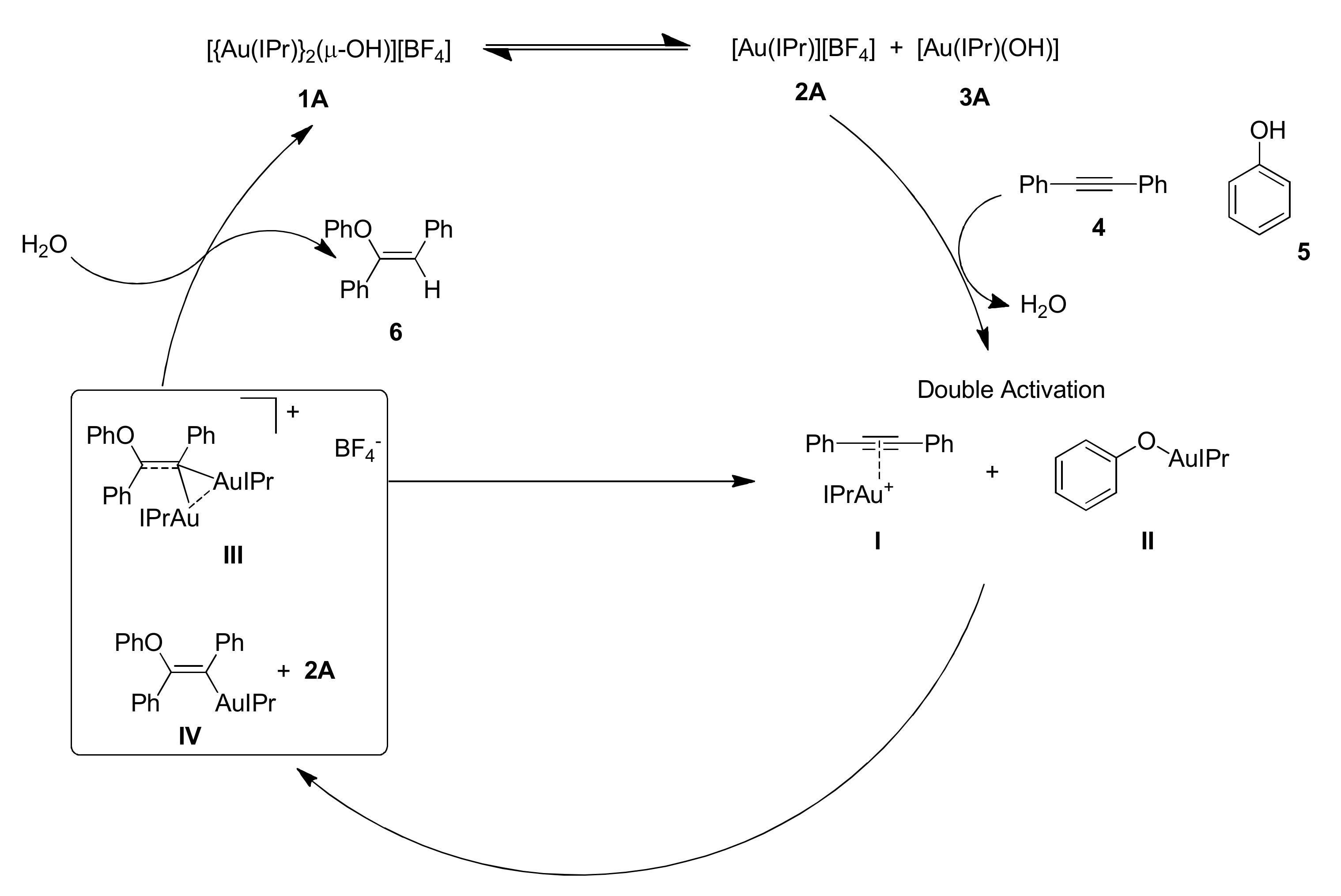
The proposed mechanism is reported in
Scheme 11. Complex [{Au(IPr)}
2(
μ-OH)][BF
4] (
1A) is in equilibrium with species
2A and
3A that are a Lewis acid and a Brønsted base, respectively. Compounds
2A and
3A in the presence of diphenylacetylene and phenol, lead to a simultaneous activation of the alkyne and of the alcohol:
2A reacts with the alkyne forming the corresponding π-coordinated compound
I,
3A reacts with phenol affording compound
II. Subsequently species
I and
II reacts to give the
gem-diaurated compound
III, in equilibrium with the mononuclear gold-vinyl compound
IV. Finally species
IV can undergo the protonolysis step forming the product, the vinyl ether
6 and the starting form of the catalyst.
An interesting evidence of the cooperativity of the two gold centers lies in the fact that isolated compound I reacts with phenol affording only 8% product yield. Preformed compound II does not react at all with diphenylacetylene. However, reaction between species I and II leads to quantitative product formation.
This cooperative mechanism is nevertheless advantageous only with phenols. It has been demonstrated, both with experimental and with computational mechanistic studies, that the use of such dinuclear gold complexes as catalysts for the intermolecular alkyne hydroalkylation with aliphatic alcohols does not provide any substantial advantage over the use of simple mononuclear complexes as catalysts [
56,
69].
5. Hydroamination
Efficient gold-catalyzed intermolecular alkyne hydroamination (
Scheme 12) has been reported for the first time in the same years in which alkyne hydration and hydroalkoxylation were originally developed [
70]. Compared to these reactions, though, alkyne hydroamination is a much broader reaction class, since there is a plethora of molecules containing a nucleophilic NH function that can be used as reactants. It is not within the scope of this review to broadly cover all the possible nitrogen-containing nucleophilic substrates that have been employed in this reaction, and the interested reader is referred to other more comprehensive contributions on this subject [
71,
72]. Here, we will focus on the nitrogen nucleophiles that have been most frequently employed for mechanistic studies, in order to shed light on the key parameters that need to be optimized for achieving best catalytic performance.
Quite obviously, the nature of the nitrogen nucleophile can have a very strong impact on the reaction, and both electronic and steric effects come into play. Concerning electronic effects, the Lewis basicity of these compounds is often markedly higher than that of water or alcohols. Consequently, these nitrogen nucleophiles will have a stronger tendency to compete with the alkyne for coordination to the gold center. Furthermore, the higher Brønsted basicity of nitrogen nucleophiles may promote the deprotonation of coordinated terminal alkynes and their consequent transformation into σ-bound gold alkynyls and related dinuclear gold complexes of type
C (
Scheme 2). Indeed, the groups of Corma and Widenhofer gathered convincing experimental proofs on the in situ predominant formation of σ,π-dinuclear gold alkynyl complexes when the hydroamination of phenylacetylene with aniline was considered [
14,
15,
21]. However, a subsequent computational study performed on the same reaction, while confirming on the one hand that the formation of such dinuclear gold alkynyl complexes was favored, also concluded that they were not the catalytically competent species [
73]. Instead, the catalytic cycle matched the one presented in
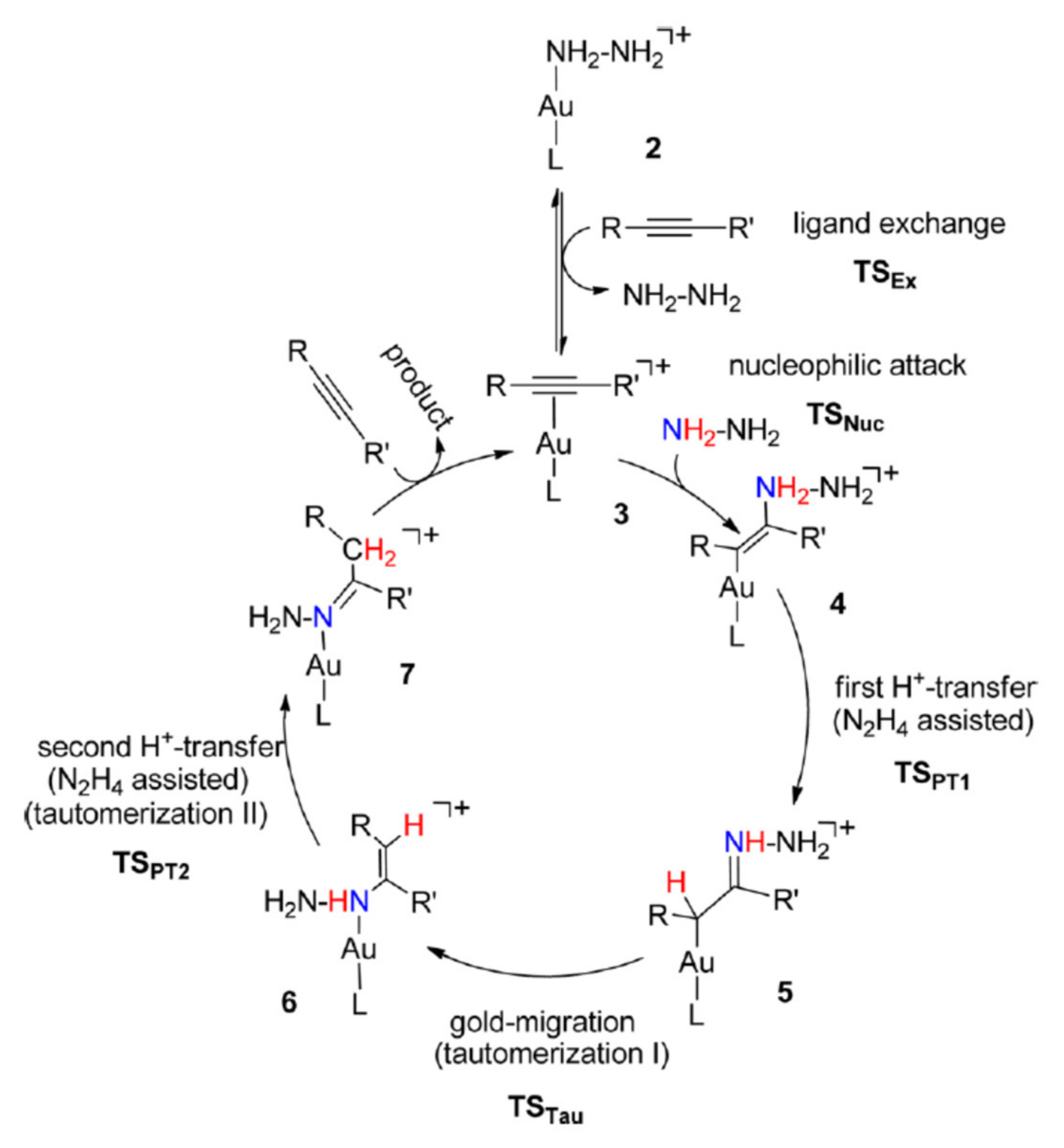
Scheme 2, and other thorough mechanistic studies performed using ammonia or hydrazine as a nucleophile (
Scheme 13) [
74,
75] confirmed this substantial similarity, at least for what it concerns the initial steps of the catalytic cycle. Thus, in spite of the strong coordinating power possessed in particular by ammonia, the reaction goes through an external nucleophilic attack of the nitrogen nucleophile to a mononuclear, cationic-complex of the alkyne with gold.
The main differences with the general mechanism, apart from the fact that a cationic LAu-nitrogen nucleophile complex may represent the catalyst resting state, is that it is the nucleophile that assists proton transfers, instead of the counteranion/solvent (though it should be remembered that both water and alcohols can do the same when they are employed as nucleophiles in stoichiometric excess, see
Section 3 and
Section 4) and especially that in the case of ammonia or hydrazine (but perhaps also of other primary nitrogen nucleophiles) two successive proton transfers take place after nucleophilic attack. Indeed, simple protonolysis to liberate the enamine product does not take place; instead, a gold-bound alkyliminium ion
5 is formed (
Scheme 13), which undergoes tautomerization via gold migration to an N-bound enamine
6. The enamine then tautomerizes to the imine
7 within the gold coordination sphere through a second, rate-determining proton transfer (see also below). The rate of these proton transfers will be obviously influenced by the Brønsted basicity of the nitrogen nucleophile: the more basic is the nucleophile, the more reluctant its conjugated acid will be to transfer the proton to the vinyl carbon atom.
Steric effects also contribute to define the reactivity of the nitrogen nucleophile, since steric bulk may render the nucleophile less prone to attack at the coordinated alkyne: for example, secondary amines are generally much less reactive than primary amines. Furthermore, as for the previous reactions, internal alkynes react more sluggishly and generally need higher catalyst amounts and higher temperatures to be converted at a synthetically useful rate.
The selectivity issue regarding this reaction is also much more intricate than in the case of alkyne hydration and hydroalkoxylation. First of all, the reaction regioselectivity follows the general trends already outlined for the previous reactions: thus, reaction with terminal alkynes generally exhibits Markovnikov selectivity, whereas lower selectivity is observed with unsymmetrically substituted internal alkynes, unless substituents are present on the alkyne substrate which strongly differ for their stereoelectronic properties or which possess functional groups that direct the nucleophilic attack preferentially toward one of the carbon atoms [
76,
77,
78]. Another important selectivity issue arises when nucleophiles with stoichiometry RNH
2 or RR’NH are employed (
Scheme 12), since only the former generates the corresponding imine, whereas the product of the latter is the corresponding enamine. These obviously different products have their own chemical behavior and can also evolve differently under reaction conditions. For example, imines are prone to hydrolysis and in the presence of traces of water provide the corresponding ketone; this reaction sequence can be conceived as a strategy to develop an amine-assisted hydration of alkynes [
78]. Furthermore, it has been demonstrated that in the presence of excess alkyne the hydroamination product can undergo further reactions such as multiple hydroamination (with the enamine as HNu) [
79], or, in the case of terminal alkynes, cycloaddition reactions (
Scheme 14) [
80].
Additional rearrangement options can be present whenever suitably functionalized alkynes are employed as reagents [
78].
Apart from the regioselectivity and the chemoselectivity (particularly in the case of possible consecutive reactions of the hydroamination product), the stereoselectivity of the reaction is also an issue in the case of enamine formation. Indeed, from the general reaction mechanism outlined in
Scheme 2, the exclusive formation of enamine products of formal
anti addition of H-NRR’ to the alkyne can be postulated. However, opposite stereoselectivity has been reported with secondary amines [
77]. This led several scientists to favor a mechanism based on an internal nucleophilic attack of the secondary nitrogen compound to the alkyne, proceeding through a tricoordinated cationic gold(I) complex. Later studies however pointed out that it is not necessary to postulate an internal nucleophilic attack, since a mechanism like the one detailed in
Scheme 13 could already account for the observed stereochemistry. Indeed, the alkyliminiun intermediate
5 in
Scheme 13 bears a single C-C bond through which the organic fragment can rotate, thereupon inverting the stereochemistry at the double bond. It has been experimentally proved that such an intermediate is accessible also from the free enamine plus the catalytic species LAu
+ [
81]. In this way, a gold-catalyzed isomerization of the initially formed enamine product takes place, similar to the isomerization of enol ethers described in
Section 4, but presumably more efficient, given the greater ease of formation of the gold-bound iminium cation with the respect to the oxonium cation. Consequently, since the same gold catalyst employed for the hydroamination reaction is also able to efficiently promote the isomerization of the enamine product, the overall result of the reaction will be the formation of the thermodynamically more favored stereoisomer, and not necessarily of the one directly resulting from the
anti addition of the amine to the alkyne.
Apart from the reaction selectivity, the main problem in using nitrogen nucleophiles in this reaction is the Lewis basicity of the nitrogen compounds, which may render them reasonably good ligands for gold, thereupon deactivating the catalyst. For this reason, for example, ammonia and aliphatic amines are much more rarely employed compared to aromatic amines [
76,
77,
82,
83]. This might be also the reason why, in contrast to the reactions treated before, ligand electronic effects come into play beside steric ones, as it will be discussed below.
5.1. Effect of the Ligand
Some alkyne hydroamination reactions, such as the reaction of arylacetylenes with anilines, have become in recent years quite popular test reactions for quickly evaluating the reactivity of gold complexes. Consequently, there is a great deal of data available in the literature, including several quite systematic studies regarding both the optimization of the catalytic activity and the substrate scope of the reaction. From these results, it is quite evident that the dependence of the catalytic performance on the nature of the employed ligand to gold is less straightforward than in the case of alkyne hydration or hydroalkoxylation discussed in the previous sections; steric bulk of the ligand is still important, but ligand electronic properties become important as well, and the general trend is that there is a direct correlation between electron donating power of the ligand and catalytic activity.
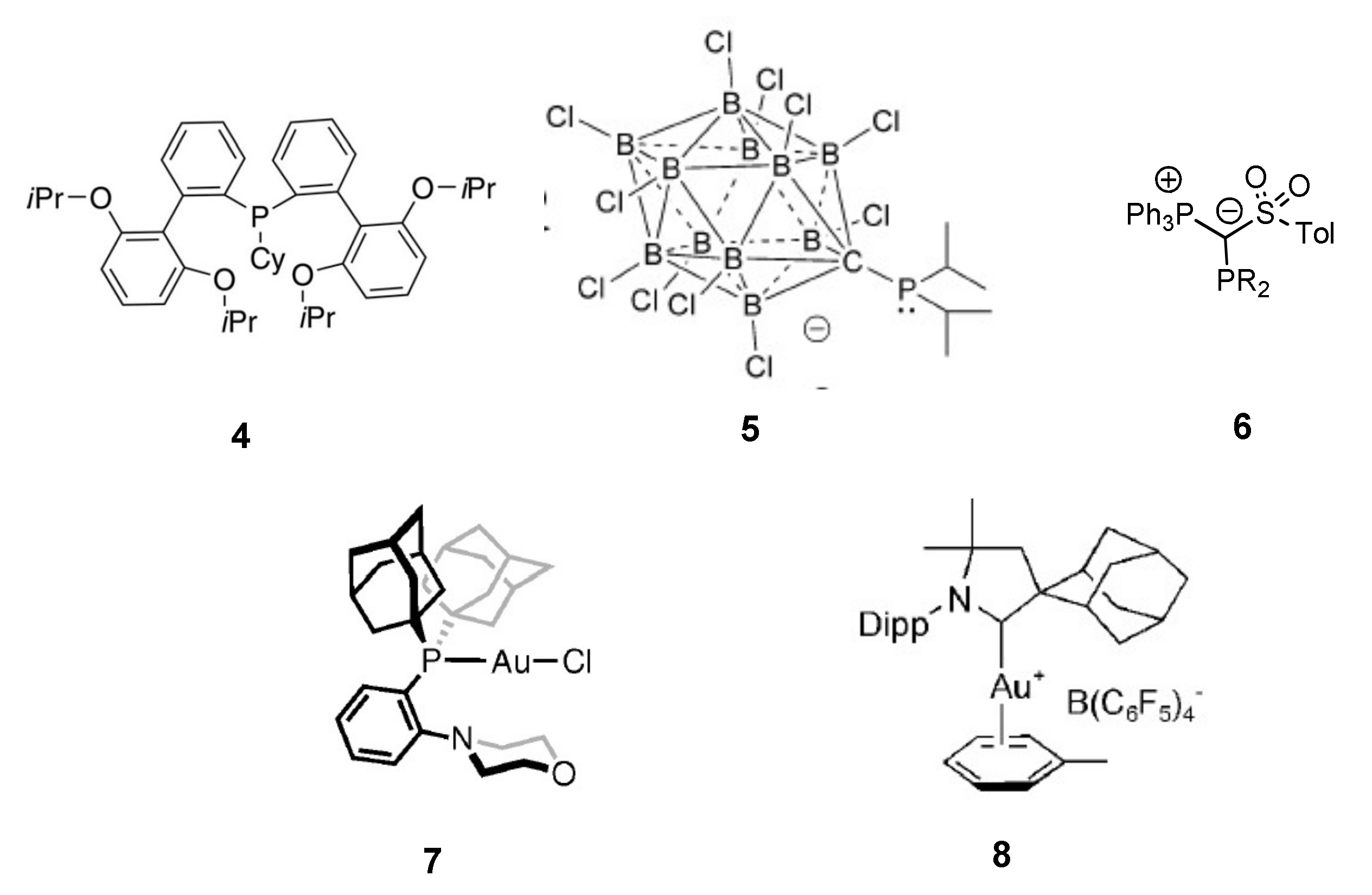
Xu and Hammond screened several ligands for this reaction [
23]; they concluded that electron-donating ligands favored the process, but that steric bulk was also important to prevent catalyst decomposition. On the basis of their results, they developed the new ligand
4 (
Scheme 15), a phosphine bearing two sterically encumbered biphenyl groups and an electron-donating cyclohexyl group [
84]. The corresponding gold(I) complex with triflate as counteranion was one order of magnitude more active than the benchmark NHC-Au-OTf complex derived from complex
1 and, upon addition of H
3PW
12O
40 as acid promoter, a catalyst loading down to only 0.0025 mol% at 50 °C could be proficiently employed. In the same year, the group of Lavallo reported on gold complexes with ligand
5, which features a phosphine ligand with a bulky, anionic carboranyl substituent; the resulting zwitterionic gold(I) complex required no activation by silver salts and allowed to reach full conversions with as low as 0.001 mol% Au at 50 °C (TONs up to 95,000) [
85]. An internal alkyne such as diphenylacetylene was also readily converted, although in this case a much higher catalyst amount (0.1 mol%) and a higher reaction temperature (80 °C) were needed for efficient catalysis. More recently, gold(I) complexes with strongly electron donating ylide-functionalized phosphines of type
6 [
86] were proposed: they exhibited high activity in the simple reaction of phenylacetylene and aniline (TON up to 8000 at 50 °C, 14,400 at 80 °C) and were also able to react secondary aromatic amines as well as internal alkynes, though again at a higher temperature. Comparable activity was also obtained with bulky NHC ligands bearing a barbiturate moiety as wingtip substituent [
87].
All these catalysts shared the same general ligand features, namely strong electron-donating character and steric bulkiness, ensuring both high activity and robustness in order to sustain the harsher reaction conditions needed for the conversion of internal alkynes and secondary amines. The presence of a negative charge on the ligand in close proximity to the gold center also seems to exert a positive effect on the catalytic performance. On the other hand, the quest for catalysts capable of efficiently converting more electron-rich nitrogen nucleophiles, such as ammonia or aliphatic amines, was up to now much less successful. The most notable examples include the functionalized biphenyl phosphine complex
7 described by Stradiotto [
77] and the CAAC complex
8 developed by the group of Bertrand [
82,
83]. Both these systems were able to catalytically convert aliphatic amines such as morpholine and piperidine, albeit at somewhat higher reaction temperatures, and complex
8 was also the first reported catalyst for hydroaminations with ammonia. The catalyst features promoting such a reactivity with ammonia and aliphatic amines, beside its robustness and resistance at the required high reaction temperatures, remain however unclear and merit further investigation.
5.2. Effect of the Counteranion
The effect of the counteranion in gold-catalyzed hydroamination reactions has been much less extensively studied than in the case of hydration or hydroalkoxylation. This effect is however expectedly less pronounced than in the former cases, since it is less probable that the counteranion plays in this reaction all the roles that have been outlined in the preceding sections. In particular, the presence of a basic nitrogen nucleophile in great excess with respect to the counteranion will make the former more likely to play a role in the proton transfer and protonolysis steps of the reaction. Indeed, computational studies favor this hypothesis [
75]. Consequently, the only effect that the counteranion will exert will be in the competition for coordination to gold, and indeed in the few cases in which this effect has been considered a simple inverse correlation was observed between the coordinating power of the anion and the catalytic performance [
88].
5.3. Effect of the Gold Oxidation State
After the pioneering studies by Utimoto et al. in the 1980s on NaAuCl
4 as a catalyst for alkyne hydrofunctionalization reactions, which also included examples of intramolecular hydroamination [
89], few further examples have been reported on the use of gold(III) catalysts for the hydroamination of alkynes, and the catalytic performance displayed by these complexes in the reaction was not particularly remarkable compared to benchmark gold(I) compounds [
90,
91]. As already discussed in
Section 3, gold(III) probably slows down the proton transfer and possibly also the subsequent tautomerization reactions that ultimately liberate the hydroamination product (
Scheme 13).
Alkyne hydroamination is also the hydrofunctionalization reaction that has been chiefly investigated in the context of catalysis by Au metal nanoparticles. Catalytic efficiency has been demonstrated at high temperature (80–100 °C) and with standard, reactive substrates such as phenylacetylene and aniline derivatives [
26,
27]; aliphatic amines and internal alkynes were found to be unreactive, and catalyst deactivation was also observed because of the deposition of alkyne oligomers on the surface of the gold nanoparticles [
92]. On the other hand, it has been also demonstrated that the catalyst can be photoactivated, when, for example, the Au nanoparticles are deposited on a photoactive oxide material such as TiO
2 [
93]. In these conditions, standard hydroamination reactions have been successfully carried out at room temperature. Similarly, micelle-stabilized gold clusters made out of just a few gold atoms are also active catalysts at room temperature [
94]. More work is needed to shed light on the reaction mechanism in these cases, and consequently on the best way to further optimize the catalytic performance.
5.4. Effect of the Reaction Conditions
The reaction conditions for intermolecular gold-catalyzed alkyne hydroaminations have been extensively varied. Several solvents were employed; whereas coordinating solvents expectedly slowed down the reaction, noncoordinating ones performed better; use of ionic liquids as solvents provided further rate acceleration, presumably because of facilitated proton transfer [
87], although they could also hamper the solubility of the substrates and have consequently a detrimental effect on the catalytic performance. Actually, solventless conditions proved in several instances to be the most useful to reach high catalytic activities [
76,
82,
85,
86,
87]. Addition of salts with slightly basic, poorly coordinating anions such as triflate proved beneficial, particularly when solventless conditions and weakly basic nitrogen nucleophiles were employed [
95]; again, this is presumably due to facilitated proton transfer within the catalytic cycle. On the other hand, the effect of the addition of acids is controversial. Beside reports in which acid addition is necessary for the generation of the catalytically competent species [
70], there are authors who recorded no effect at all and other reports in which the addition of acid proved instead beneficial [
84]. Possibly the fine features of the reaction mechanism with different substrates and catalysts and the consequent varying importance of proton transfer/protonolysis in the overall reaction kinetics explain this behavior.
5.5. Dual Gold Catalysis
As discussed above, it is generally believed that dinuclear gold(I) alkynyl complexes play no specific role in the mechanism of hydroamination but instead act as reservoir of catalytically active species [
73]. Nevertheless, some investigations have been carried out on the use of di- or polynuclear gold complexes as catalysts, in the hope of disclosing systems that can coordinate both reaction substrates, activating them and bringing them in spatial proximity to facilitate reaction. Nolan et al. have extended the application of the dinuclear, hydroxyl-bridged gold(I) NHC complexes that they successfully developed for the hydrophenoxylation of alkynes (
Scheme 11) to hydroamination reactions employing azoles as the NH nucleophiles [
95]. Indeed, the dinuclear complexes proved again superior to analogous mononuclear ones, and the authors explain their behavior in similar terms, i.e., through the simultaneous activation of the alkyne (by π-coordination to a cationic gold center) and of the azole (by interaction with a gold hydroxide complex with subsequent deprotonation and coordination).
Our group has also observed a cooperative effect with dinuclear complexes of type
9 (
Scheme 16), with which we have investigated the catalytic efficiency in standard hydroamination reactions in dependence from parameters such as the steric bulk of the wingtip substituents and the length of the bridge between the NHC units [
88]. Interestingly, we observed on the one hand increased catalytic activity by replacing a bulky aryl wingtip substituent with a methyl one, but also a lower stability of the complex preventing e.g., reaction with internal alkynes or secondary aromatic amines, which have to be carried out at higher temperature. On the other hand, we also observed a significantly higher catalytic activity for the complex bearing a propylene bridge between the NHC units; we know from previous investigations that this bridge allows the gold centers to come in close proximity [
96], hence we speculate that in this case a cooperative reaction mechanism can be present to some extent, in which one gold center in a dinuclear complex coordinates the alkyne and the other one coordinates the amine, subsequently directing its attack to the coordinated alkyne. Analogous observations have been made by other groups in studies on polynuclear gold(I) complexes [
97]. Apparently, a similar mechanism is instead not operative in the case of alkyne hydroalkoxylation, where we record similar activity irrespective of the length of the bridge [
51]; this obviously correlates with the much lower tendency of an alcohol substrate to coordinate to gold.
6. Hydroarylation
The intermolecular addition of aromatic C-H bonds across the triple bond of an alkyne, i.e., alkyne hydroarylation [
98], is the counterpart to alkyne hydroamination, in the sense that in the former case a particularly weak nucleophile is employed, instead of a relatively strong one. Whereas intramolecular, gold-catalyzed alkyne hydroarylation reactions, often simply termed “cyclization” reactions, are quite well-known in the literature [
99], studies on the intermolecular variant are instead rather sparse [
100], and very few dedicated mechanistic studies have been reported, which will be discussed in the following.
As in the related hydrofunctionalization reactions discussed in this article, the general mechanism for intermolecular alkyne hydroarylation is supposed to comprise the two key steps outlined in
Scheme 2, namely the external nucleophilic attack on the gold-coordinated alkyne, and the proton transfer/protonolysis to release the arylalkene product and the gold catalyst. Consequently, products of formal
anti addition of H-Nu to the alkyne are expected also in this case. On the other hand, the regioselectivity of the hydroarylation reaction can vary quite extensively, depending in particular on the electronic properties of the alkyne substituents. For example, when the electron-poor ethyl propiolate is the substrate, complete
anti-Markovnikov regioselectivity is observed and the reaction yields the
E isomer of the-arylated olefin as the major product (
Scheme 17, Reaction 1). Conversely, when the relatively electron-rich phenylacetylene is reacted with mesitylene, the regioselectivity is reversed and the α-arylated product is formed (
Scheme 17, Reaction 2). In the following section, we will summarize the most relevant findings pertaining to the mechanism of such reactions.
The pioneering studies conducted by the groups of Reetz [
101] and He [
102], in 2003 and 2004 respectively, utilized simple gold salts to catalyze intermolecular hydroarylation of alkynes. In the report by Reetz, gold(III) chloride, in combination with certain silver(I) salts (SbF
6− and BF
4−), catalyzes the hydroarylation of phenylacetylene by mesitylene in nitromethane with good yields. With more electron-poor alkynes such as ethyl propiolate, AuCl
3 was surpassed in its effectiveness by gold(I) complexes such as Ph
3PAuCl, and the Lewis Acid BF
3·OEt
2 was shown to be an excellent cocatalyst, as an alternative to silver(I) salts. The authors reported that exclusion of moisture is needed to maximize yields.
The group of He reported instead on the hydroarylation of electron-poor alkynes using the AuCl3/AgOTf catalyst/cocatalyst combination. At room temperature, with 1,2-dichloroethane as a solvent and generally longer reaction times, only one equivalent arene (instead of at least two equivalences used by Reetz) was used to produce the hydroarylation products in good to excellent yields. With some substrates, solventless conditions were shown to drastically reduce reaction times. All reactions were performed under air.
Subsequent work by our group [
103] and others [
104,
105,
106] has expanded on such reactions using well-defined gold catalysts. N-heterocyclic carbene (NHC) complexes of Au(I) are shown to catalyze Reaction 2 at low catalyst loadings (0.1 mol%) at room temperature. In addition to the AgBF
4 cocatalyst, an acid (1 equiv HBF
4) is also used. Replacing HBF
4 with trifluoroacetic acid (HTFA) leads to a lower yield, although some reactivity remains even under neutral conditions. Consistent with the report by Reetz, an Au(I) NHC complex outperforms its Au(III) counterpart in these catalytic reactions. Using slightly milder reaction conditions (0.1 mol% catalyst/AgBF
4, 10 mol% HBF
4, dichloroethane, room temperature), Hahn and coworkers [
106] used an Au(I) phosphine with a tethered phosphine oxide moiety as catalyst for the same reaction and recorded improved results.
As part of their work to develop cationic phosphine ligands, Alcarazo and coworkers [
107] showed that Au(I) complexes with cationic phosphine ligands such as
9 (
Scheme 18) possessed superior catalytic activity for Reaction 1 compared with simple Au(I) complexes of PPh
3 or P(OPh)
3. Based on reduction potential measurements, the authors suggested that the cationic phosphines are weaker donors than phosphites and should account for the enhanced acceptor properties/electrophilicity of the resulting Au(I) compounds. However, catalyst stability issues cap the conversions at around 60%. Subsequent DFT studies by Fernandez and coworkers [
108] predicted that the cationic phosphine ligand is responsible for a 9 kcal/mol decrease in the activation barrier (compared to PPh
3) for the nucleophilic attack step, presumably the rate-determining-step of the reaction.
In the only example where the scope of a well-defined Au(III) catalysts rivals that of AuCl
3, the group of Bourissou [
109] synthesized a C-P cyclized Au(III) complex
10 (
Scheme 18) and showed its catalytic activity for both Reaction 1 and a close analogue of Reaction 2. The reactions proceeded in CH
2Cl
2/TFA at room temperature under 5 mol% catalyst loading, and achieved 69% yield for reaction 1 in 5 minutes. While ethyl propiolate was not tested, reaction of mesitylene with methyl propiolate achieved 99% yield within 30 minutes. Using a benchmark reaction with the more activated alkene 1,3,5-trimethoxybenzene and diphenylacetylene, the Au(III) complex showed superior activity to its C-N, P-C-P pincer, and Au(I)-phosphine counterparts. AuCl
3/3AgTFA was also shown to be less active, highlighting the importance of the specific ligand system. HTFA was necessary, as replacing it with acetic acid completely shuts down catalysis. Interestingly, added silver has no effect on this silver-free system, and conducting the reaction in air using technical solvents also had minimal effect on the benchmark reaction.
In our most recent contributions to this topic, [
110,
111] ionic liquids (IL) are adopted as the solvent system in Au(I) catalysis of Reaction 2. Under neutral conditions in an IL, IPrAuCl catalyzed Reaction 2 at low catalyst loading to achieve full conversion of the alkyne within one hour, a significant improvement over the same system in dichloroethane. Intriguingly, a study on the ligand effects showed no clear pattern—electron-rich phosphines and IPr performed worse than PPh
3, whereas using an electron-poor triaryl phosphite ligand led to low activity. The anion component of the IL is shown to influence the reactivity, as bis(trifluoromethylsulfonyl)imide (NTf
2) paired with 1-butyl-3-methylimidazolium significantly outperformed its PF
6− analogue and the BF
4− version shuts down catalysis altogether. While the IL solvent system benefits the catalytic Reaction 2, such system is inactive for the Au(I)-catalysis of Reaction 1.
6.1. Mechanistic Considerations: Rate-Determining Step
Based on the available literature data, it is plausible that either one of the key steps of the catalytic cycle could be the rate-determining step, varying based on the choice of substrates and the oxidation state of the catalyst. In the case of Au(I)-catalyzed Reaction 1, the electron-rich alkyne raises the activation barrier for the nucleophilic attack, and only in cases where the associated ligand withdraws enough electron density from the alkyne, as in the case of Alcarazo’s system, does the catalytic reaction proceed to a significant extent. In the case of Au(I)-catalyzed Reaction 2, the electron-poor alkyne makes nucleophilic attack on the Au-bound alkyne more favorable. In turn, conditions that favor the proton transfer/protonolysis process, such as the presence of an acid, become important. Indeed, the importance of a Brønsted acid to enhance catalytic activity of Reaction 2 is shown in our [
103] and Hahn’s [
106] work. The ability of IL to enhance catalytic performance can also be a result of IL providing a more favorable environment for proton transfer/protonolysis, thus lowering the barrier of the step and leading to faster alkyne hydroarylation. This mechanistic hypothesis is further corroborated by a recent systematic study by the group of Hashmi on the ligand and counteranion effects in several gold(I)-catalyzed reactions [
112], including an intramolecular hydroarylation of an electron-rich terminal alkyne group. Results proved in first instance that catalytic performance exhibited an inverse correlation with the basicity of the anion. Second, although the effect of the ligand was somewhat blurred by the comparatively low stability of some complexes under reaction conditions, poorly electron-donating ligands clearly stood out as the best choice. These observations confirm previous ones and indicate in this case the nucleophilic attack of the arene as the rate-determining step of the process.
For Au(III)-catalyzed hydroarylation, the proton-transfer step exhibits a larger barrier as the Au(III)-C bond is more resistant to protonolysis [
113], as pointed out also in
Section 3 in the context of alkyne hydration [
47]. As a result, the discrete Au(III) catalysts for hydroarylation present in the recent literature [
103,
109] required the presence of acid. The exceptions to the above picture are the AuCl
3-catalyzed reactions, some of which were shown to work in the absence of any added acid [
101,
102]. However, as suggested by Reetz, the oxidation state of such Au/Ag systems are ill-defined and alternative pathways might be at work.
Interestingly, using a bifunctional ligand
11 to allow outer-sphere H-bonding between a tethered amide group with an incoming arene nucleophile, the group of Li and Zhang recently developed a versatile Au(I)-based system for alkyne hydroarylation by β-naphthol (
Scheme 19) [
114]. The system is highly selective for hydroarylation and no hydrophenoxylation reaction (
Section 3) is observed. It is also specific to naphthol as a nucleophile, as no reactivity is observed when phenol is used instead, presumably because the latter is less nucleophilic. The reaction is effective even at the gram scale, and a broad scope of alkynes can be hydroarylated. It is remarkable that the directing/activating effect of the functional group on the ligand allows to overcome the kinetic barrier even with alkynes, such as phenylacetylene, that are not easily activated by gold(I) for this reaction.
Interestingly, NaBArF served as a better cocatalyst than AgOTf, and no acid cocatalyst was required even under a low catalyst loading and temperature. Control experiments further showed the presence of the hydroxyl group as an H-bonding donor to be important, as when the hydroxy group in 2-napthol was replaced by a methoxy or an acetoxy group, no reaction was observed. The results are consistent with the nucleophilic attack being the rate-determining step in Au(I)-catalyzed hydroarylation of electron-rich alkynes.
6.2. Comparison of the Au(I) and Au(III) Oxidation States
Alkyne hydroarylation presents a rare case where the catalytic activity of well-defined Au(I) and Au(III) systems can be compared. In our hands [
103], an Au(I)-NHC catalyst was systematically superior to its Au(III) analogue in catalyzing Reaction 2 and related reactions involving electron-poor alkynes. However, this system is inactive in catalyzing Reaction 1 with a more electron-rich alkyne. In fact, no Au(I)-based system has yet been demonstrated to be active for both Reaction 1 and Reaction 2, as have been shown for AuCl
3 and Bourissou’s Au(III) catalysts.
In the case of Bourissou’s Au(III)-based C-P bidentate complexes, no Au(I) analogues exists for the featured C-P bidentate ligand, and such analogue will likely block both coordination sites for the Au(I) center. However, the group showed in a set of control experiments that the high hydroarylation reactivity is ligand specific and does not extend to related Au(III) complexes with different ligand motifs.
6.3. Intermolecular Hydroarylation with Heteroarenes
Alkyne hydroarylation by heteroarenes are in general more facile because of the increased nucleophilicity of the heterocycles. Oftentimes, the hydroarylation with these substrates is combined with subsequent cyclization reactions to form products such as carbazoles, indolizines, and phenols. Since such cascade reactions are already summarized in previous reviews [
98,
99], we will focus instead on the mechanistic aspects of the key hydroheteroarylation step.
The efficiency of Au-catalyzed alkyne hydroarylation is significantly higher with heteroarenes than with regular arenes. The group of He [
115] reported AuCl
3-catalyzed hydroarylation of ethyl propiolate by indole or 2,3-benzofuran within minutes. In comparison, the same group reported that AuCl
3-catalyzed hydroarylation of the same alkyne by mesitylene requires days and the addition of a silver(I) salt. In our hands [
111], Au(I)-catalyzed hydroarylation of ethyl propiolate by heteroarenes in ionic liquids achieves similar conversion at room temperature instead of 60 °C, under otherwise identical conditions. Furthermore, in contrast with hydroarylation with regular arenes (see
Section 6.1), hydroarylation of both electron-rich phenylacetylene and electron-deficient ethyl propriolate can be effectively catalyzed by Au(I)-NHC and Au(I)-phosphine complexes.
6.4. Selectivity: Monoarylation versus Diarylation
An interesting aspect of alkyne hydroarylation by heteroarenes lies in the facile second hydroarylation of the heteroarylalkene product to form the diarylated product (
Scheme 1). In fact, most of the literature on alkyne hydroarylation by heteroarenes reported selectivity for the diarylated product [
115,
116,
117,
118,
119]. Remarkably, products of double hydroarylation of the triple bond are generally not observed when regular arenes are employed as substrates. A computational study by the group of Ariafard [
120], using the Me
3PAu
+-catalyzed hydroarylation of propyne by pyrrole in the presence of acetic acid as a model system, suggested multiple, competitive pathways for the double hydroarylation product with overall barrier similar to that of the first hydroarylation. In all these pathways, the gold center is not directly involved in the second hydroarylation process, but it rather acts indirectly by promoting the generation of protons which in turn catalyze the hydroarylation. Au-complexed acetic acid, Au-complexed alkyne, and the Au vinyl intermediate (compound
B,
Scheme 2) all could serve as a viable proton source to generate a crucial carbocation intermediate, which in turn is transformed into the doubly hydroarylated product (
Scheme 20).
A subsequent kinetic study by Hashmi on the Au(I)-catalyzed hydroarylation of phenylacetylene by heterocycles indicated that phenylacetylene served as a source of proton for the second hydroarylation observed in such reactions [
121]. In the absence of phenylacetylene as a proton source, Au(I) is unable to catalyze further conversion of the monoarylated product (
Scheme 21, reaction (b)); on the other hand, a proton source by itself, such as 5% HNTf
2 (bis(trifluoromethanesulfonyl)imine) proves adequate to catalyze the second hydroarylation in the absence of gold. The fact that the second hydroarylation is proton-driven leads to the counterintuitive observation that use of excess pyrrole provides higher selectivity for the monohydroarylated product over the dihydroarylated one. Use of a higher number of equivalents of the heteroarene leads to faster consumption of the phenylacetylene before it can form gold-bound alkynyls with liberation of protons, thus limiting the production of the diarylated product. To drive selectivity toward the diarylated product, addition of a Brønsted acid in a second step (
Scheme 21, reaction (a)) results in the complete conversion to the diarylated product in minutes, which can be put to advantage in the production of double hydroarylation products bearing two different heteroaryl substituents.
Very recently, it has been established by the group of Lee that in the case of 2,3 or 4-substituted indoles the gold-catalyzed hydroarylation reaction of arylacetylenes conveniently stops at the monohydroarylation stage using a defect of indole [
122]. Steric effects are invoked to explain the lack of further reactivity. Nevertheless, deliberate addition of an acid allows also in this case to effect a second hydroarylation with a different heterocycle, thus opening the way to the preparation of more complex products.
7. Addition of Acids
Alkyne hydrofunctionalization by reaction with Brønsted acids as HNu is a very peculiar reaction among those that are treated in this review. Acids in neutral form are generally poor nucleophiles, but their acid properties may efficiently promote proton transfer/protonolysis processes that take place after nucleophilic attack of the acid to the coordinated alkyne, according to the general mechanism outlined in
Scheme 2. Furthermore, the conjugated bases of these acids are stronger nucleophiles but also potential ligands for gold centers, hence their presence in high concentrations deactivates the catalyst. This impairs the reactivity in this context of strong acids with strongly coordinating conjugated bases, such as hydrochloric acid. Indeed, hydrofunctionalization using this acid as nucleophile has been achieved only recently, employing gold(I) complexes with Buchwald biphenylphosphines as ligands and using HCl/DMPU as the HCl source [
123]; use of this peculiar HCl source enhances its nucleophilicity upon the establishment of a strong hydrogen bond to DMPU without the formation of high concentrations of free chloride. The process requires 2 mol% Au and delivers mainly the
trans hydrochlorinated product with high Markovnikov regioselectivity in the case of terminal alkynes. Interestingly, following the success of gas-phase acetylene hydrochlorination using supported gold catalysts [
124], heterogeneous gold metal catalysts such as Au/TiO
2 have been also successfully employed for alkyne hydrochlorination reactions in solution, using HCl/DMPU or HCl in 1,4-dioxane as reagent [
29,
30]. Formation of
cis-hydrochlorinated products with Markovnikov regioselectivity has been observed in these cases, which points toward a mechanism fundamentally different from the one outlined in
Scheme 2; indeed, the mechanism has been investigated both experimentally and computationally by the group of Corma and has been rationalized through a surface reaction involving dissociative adsorption of HCl at the gold surface followed by regioselective, stepwise addition to an adsorbed alkyne molecule (
Scheme 22) [
30]. The efficiency of this process, though, needs to be further optimized, since it still requires 2–5 mol% Au and temperatures in the range 80–120 °C for several hours; furthermore, catalyst deactivation by surface deposition of alkyne oligomers was observed also in this case.
The reaction is more facile with hydrofluoric acid, mainly because of the lower coordinating ability of fluoride toward gold(I). Hydrofluorination of internal alkynes with Et
3N·3HF was reported already in 2007 by the group of Sadighi [
125]. The reaction required an acid cocatalyst, since the acidity of the employed Et
3N·3HF hydrofluorinating reagent was too low to enable efficient final protonolysis, and was efficiently promoted by 2.5 mol% gold(I) complexes with bulky NHC ligands. After the seminal report by Sadighi, several other examples of application of this synthetic methodology followed. In 2014, the group of Xu and Hammond introduced HF/DMPU as a reagent for alkyne hydrofluorination reactions [
126]. With this reagent, no acid cocatalyst was needed, and the reaction scope could be extended to terminal alkynes. The catalyst of choice was in this case a JohnPhos-gold(I) complex, still employed at 2.5 mol% concentration. As usual, products of
anti-addition of HF were obtained with all these homogeneous gold(I) catalysts, and complete Markovnikov regioselectivity was observed with terminal alkynes, whereas with internal alkynes electronic or otherwise directing effects of the substituents at the triple bond determined the regioselectivity [
125,
126,
127]. By employing suitable additives and reaction conditions, the resulting vinyl fluoride product can sum another equivalent of HF forming
gem-alkyl difluorides, in a process that is however not gold-catalyzed [
126]. No systematic optimization study has been carried out for this reaction for what it concerns the ligand at gold as well as its counteranion. Whereas bulky, electron-rich ligands are seemingly favored also in this case, the employed counteranions range from noncoordinating anions introduced through exchange with silver salts [
125,
127] to imidate [
126] or bifluoride [
128] anions; the latter require no activation of the complex by silver salts.
Finally, carboxylic acids have been also employed as nucleophiles in gold-catalyzed alkyne intermolecular hydrofunctionalization reactions to conveniently produce vinylesters (
Scheme 23). The first report was published in 2010 [
129] and showcased 5 mol% in situ formed Ph
3PAu
+ as catalyst. As in several other gold-catalyzed intermolecular hydrofunctionalization reactions, also in this case internal alkynes reacted more sluggishly than terminal ones; consequently, subsequent research concentrated on the activation of internal alkynes for this process. Use of water as a reaction solvent and of acetate as counteranion for Au apparently resulted in improved yields, though the required catalyst concentration still remained high [
130]. Furthermore, the group of Nolan applied to this reaction the same dinuclear catalytic system that proved very useful in alkyne hydrophenoxylations reactions (
Scheme 11), and indeed the approach proved successful, allowing to use one order of magnitude less catalyst compared to mononuclear complexes [
131]. The authors postulated for this reaction the same cooperative mechanism discussed in
Section 4.2, with simultaneous activation of the alkyne (by-coordination to gold) and of the carboxylic acid (by deprotonation and coordination). From the point of view of catalytic activity, by far the most successful catalytic system up to now was the one developed by Li and Zhang [
132]. They employed as catalyst a gold(I) complex with the same bifunctional ligand
11 that was employed by the group also for hydroarylations with β-naphtols (see
Section 6.1). The remote functional group on the ligand was capable of establishing a hydrogen bond with an incoming carboxylic acid, thereupon exerting a directing and activating effect on the nucleophile. The resulting catalytic productivities were extremely high, reaching TONs up to 10
5, and the catalyst was able to convert both terminal and internal alkynes with high regio- and stereoselectivity. Interestingly, the catalytic system was found to be efficient also for alkyne hydration reactions and hydroaminations with aniline.




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
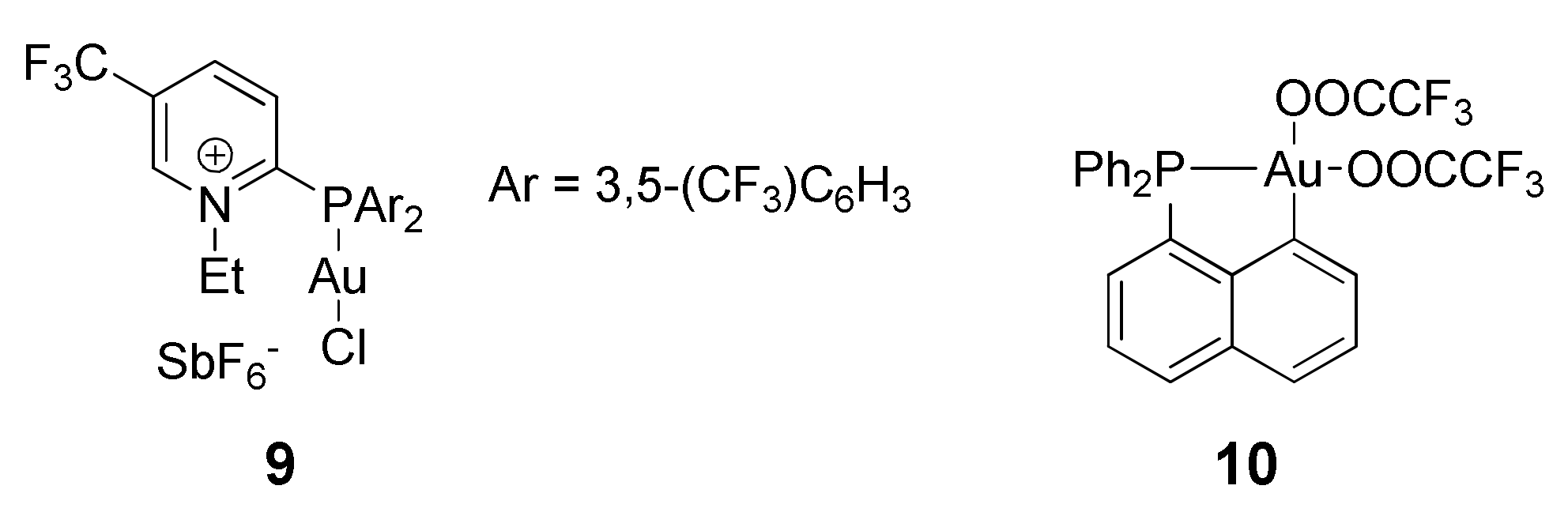{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
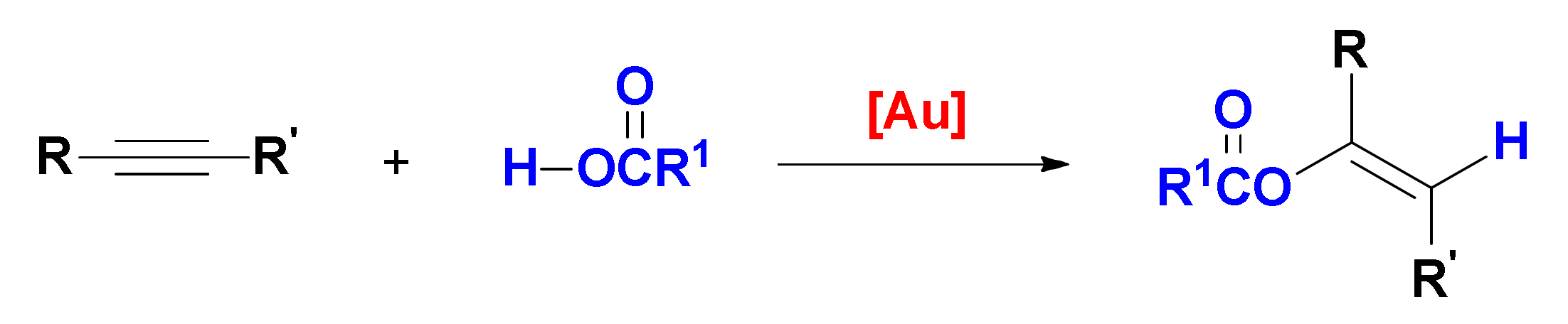{kind=link}