K-Modified Co–Mn–Al Mixed Oxide—Effect of Calcination Temperature on N2O Conversion in the Presence of H2O and NOx

 ,
, 
Abstract
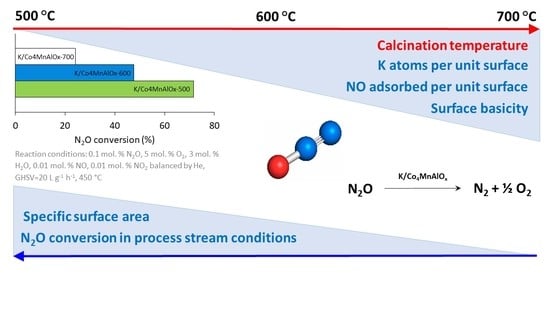
1. Introduction
2. Results and Discussion
2.1. Chemical, Structural and Textural Properties of Catalysts
2.2. XPS
2.3. TPR-H2
2.4. TPD-CO2
2.5. TPD-NO
2.6. N2O Catalytic Decomposition
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. N2O Catalytic Decomposition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Konsolakis, M. Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. [Google Scholar] [CrossRef]
- Karásková, K.; Obalová, L.; Jirátová, K.; Kovanda, F. Effect of promoters in Co-Mn-Al mixed oxide catalyst on N2O decomposition. Chem. Eng. J. 2010, 160, 480–487. [Google Scholar] [CrossRef]
- Obalová, L.; Jirátová, K.; Kovanda, F.; Pacultová, K.; Lacný, Z.; Mikulová, Z. Catalytic decomposition of nitrous oxide over catalysts prepared from Co/Mg-Mn/Al hydrotalcite-like compounds. Appl. Catal. B Environ. 2005, 60, 289–297. [Google Scholar] [CrossRef]
- Obalová, L.; Kovanda, F.; Jirátová, K.; Pacultová, K.; Lacný, Z. Application of calcined layered double hydroxides as catalysts for abatement of N2O emissions. Collect. Czech. Chem. Commun. 2008, 73, 1045–1060. [Google Scholar] [CrossRef]
- Piskorz, W.; Zasada, F.; Stelmachowski, P.; Kotarba, A.; Sojka, Z. Decomposition of N2O over the surface of cobalt spinel: A DFT account of reactivity experiments. Catal. Today 2008, 137, 418–422. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Ciura, K.; Grzybek, G. Morphology-dependent reactivity of cobalt oxide nanoparticles in N2O decomposition. Catal. Sci. Technol. 2016, 6, 5554–5560. [Google Scholar] [CrossRef]
- Wojcik, S.; Grzybek, G.; Stelmachowski, P.; Sojka, Z.; Kotarba, A. Bulk, Surface and Interface Promotion of Co3O4 for the Low-Temperature N2O Decomposition Catalysis. Catalysts 2020, 10, 41. [Google Scholar] [CrossRef]
- Wojcik, S.; Thersleff, T.; Gebska, K.; Grzybek, G.; Kotarba, A. Atomic-Level Dispersion of Bismuth over Co3O4 Nanocrystals-Outstanding Promotional Effect in Catalytic DeN2O. Catalysts 2020, 10, 51. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Vaccari, A. Clays and catalysis: A promising future. Appl. Clay Sci. 1999, 14, 161–198. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Overeijnder, J.; Kapteijn, F.; Moulijn, J.A. Structural promotion and stabilizing effect of Mg in the catalytic decomposition of nitrous oxide over calcined hydrotalcite-like compounds. Appl. Catal. B Environ. 1999, 23, 59–72. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Jirátová, K.; Kovanda, F. Effect of potassium in calcined Co-Mn-Al layered double hydroxide on the catalytic decomposition of N2O. Appl. Catal. B Environ. 2009, 90, 132–140. [Google Scholar] [CrossRef]
- Klyushina, A.; Pacultová, K.; Karásková, K.; Jirátová, K.; Ritz, M.; Fridrichová, D.; Volodarskaja, A.; Obalová, L. Effect of preparation method on catalytic properties of Co-Mn-Al mixed oxides for N2O decomposition. J. Mol. Catal. A Chem. 2016, 425, 237–247. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Wach, A.; Kustrowski, P.; Mamulová-Kutláková, K.; Michalik, S.; Jirátová, K. Alkali metals as promoters in Co-Mn-Al mixed oxide for N2O decomposition. Appl. Catal. A Gen. 2013, 462, 227–235. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Soliman, S.A.; Abdellah, S.E. Enhanced direct N2O decomposition over CuxCo1−xCo2O4 (0.0 ≤ x ≤ 1.0) spinel-oxide catalysts. J. Ind. Eng. Chem. 2015, 21, 814–821. [Google Scholar] [CrossRef]
- De Roy, A.; Forano, C.; Besse, J.P. Layered Double Hydroxides: Present and Future; Nova Science Publishers: New York, NY, USA, 2001; pp. 1–39. [Google Scholar]
- Chmielarz, L.; Rutkowska, M.; Kustrowski, P.; Drozdek, M.; Piwowarska, Z.; Dudek, B.; Dziembaj, R.; Michalik, M. An influence of thermal treatment conditions of hydrotalcite-like materials on their catalytic activity in the process of N2O decomposition. J. Therm. Anal. Calorim. 2011, 105, 161–170. [Google Scholar] [CrossRef]
- Kovanda, F.; Rojka, T.; Dobešová, J.; Machovič, V.; Bezdička, P.; Obalová, L.; Jirátová, K.; Grygar, T. Mixed oxides obtained from Co and Mn containing layered double hydroxides: Preparation, characterization, and catalytic properties. J. Solid State Chem. 2006, 179, 812–823. [Google Scholar] [CrossRef]
- Roman-Martinez, M.C.; Kapteijn, F.; Cazorla-Amoros, D.; Perez-Ramirez, J.; Moulijn, J.A. A TEOM-MS study on the interaction of N2O with a hydrotalcite-derived multimetallic mixed oxide catalyst. Appl. Catal. A Gen. 2002, 225, 87–100. [Google Scholar] [CrossRef]
- Tao, Y.X.; Yu, J.J.; Liu, C.C.; Hao, Z.P. N2O catalytic decomposition over mixed oxides derived from Co-Mg/Al hydrotalcite-like compounds. Acta Phys. Chim. Sin. 2007, 23, 162–168. [Google Scholar]
- Xu, X.-L.; Xu, X.-F.; Zhang, G.-T.; Niu, X.-J. Preparation of Co-Al mixed oxide-supported gold catalysts and their catalytic activity for N2O decomposition. J. Fuel Chem. Technol. 2009, 37, 595–600. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, Y.; Wang, A.; Li, L.; Wang, X.; Zhang, T. N2O decomposition over K-promoted Co-Al catalysts prepared from hydrotalcite-like precursors. Appl. Catal. B Environ. 2009, 89, 391–397. [Google Scholar] [CrossRef]
- Pacultová, K.; Bílková, T.; Klegova, A.; Karásková, K.; Fridrichová, D.; Jirátová, K.; Kiška, T.; Balabánová, J.; Koštejn, M.; Kotarba, A.; et al. Co-Mn-Al Mixed Oxides Promoted by K for Direct NO Decomposition: Effect of Preparation Parameters. Catalysts 2019, 9, 593. [Google Scholar] [CrossRef]
- Grzybek, G.; Wójcik, S.; Legutko, P.; Gryboś, J.; Indyka, P.; Leszczyński, B.; Kotarba, A.; Sojka, Z. Thermal stability and repartition of potassium promoter between the support and active phase in the K-Co2.6Zn0.4O4|α-Al2O3 catalyst for N2O decomposition: Crucial role of activation temperature on catalytic performance. Appl. Catal. B Environ. 2017, 205, 597–604. [Google Scholar] [CrossRef]
- Obalová, L.; Jirátová, K.; Kovanda, F.; Valášková, M.; Balabánová, J.; Pacultová, K. Structure–activity relationship in the N2O decomposition over Ni-(Mg)-Al and Ni-(Mg)-Mn mixed oxides prepared from hydrotalcite-like precursors. J. Mol. Catal. A Chem. 2006, 248, 210–219. [Google Scholar] [CrossRef]
- Obalová, L.; Pacultová, K.; Balabánová, J.; Jirátová, K.; Bastl, Z.; Valášková, M.; Lacný, Z.; Kovanda, F. Effect of Mn/Al ratio in Co–Mn–Al mixed oxide catalysts prepared from hydrotalcite-like precursors on catalytic decomposition of N2O. Catal. Today 2007, 119, 233–238. [Google Scholar] [CrossRef]
- Obalová, L.; Maniak, G.; Karásková, K.; Kovanda, F.; Kotarba, A. Electronic nature of potassium promotion effect in Co-Mn-Al mixed oxide on the catalytic decomposition of N2O. Catal. Commun. 2011, 12, 1055–1058. [Google Scholar] [CrossRef]
- Pacultová, K.; Karásková, K.; Kovanda, F.; Jirátová, K.; Šramek, J.; Kustrowski, P.; Kotarba, A.; Chromčáková, Ž.; Kočí, K.; Obalová, L. K-doped Co-Mn-Al mixed oxide catalyst for N2O abatement from nitric acid plant waste gases: Pilot plant studies. Ind. Eng. Chem. Res. 2016, 55, 7076–7084. [Google Scholar] [CrossRef]
- Kovanda, F.; Grygar, T.; Dorničák, V.; Rojka, T.; Bezdička, P.; Jirátová, K. Thermal behaviour of Cu–Mg–Mn and Ni–Mg–Mn layered double hydroxides and characterization of formed oxides. Appl. Clay Sci. 2005, 28, 121–136. [Google Scholar] [CrossRef]
- Kannan, S.; Swamy, C.S. Catalytic decomposition of nitrous oxide over calcined cobalt aluminum hydrotalcites. Catal. Today 1999, 53, 725–737. [Google Scholar] [CrossRef]
- Atribak, I.; Guillén-Hurtado, N.; Bueno-López, A.; García-García, A. Influence of the physico-chemical properties of CeO2–ZrO2 mixed oxides on the catalytic oxidation of NO to NO2. Appl. Surf. Sci. 2010, 256, 7706–7712. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Hagelin-Weaver, H.A.E.; Hoflund, G.B.; Minahan, D.A.; Salaita, G.N. Electron energy loss spectroscopic investigation of Co metal, CoO, and Co3O4 before and after Ar+ bombardment. Appl. Surf. Sci. 2004, 235, 420–448. [Google Scholar] [CrossRef]
- Chromčáková, Ž.; Obalová, L.; Kovanda, F.; Legut, D.; Titov, A.; Ritz, M.; Fridrichová, D.; Michalik, S.; Kuśtrowski, P.; Jirátová, K. Effect of precursor synthesis on catalytic activity of Co3O4 in N2O decomposition. Catal. Today 2015, 257, 18–25. [Google Scholar] [CrossRef]
- Langell, M.A.; Gevrey, F.; Nydegger, M.W. Surface composition of MnxCo1-xO solid solutions by X-ray photoelectron and Auger spectroscopies. Appl. Surf. Sci. 2000, 153, 114–127. [Google Scholar] [CrossRef]
- Iablokov, V.; Barbosa, R.; Pollefeyt, G.; Van Driessche, I.; Chenakin, S.; Kruse, N. Catalytic CO Oxidation over Well-Defined Cobalt Oxide Nanoparticles: Size-Reactivity Correlation. ACS Catal. 2015, 5, 5714–5718. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Hu, X.B.; Zheng, K.; Wei, X.H.; Zhao, Y.X. Effect of SnO2 on the structure and catalytic performance of Co3O4 for N2O decomposition. Catal. Commun. 2018, 111, 70–74. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Bawaked, S.M.; Kosa, S.A.; Schwieger, W. Effect of microwave power on the thermal genesis of Co3O4 nanoparticles from cobalt oxalate micro-rods. Appl. Surf. Sci. 2015, 351, 600–609. [Google Scholar] [CrossRef]
- Lykaki, M.; Papista, E.; Kaklidis, N.; Carabineiro, S.A.C.; Konsolakis, M. Ceria Nanoparticles’ Morphological Effects on the N2O Decomposition Performance of Co3O4/CeO2 Mixed Oxides. Catalysts 2019, 9, 233. [Google Scholar] [CrossRef]
- Kim, S.C.; Shim, W.G. Catalytic combustion of VOCs over a series of manganese oxide catalysts. Appl. Catal. B Environ. 2010, 98, 180–185. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbookof X-ray Photoeletron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1995. [Google Scholar]
- Dupin, J.C.; Gonbeau, D.; Vinatier, P.; Levasseur, A. Systematic XPS studies of metal oxides, hydroxides and peroxides. Phys. Chem. Chem. Phys. 2000, 2, 1319–1324. [Google Scholar] [CrossRef]
- Piumetti, M.; Bensaid, S.; Russo, N.; Fino, D. Nanostructured ceria-based catalysts for soot combustion: Investigations on the surface sensitivity. Appl. Catal. B Environ. 2015, 165, 742–751. [Google Scholar] [CrossRef]
- Chuang, T.J.; Brundle, C.R.; Rice, D.W. Interpretation of X-ray Photoemission spectra of cobalt oxides and cobalt oxide surfaces. Surf. Sci. 1976, 59, 413–429. [Google Scholar] [CrossRef]
- Li, W.; Yang, S.S.; Wang, K.L.; Tu, S.H.; Lu, M.J.; Wu, P.X. Evaluation of the physiochemical properties and catalytic performance of CuCoMnAl mixed oxides derived from layered double hydroxides precursors with different mole ratios of Cu/Co on the oxidation of toluene. React. Kinet. Mech. Catal. 2019, 128, 965–977. [Google Scholar] [CrossRef]
- Chen, H.H.; Yang, M.; Tao, S.; Chen, G.W. Oxygen vacancy enhanced catalytic activity of reduced Co3O4 towards p-nitrophenol reduction. Appl. Catal. B Environ. 2017, 209, 648–656. [Google Scholar] [CrossRef]
- Luo, Y.J.; Zheng, Y.B.; Zuo, J.C.; Feng, X.S.; Wang, X.Y.; Zhang, T.H.; Zhang, K.; Jiang, L.L. Insights into the high performance of Mn-Co oxides derived from metal organic frameworks for total toluene oxidation. J. Hazard. Mater. 2018, 349, 119–127. [Google Scholar] [CrossRef]
- Kaczmarczyk, J.; Zasada, F.; Janas, J.; Indyka, P.; Piskorz, W.; Kotarba, A.; Sojka, Z. Thermodynamic Stability, Redox Properties, and Reactivity of Mn3O4, Fe3O4, and Co3O4 Model Catalysts for N2O Decomposition: Resolving the Origins of Steady Turnover. ACS Catal. 2016, 6, 1235–1246. [Google Scholar] [CrossRef]
- Jung, D.H.; Umirov, N.; Kim, T.; Bakenov, Z.; Kim, J.S.; Kim, S.S. Thermal and Structural Stabilities of LixCoO2 Cathode for Li Secondary Battery Studied by a Temperature Programmed Reduction. Eurasian Chem. Technol. J. 2019, 21, 3–12. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chen, Y.W. The mechanism of reduction of cobalt by hydrogen. Mater. Chem. Phys. 2004, 85, 171–175. [Google Scholar] [CrossRef]
- Calgaro, C.O.; Perez-Lopez, O.W. Decomposition of methane over Co3-xAlxO4 (x = 0-2) coprecipitated catalysts: The role of Co phases in the activity and stability. Int. J. Hydrog. Energy 2017, 42, 29756–29772. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, Z.; Shangguan, W. Simultaneous catalytic removal of NOx and soot particulate over Co–Al mixed oxide catalysts derived from hydrotalcites. Catal. Commun. 2007, 8, 1659–1664. [Google Scholar] [CrossRef]
- Arnoldy, P.; van Oers, E.M.; Bruinsma, O.S.L.; de Beer, V.H.J.; Moulijn, J.A. Temperature-Programmed Reduction of Al2O3-, SiO2-, and carbon-supported Re2O7 catalysts. J. Catal. 1985, 93, 231–245. [Google Scholar] [CrossRef]
- Wu, M.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, Y.; Wang, L.; Lu, G. An effective Mn-Co mixed oxide catalyst for the solvent-free selective oxidation of cyclohexane with molecular oxygen. Appl. Catal. A Gen. 2016, 523, 97–106. [Google Scholar] [CrossRef]
- Kapteijn, F.; Singoredjo, L.; Andreini, A.; Moulijn, J.A. Activity and selectivity of pure manganese oxides in the selective catalytic reduction of nitric-oxide with ammonia. Appl. Catal. B Environ. 1994, 3, 173–189. [Google Scholar] [CrossRef]
- Arena, F.; Torre, T.; Raimondo, C.; Parmaliana, A. Structure and redox properties of bulk and supported manganese oxide catalysts. Phys. Chem. Chem. Phys. 2001, 3, 1911–1917. [Google Scholar] [CrossRef]
- Royer, S.; Bérubé, F.; Kaliaguine, S. Effect of the synthesis conditions on the redox and catalytic properties in oxidation reactions of LaCo1−xFexO3. Appl. Catal. A Gen. 2005, 282, 273–284. [Google Scholar] [CrossRef]
- Smoláková, L.; Frolich, K.; Troppová, I.; Kutálek, P.; Kroft, E.; Čapek, L. Determination of basic sites in Mg-Al mixed oxides by combination of TPD-CO2 and CO2 adsorption calorimetry. J. Therm. Anal. Calorim. 2017, 127, 1921–1929. [Google Scholar] [CrossRef]
- Kotarba, A.; Bieniasz, W.; Kuśtrowski, P.; Stadnicka, K.; Sojka, Z. Composite ferrite catalyst for ethylbenzene dehydrogenation: Enhancement of potassium stability and catalytic performance by phase selective doping. Appl. Catal. A Gen. 2011, 407, 100–105. [Google Scholar] [CrossRef]
- Kotarba, A.; Kruk, I.; Sojka, Z. Energetics of Potassium Loss from Styrene Catalyst Model Components: Reassignment of K Storage and Release Phases. J. Catal. 2002, 211, 265–272. [Google Scholar] [CrossRef]
- Pinho, P.V.B.; Grybos, J.; Hudy, C.; Janas, J.; Gora-Marek, K.; Zasada, F.; Sojka, Z. Interaction of nitric oxide with the (100) surface of cobalt spinel nanocubes—A comprehensive DFT, atomistic thermodynamic, IR and TPD account. Appl. Surf. Sci. 2020, 513, 10. [Google Scholar] [CrossRef]
- Pacultová, K.; Klegová, A.; Karásková, K.; Fridrichová, D.; Bílková, T.; Obalová, L. Oxygen effect in NO direct decomposition over K/Co-Mg-Mn-Al mixed oxide catalyst-temperature programmed desorption study. J. Catal. 2020. submitted. [Google Scholar]
- Bílková, T.; Pacultová, K.; Karásková, K.; Fridrichová, D.; Obalová, L.; Haneda, M. Reaction mechanism of NO direct decomposition over K-promoted Co-Mn-Al mixed oxides–DRIFTS, TPD and transient kinetic studies. Appl. Surf. Sci. submitted. 2020. [Google Scholar]
- Alvarez, P.; Aguila, G.; Guerrero, S.; Araya, P. Thermal stability of the Cu-CeO2 interface on silica and alumina, and its relation with activity in the oxidation reaction of Co and the decomposition of N2O. J. Chil. Chem. Soc. 2018, 63, 4102–4108. [Google Scholar] [CrossRef]
- Pacultová, K.; Karásková, K.; Strakošová, J.; Jirátová, K.; Obalová, L. Supported Co–Mn–Al mixed oxides as catalysts for N2O decomposition. Comptes Rendus Chim. 2015, 18, 1114–1122. [Google Scholar] [CrossRef]
- Obalová, L.; Fíla, V. Kinetic analysis of N2O decomposition over calcined hydrotalcites. Appl. Catal. B Environ. 2007, 70, 353–359. [Google Scholar] [CrossRef]
- Zasada, F.; Stelmachowski, P.; Maniak, G.; Paul, J.F.; Kotarba, A.; Sojka, Z. Potassium Promotion of Cobalt Spinel Catalyst for N2O Decomposition-Accounted by Work Function Measurements and DFT Modelling. Catal. Lett. 2009, 127, 126–131. [Google Scholar] [CrossRef]
- Del Río, L.; Marbán, G. Stainless steel wire mesh-supported potassium-doped cobalt oxide catalysts for the catalytic decomposition of nitrous oxide. Appl. Catal. B Environ. 2012, 126, 39–46. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Zasada, F.; Piskorz, W.; Kotarba, A.; Paul, J.-F.; Sojka, Z. Experimental and DFT studies of N2O decomposition over bare and Co-doped magnesium oxide—Insights into the role of active sites topology in dry and wet conditions. Catal. Today 2008, 137, 423–428. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B Environ. 2008, 78, 242–249. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Del Valle, S.P.; Marie, O. NOx adsorption on K and Ba loaded on zirconia-titania NSR catalysts: A comparative study by in situ and operando IR spectroscopy. Appl. Catal. B Environ. 2018, 231, 391–399. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | K/Co4MnAlOx-500 | K/Co4MnAlOx-600 | K/Co4MnAlOx-700 |
|---|---|---|---|
| Co (wt.%) | 52.2 | 54.5 | 56.1 |
| Mn (wt.%) | 11.0 | 11.5 | 11.8 |
| Al (wt.%) | 5.0 | 5.2 | 5.2 |
| K (wt.%) | 2.2 | 2.3 | 2.3 |
| SBET (m2 g−1) | 98 | 77 | 71 |
| Vmeso (cm3 g−1) | 0.37 | 0.35 | 0.36 |
| Lc a (nm) | 8.7 | 9.8 | 11.7 |
| TPR-H2 (25–1000 °C) (mmol g−1) | 10.9 | 13.6 | 12.4 |
| TPR-H2 (25–500 °C) (mmol g−1) | 3.6 | 4.5 | 3.8 |
| Tmax b (°C) | 173; 387; 589; 776 | 218; 434; 604; 754; 856 | 229; 434; 625; 759; 856 |
| (Co + Mn) mean oxidation state | 2.4 | 2.8 | 2.5 |
| TPD-CO2 (28–650 °C) (mmol g−1) | 1.9 | 3.2 | 3.7 |
| TPD-CO2 (28–650 °C) (mmol m−2) | 0.02 | 0.04 | 0.05 |
| TPD-NO (50–650 °C) (a.u. g−1) c | 2.4; 8.3; 7.0; 11.8; 1.4 | n.d. | 1.2; 5.5; 14.7; 6.1 |
| TPD-NO (50–650 °C) (a.u. m−2) | 0.30 | n.d. | 0.39 |
| Ea (J mol−1) d | 104,423 e 117,626 f 188,013 g | 88,452 e 107,907 f 160,576 g | 97,315 e 136,848 f 167,477 g |
| Sample | Co 2p3/2 a | Mn 2p3/2 b | O 1s | O 1s | Co2+/Co3+ | Mn3+/Mn4+ |
|---|---|---|---|---|---|---|
| K/Co4MnAlOx-500 | 780.0 | 641.9 | 530.1 | 531.8 | 1.32 | 2.88 |
| K/Co4MnAlOx-600 | 779.9 | 641.6 | 529.7 | 531.3 | 1.30 | 2.72 |
| K/Co4MnAlOx-700 | 779.9 | 641.5 | 529.7 | 531.4 | 1.26 | 2.68 |
| Sample | K/Co4MnAlOx-500 | K/Co4MnAlOx-600 | K/Co4MnAlOx-700 |
|---|---|---|---|
| Co 2p (at. %) | 13.16 | 12.56 | 11.78 |
| Mn 2p (at. %) | 3.76 | 5.05 | 5.24 |
| Al 2p(at. %) | 7.72 | 7.44 | 7.86 |
| O 1s (at. %) | 55.08 | 51.80 | 51.68 |
| C 1s (at. %) | 13.50 | 18.68 | 18.21 |
| K 2p (at. %) | 4.04 | 3.82 | 4.48 |
| K (atoms nm−2) | 6.4 | 7.6 | 9.7 |
| Na 1s (at. %) | 1.24 | 0.64 | 0.75 |
| N 1s (at. %) | 1.51 | 0 | 0 |
| Surface/Bulk Molar Ratio | K/Co4MnAlOx-500 | K/Co4MnAlOx-600 | K/Co4MnAlOx-700 |
|---|---|---|---|
| Co | 0.5 | 0.4 | 0.4 |
| Mn | 0.6 | 0.7 | 0.7 |
| Al | 1.3 | 1.2 | 1.2 |
| K | 2.3 | 2.0 | 2.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karásková, K.; Pacultová, K.; Jirátová, K.; Fridrichová, D.; Koštejn, M.; Obalová, L. K-Modified Co–Mn–Al Mixed Oxide—Effect of Calcination Temperature on N2O Conversion in the Presence of H2O and NOx. Catalysts 2020, 10, 1134. https://doi.org/10.3390/catal10101134
Karásková K, Pacultová K, Jirátová K, Fridrichová D, Koštejn M, Obalová L. K-Modified Co–Mn–Al Mixed Oxide—Effect of Calcination Temperature on N2O Conversion in the Presence of H2O and NOx. Catalysts. 2020; 10(10):1134. https://doi.org/10.3390/catal10101134
Chicago/Turabian StyleKarásková, Kateřina, Kateřina Pacultová, Květuše Jirátová, Dagmar Fridrichová, Martin Koštejn, and Lucie Obalová. 2020. "K-Modified Co–Mn–Al Mixed Oxide—Effect of Calcination Temperature on N2O Conversion in the Presence of H2O and NOx" Catalysts 10, no. 10: 1134. https://doi.org/10.3390/catal10101134
APA StyleKarásková, K., Pacultová, K., Jirátová, K., Fridrichová, D., Koštejn, M., & Obalová, L. (2020). K-Modified Co–Mn–Al Mixed Oxide—Effect of Calcination Temperature on N2O Conversion in the Presence of H2O and NOx. Catalysts, 10(10), 1134. https://doi.org/10.3390/catal10101134