Promoting Effect of the Core-Shell Structure of MnO2@TiO2 Nanorods on SO2 Resistance in Hg0 Removal Process

 ,
, 

Abstract
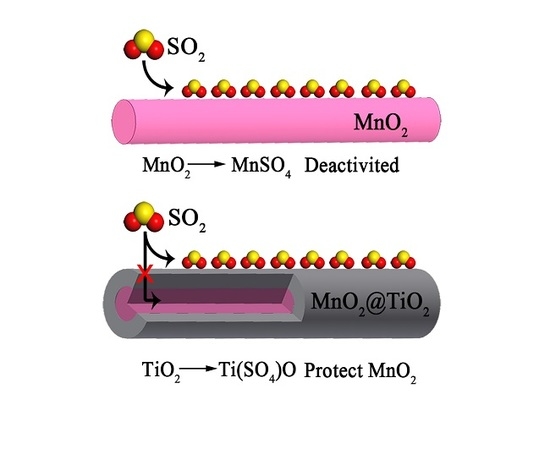
:
1. Introduction
2. Results and Discussion
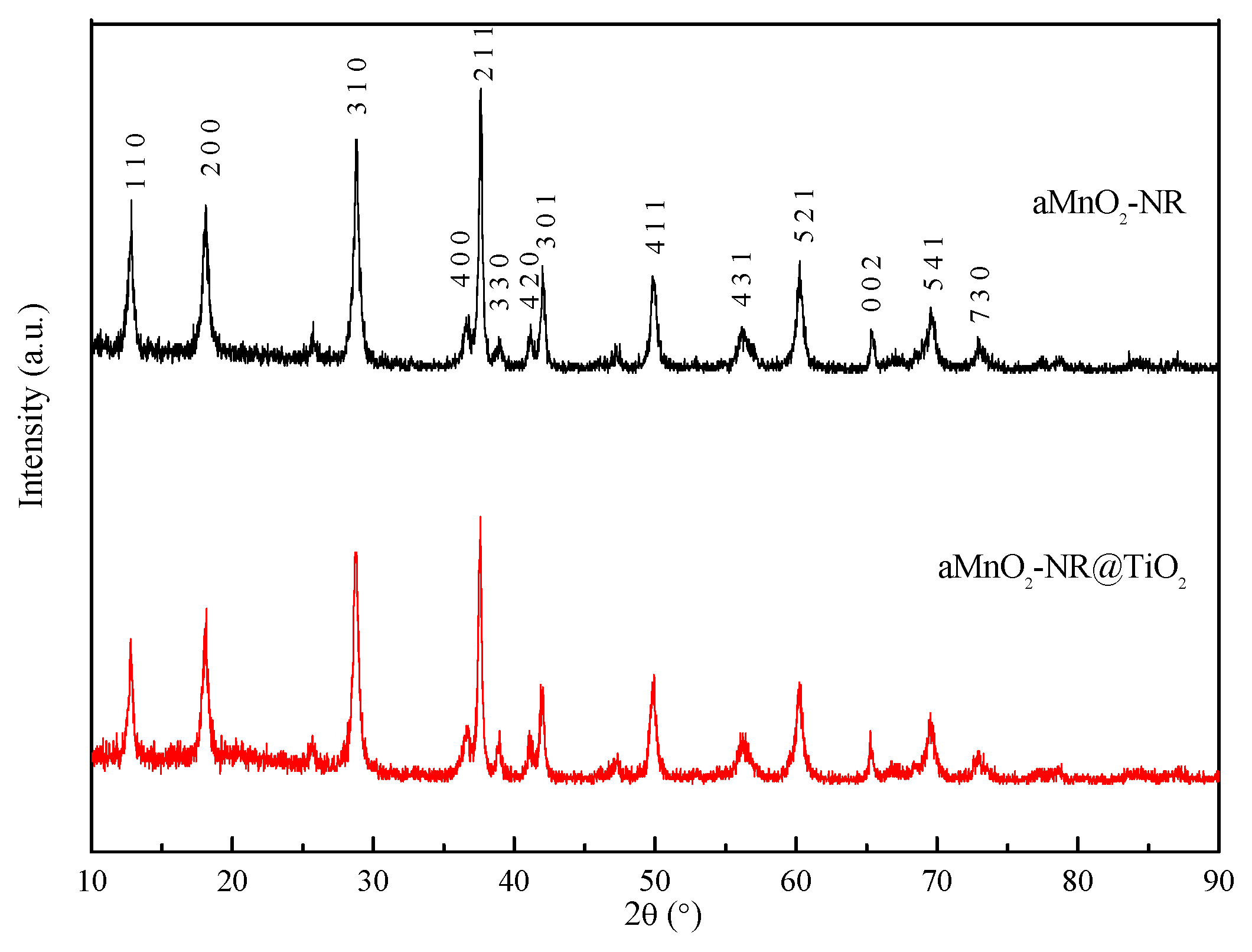
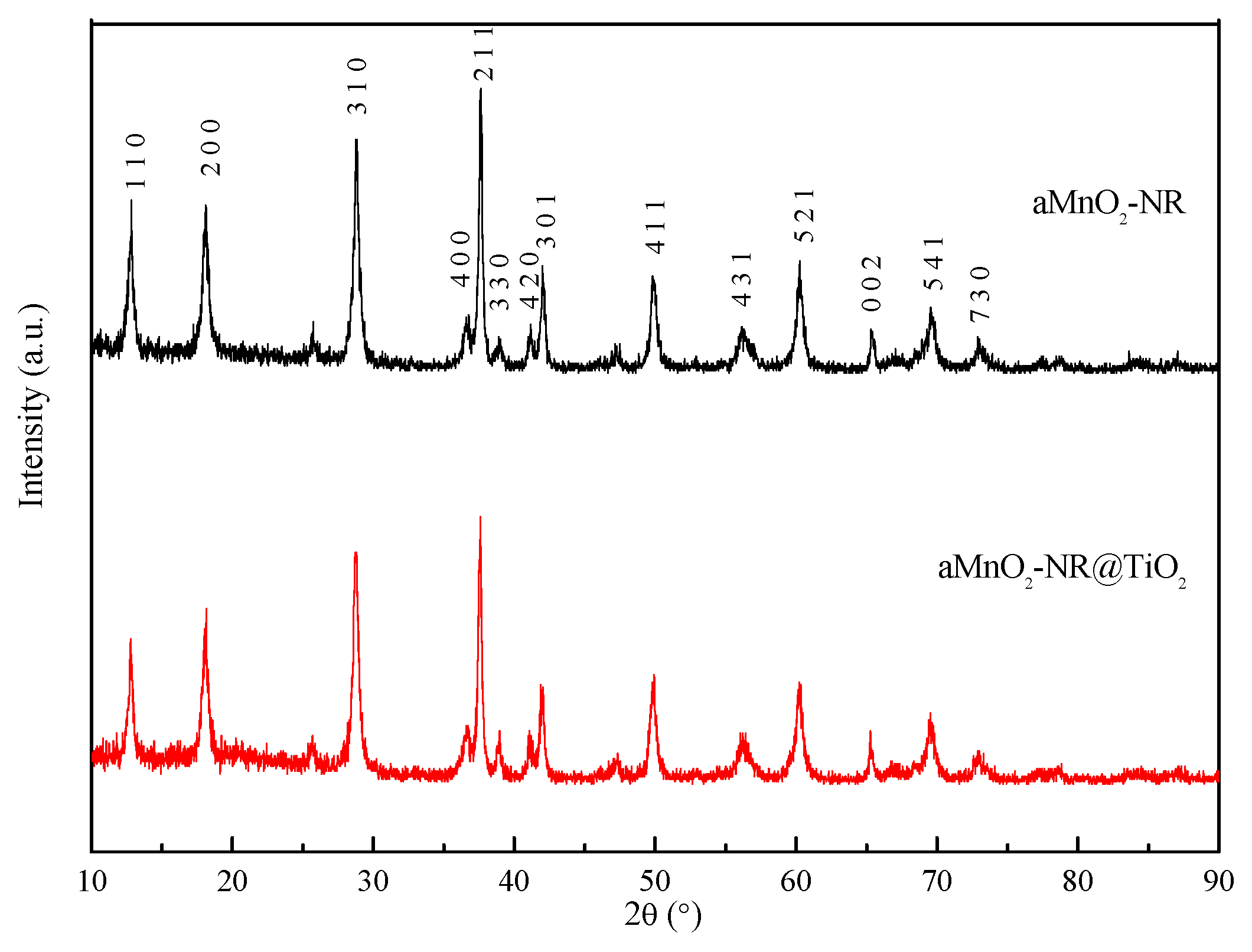
2.1. Structure Characterization
2.2. Hg0 Adsorption
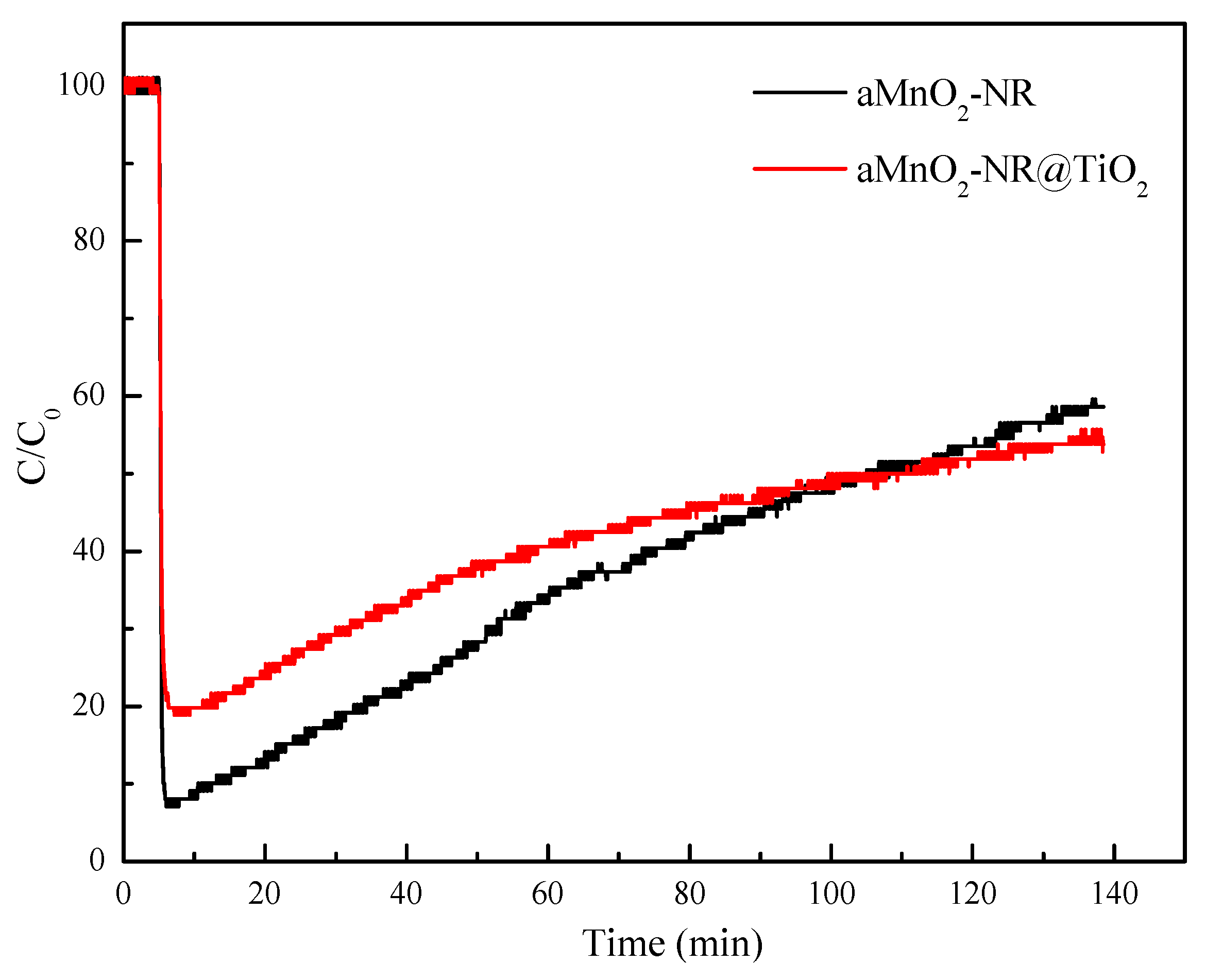
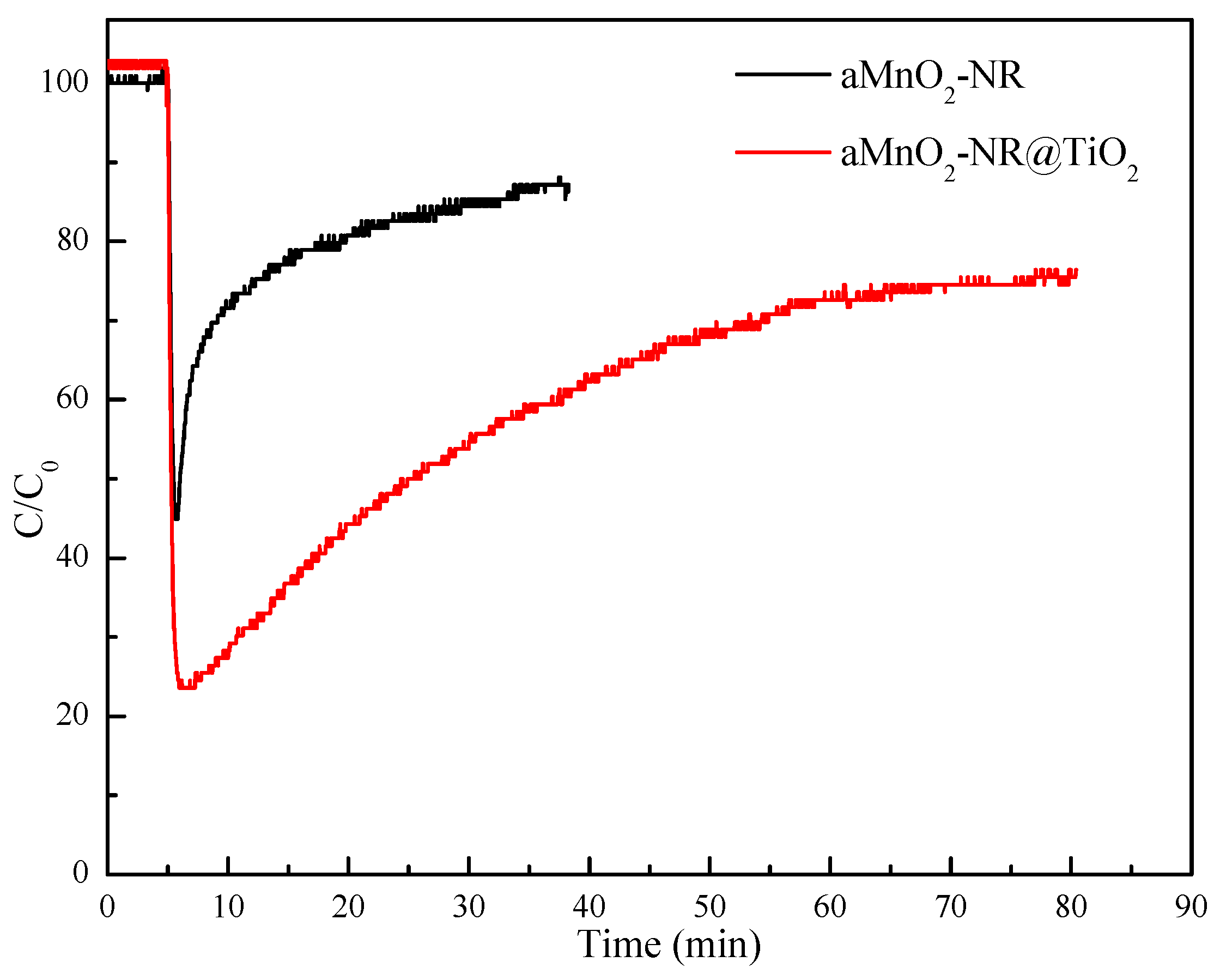
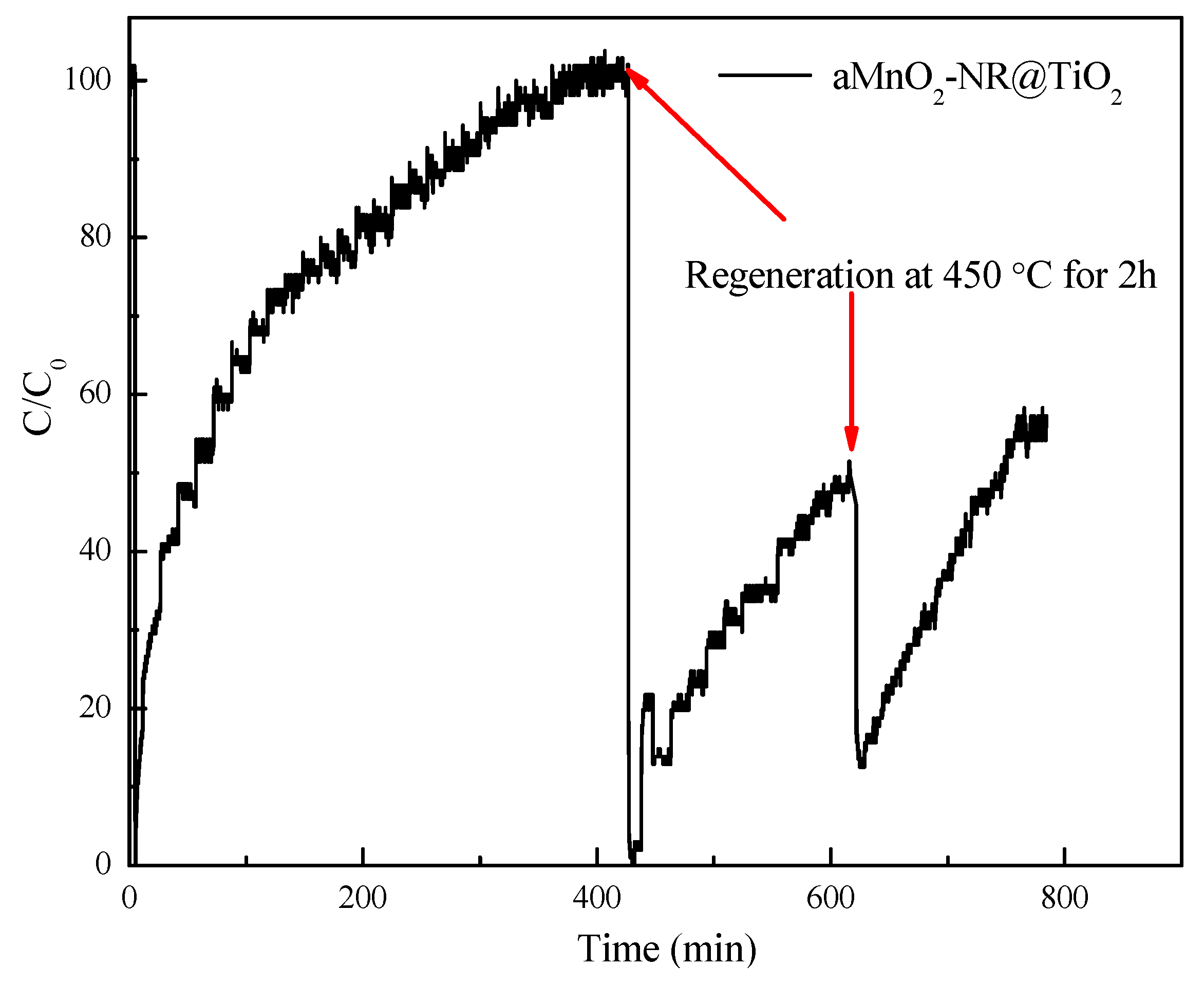
2.2.1. Hg0 Adsorption Performance
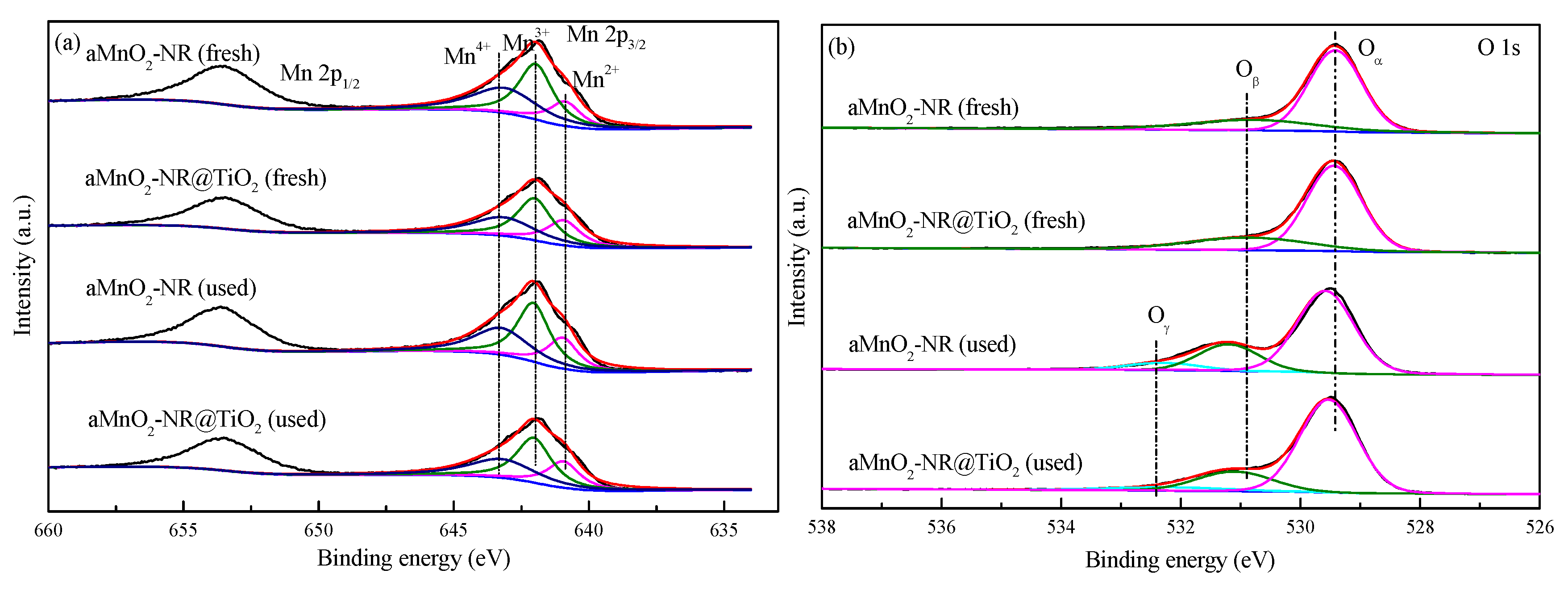
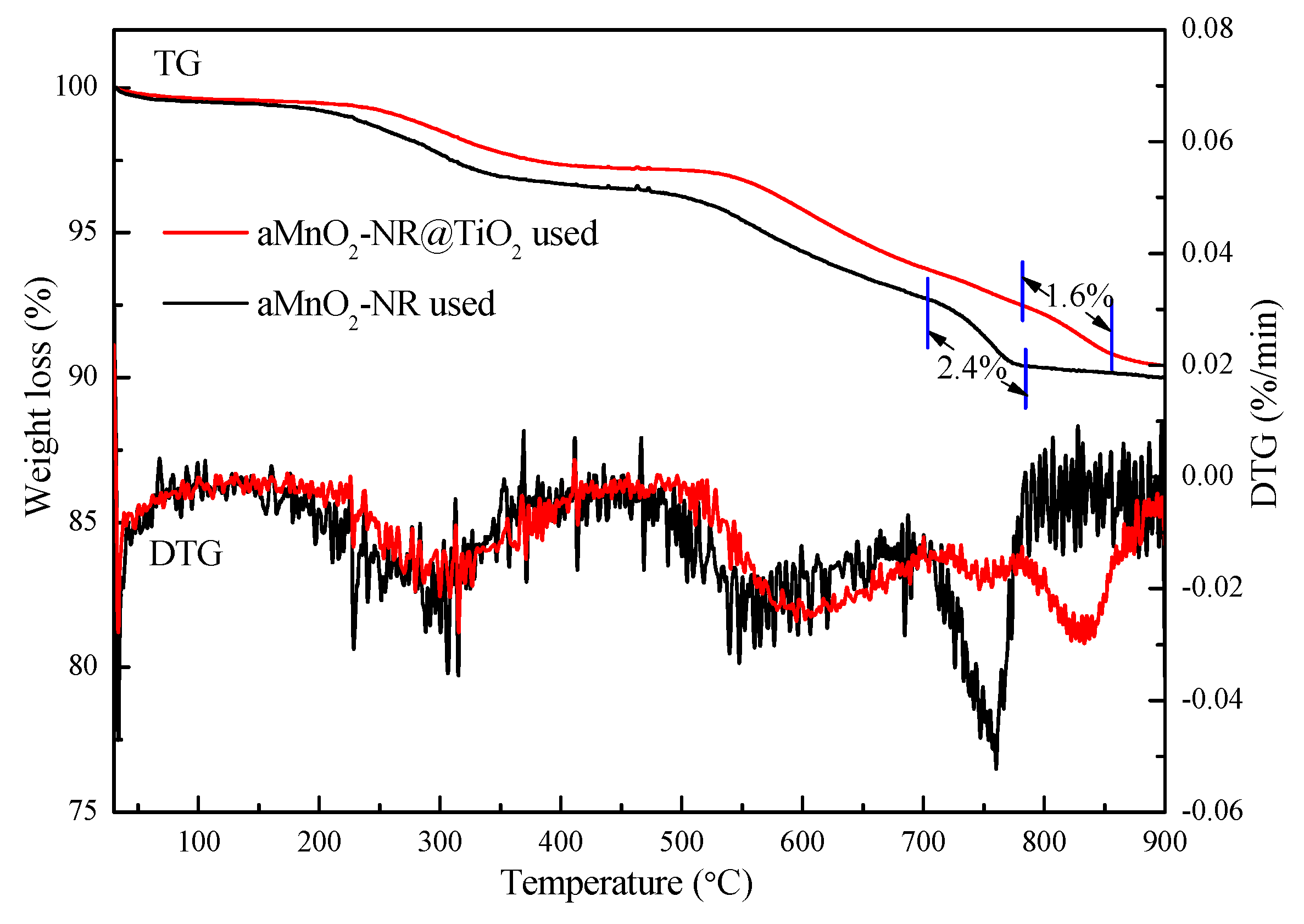
2.2.2. Structure-Activity Relationship
2.3. Models of Adsorption Kinetics
2.4. The Mechanism of SO2 Effects on Hg0 Adsorption
3. Materials and Methods
3.1. Catalysts Preparation
3.2. Hg0 Adsorption Experiments
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, H.; Mu, X.; Yang, G.; George, M.; Cao, P.; Fanady, B.; Rong, S.; Gao, X.; Wu, T. Graphene-like MoS2 containing adsorbents for Hg0 capture at coal-fired power plants. Appl. Energy 2017, 207, 254–264. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, Y.; Zhou, Q.; Zhu, C.; She, M.; Ding, W. Adsorptive removal of gas-phase mercury by oxygen non-thermal plasma modified activated carbon. Chem. Eng. J. 2016, 294, 281–289. [Google Scholar] [CrossRef]
- Zhang, B.; Zeng, X.; Xu, P.; Chen, J.; Xu, Y.; Luo, G.; Xu, M.; Yao, H. Using the Novel Method of Nonthermal Plasma To Add Cl Active Sites on Activated Carbon for Removal of Mercury from Flue Gas. Environ. Sci. Technol. 2016, 50, 11837–11843. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Duan, Y.F.; Hong, Y.G.; Zhu, C.; She, M.; Zhang, J.; Wei, H.Q. Experimental and kinetic studies of gas-phase mercury adsorption by raw and bromine modified activated carbon. Fuel Process. Technol. 2015, 134, 325–332. [Google Scholar] [CrossRef]
- Liu, W.; Vidic, R.D.; Brown, T.D. Impact of Flue Gas Conditions on Mercury Uptake by Sulfur-Impregnated Activated Carbon. Environ. Sci. Technol. 2000, 34, 154–159. [Google Scholar] [CrossRef]
- Bisson, T.M.; Xu, Z. Potential Hazards of Brominated Carbon Sorbents for Mercury Emission Control. Environ. Sci. Technol. 2015, 49, 2496–2502. [Google Scholar] [CrossRef]
- Chen, C.; Jia, W.; Liu, S.; Cao, Y. The enhancement of CuO modified V2O5-WO3/TiO2 based SCR catalyst for Hg0 oxidation in simulated flue gas. Appl. Surf. Sci. 2018, 436, 1022–1029. [Google Scholar] [CrossRef]
- Li, H.; Zhang, W.; Wang, J.; Yang, Z.; Li, L.; Shih, K. Coexistence of enhanced Hg0 oxidation and induced Hg2+ reduction on CuO/TiO2 catalyst in the presence of NO and NH3. Chem. Eng. J. 2017, 330, 1248–1254. [Google Scholar] [CrossRef]
- Liu, T.; Xue, L.; Guo, X.; Liu, J.; Huang, Y.; Zheng, C. Mechanisms of Elemental Mercury Transformation on α-Fe2O3(001) Surface from Experimental and Theoretical Study: Influences of HCl, O2, and SO2. Environ. Sci. Technol. 2016, 50, 13585–13591. [Google Scholar] [CrossRef]
- Zarei, S.; Niad, M.; Raanaei, H. The removal of mercury ion pollution by using Fe3O4-nanocellulose: Synthesis, characterizations and DFT studies. J. Hazard. Mater. 2018, 344, 258–273. [Google Scholar] [CrossRef]
- He, C.; Shen, B.; Chi, G.; Li, F. Elemental mercury removal by CeO2/TiO2-PILCs under simulated coal-fired flue gas. Chem. Eng. J. 2016, 300, 1–8. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, X.; Huang, Z.; Hou, Y.; Guo, Y.; Wu, M. Superior activity of CeO2 modified V2O5/AC catalyst for mercury removal at low temperature. Chem. Eng. J. 2018, 337, 741–749. [Google Scholar] [CrossRef]
- Xu, H.; Jia, J.; Guo, Y.; Qu, Z.; Liao, Y.; Xie, J.; Shangguan, W.; Yan, N. Design of 3D MnO2/Carbon sphere composite for the catalytic oxidation and adsorption of elemental mercury. J. Hazard. Mater. 2018, 342, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Cimino, S.; Scala, F. Removal of Elemental Mercury by MnOx Catalysts Supported on TiO2 or Al2O3. Ind. Eng. Chem. Res. 2016, 55, 5133–5138. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, Y.; Yang, J.; Zhang, J.; Zheng, C. Fe-modified MnOx/TiO2 as the SCR catalyst for simultaneous removal of NO and mercury from coal combustion flue gas. Chem. Eng. J. 2018, 348, 618–629. [Google Scholar] [CrossRef]
- Yao, T.; Duan, Y.; Zhu, C.; Zhou, Q.; Xu, J.; Liu, M.; Wei, H. Investigation of mercury adsorption and cyclic mercury retention over MnOx/γ-Al2O3 sorbent. Chemosphere 2018, 202, 358–365. [Google Scholar] [CrossRef]
- Xie, J.K.; Qu, Z.; Yan, N.Q.; Yang, S.J.; Chen, W.M.; Hu, L.G.; Huang, W.J.; Liu, P. Novel regenerable sorbent based on Zr–Mn binary metal oxides for flue gas mercury retention and recovery. J. Hazard. Mater. 2013, 261, 206–213. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Wang, S.; Wang, X.; Hu, J. Removal of elemental mercury in flue gas at lower temperatures over Mn-Ce based materials prepared by co-precipitation. Fuel 2017, 208, 576–586. [Google Scholar] [CrossRef]
- Yi, Y.; Li, C.; Zhao, L.; Du, X.; Gao, L.; Chen, J.; Zhai, Y.; Zeng, G. The synthetic evaluation of CuO-MnOx-modified pinecone biochar for simultaneous removal formaldehyde and elemental mercury from simulated flue gas. Environ. Sci. Pollut. Res. 2017, 25, 4761–4775. [Google Scholar] [CrossRef]
- Xu, H.; Qu, Z.; Zhao, S.; Mei, J.; Quan, F.; Yan, N. Different crystal-forms of one-dimensional MnO2 nanomaterials for the catalytic oxidation and adsorption of elemental mercury. J. Hazard. Mater. 2015, 299, 86–93. [Google Scholar] [CrossRef]
- Chalkidis, A.; Jampaiah, D.; Hartley, P.G.; Sabri, Y.M.; Bhargava, S.K. Regenerable α-MnO2 nanotubes for elemental mercury removal from natural gas. Fuel Process. Technol. 2019, 193, 317–327. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Wang, J.; Tan, B.; Cui, Y.; He, G. Reaction mechanism for the influence of SO2 on Hg0 adsorption and oxidation with Ce0.1-Zr-MnO2. Fuel 2017, 203, 308–315. [Google Scholar] [CrossRef]
- Yang, Z.; Li, H.; Liu, X.; Li, P.; Yang, J.; Lee, P.H.; Shih, K. Promotional effect of CuO loading on the catalytic activity and SO2 resistance of MnOx/TiO2 catalyst for simultaneous NO reduction and Hg0 oxidation. Fuel 2018, 227, 79–88. [Google Scholar] [CrossRef]
- Li, H.; Wu, C.Y.; Li, Y.; Zhang, J. Superior activity of MnOx-CeO2/TiO2 catalyst for catalytic oxidation of elemental mercury at low flue gas temperatures. Appl. Catal. B 2012, 111, 381–388. [Google Scholar] [CrossRef]
- Mitsudome, T.; Yamamoto, M.; Maeno, Z.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. One-step Synthesis of Core-Gold/Shell-Ceria Nanomaterial and Its Catalysis for Highly Selective Semihydrogenation of Alkynes. J. Am. Chem. Soc. 2015, 137, 13452–13455. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, T.; Wang, Q.; Umar, A. Morphology and chemical composition dependent synthesis and electrochemical properties of MnO2-based nanostructures for efficient hydrazine detection. Sens. Actuators B 2016, 224, 878–884. [Google Scholar] [CrossRef]
- Tabish, T.A.; Memon, F.A.; Gomez, D.E.; Horsell, D.W.; Zhang, S. A facile synthesis of porous graphene for efficient water and wastewater treatment. Sci. Rep. 2018, 8, 1817. [Google Scholar] [CrossRef]
- McKay, G.; Ho, Y.S. Pseudo-second order model for sorption processes. Process Biochem. 1999, 34, 451–465. [Google Scholar]
- Zhang, D.; Hou, L.A.; Chen, G.; Zhang, A.; Wang, F.; Wang, R.; Li, C. Cr Doping MnOx Adsorbent Significantly Improving Hg0 Removal and SO2 Resistance from Coal-Fired Flue Gas and the Mechanism Investigation. Ind. Eng. Chem. Res. 2018, 57, 17245–17258. [Google Scholar] [CrossRef]
- Hu, H.; Cai, S.; Li, H.; Huang, L.; Shi, L.; Zhang, D. Mechanistic Aspects of deNOx Processing over TiO2 Supported Co–Mn Oxide Catalysts: Structure–Activity Relationships and In Situ DRIFTs Analysis. ACS Catal. 2015, 5, 6069–6077. [Google Scholar] [CrossRef]
- Xu, H.; Qu, Z.; Zong, C.; Quan, F.; Mei, J.; Yan, N. Catalytic oxidation and adsorption of Hg0 over low-temperature NH3-SCR LaMnO3 perovskite oxide from flue gas. Appl. Catal. B 2016, 186, 30–40. [Google Scholar] [CrossRef]
- Peng, Y.; Qu, R.; Zhang, X.; Li, J. The relationship between structure and activity of MoO3-CeO2 catalysts for NO removal: Influences of acidity and reducibility. Chem. Commun. 2013, 49, 6215–6217. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mu, B.; Zhang, X.; Yuan, D.; Ma, C.; Xu, H.; Qu, Z.; Fang, S. Graphene enhanced Mn-Ce binary metal oxides for catalytic oxidation and adsorption of elemental mercury from coal-fired flue gas. Chem. Eng. J. 2019, 358, 1499–1506. [Google Scholar] [CrossRef]
- Reddy, B.M.; Sreekanth, P.M.; Yamada, Y.; Xu, Q.; Kobayashi, T. Surface characterization of sulfate, molybdate, and tungstate promoted TiO2-ZrO2 solid acid catalysts by XPS and other techniques. Appl. Catal. A 2002, 228, 269–278. [Google Scholar] [CrossRef]
- Li, H.; Zhu, L.; Wang, J.; Li, L.; Shih, K. Development of Nano-Sulfide Sorbent for Efficient Removal of Elemental Mercury from Coal Combustion Fuel Gas. Environ. Sci. Technol. 2016, 50, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Chen, D.; Zou, S.; Xiong, S.; Xiao, X.; Dang, H.; Chen, T.; Yang, S. Recyclable Naturally Derived Magnetic Pyrrhotite for Elemental Mercury Recovery from Flue Gas. Environ. Sci. Technol. 2016, 50, 10562–10569. [Google Scholar] [CrossRef]
- Yang, S.; Guo, Y.; Yan, N.; Wu, D.; He, H.; Xie, J.; Qu, Z.; Jia, J. Remarkable effect of the incorporation of titanium on the catalytic activity and SO2 poisoning resistance of magnetic Mn–Fe spinel for elemental mercury capture. Appl. Catal. B Environ. 2011, 101, 698–708. [Google Scholar] [CrossRef]
- Xu, W.; He, H.; Yu, Y. Deactivation of a Ce/TiO2 Catalyst by SO2 in the Selective Catalytic Reduction of NO by NH3. J. Phys. Chem. C 2009, 113, 4426–4432. [Google Scholar] [CrossRef]
- Yu, J.; Guo, F.; Wang, Y.; Zhu, J.; Liu, Y.; Su, F.; Gao, S.; Xu, G. Sulfur poisoning resistant mesoporous Mn-base catalyst for low-temperature SCR of NO with NH3. Appl. Catal. B Environ. 2010, 95, 160–168. [Google Scholar] [CrossRef]
- Zhang, A.; Zhang, Z.; Lu, H.; Liu, Z.; Xiang, J.; Zhou, C.; Xing, W.; Sun, L. Effect of Promotion with Ru Addition on the Activity and SO2 Resistance of MnOx–TiO2 Adsorbent for Hg0 Removal. Ind. Eng. Chem. Res. 2015, 54, 2930–2939. [Google Scholar] [CrossRef]
- Wu, Z.; Jin, R.; Wang, H.; Liu, Y. Effect of ceria doping on SO2 resistance of Mn/TiO2 for selective catalytic reduction of NO with NH3 at low temperature. Catal. Commun. 2009, 10, 935–939. [Google Scholar] [CrossRef]
- Gardy, J.; Hassanpour, A.; Lai, X.; Ahmed, M.H. Synthesis of Ti(SO4)O solid acid nano-catalyst and its application for biodiesel production from used cooking oil. Appl. Catal. A 2016, 527, 81–95. [Google Scholar] [CrossRef]
- Liang, S.; Teng, F.; Bulgan, G.; Zong, R.; Zhu, Y. Effect of phase structure of MnO2 nanorod catalyst on the activity for CO oxidation. J. Phys. Chem. C 2008, 112, 5307–5315. [Google Scholar] [CrossRef]
- Li, W.; Yang, J.; Wu, Z.; Wang, J.; Li, B.; Feng, S.; Deng, Y.; Zhang, F.; Zhao, D. A versatile kinetics-controlled coating method to construct uniform porous TiO2 shells for multifunctional core–shell structures. J. Am. Chem. Soc. 2012, 134, 11864–11867. [Google Scholar] [CrossRef]
- Zhang, X.; Cui, Y.; Wang, J.; Tan, B.; Li, C.; Zhang, H.; He, G. Simultaneous removal of Hg0 and NO from flue gas by Co0.3-Ce0.35-Zr0.35O2 impregnated with MnOx. Chem. Eng. J. 2017, 326, 1210–1222. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
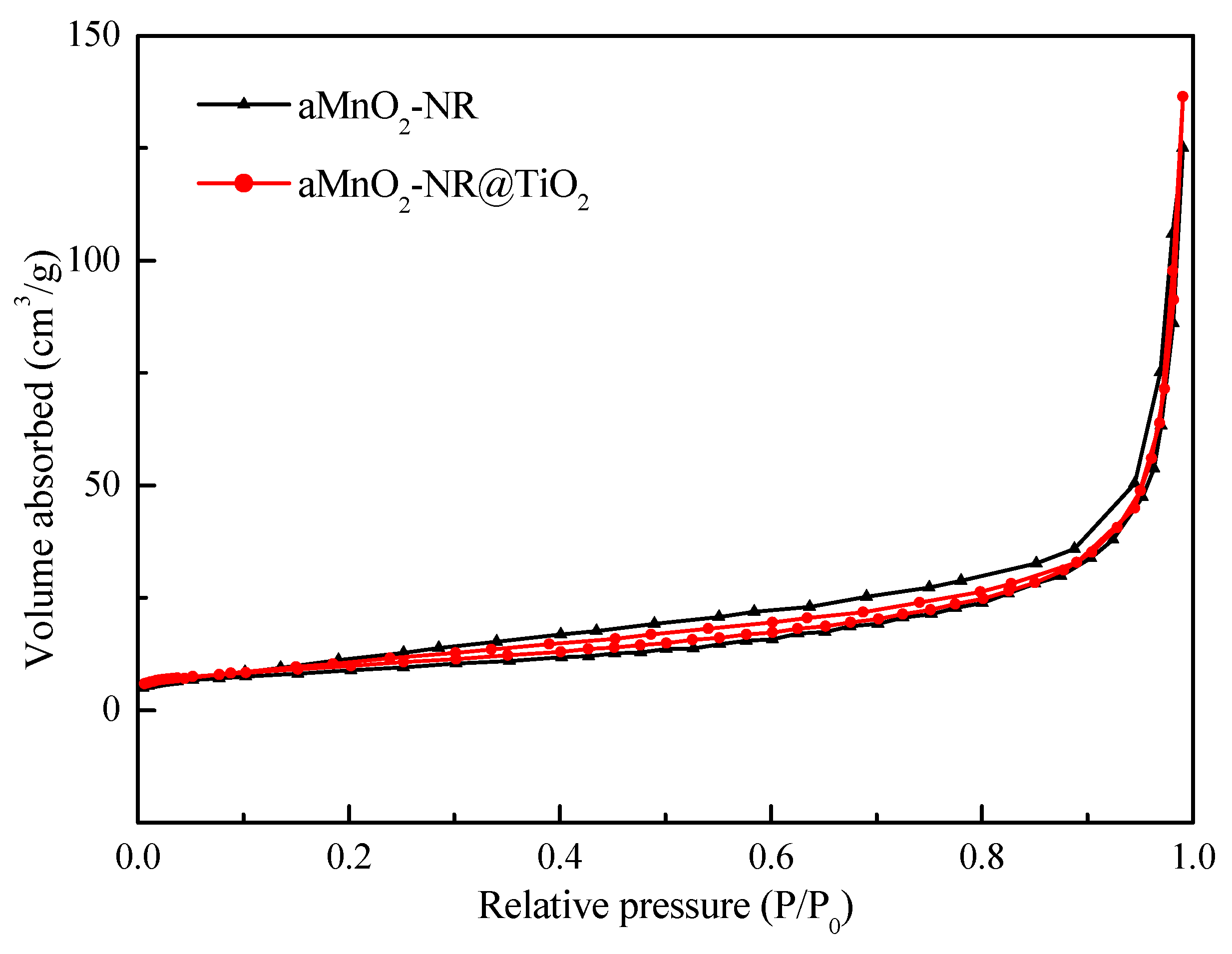
| Samples | BET Surface Area (m2/g) | Pore Volume (cm3/g) | Average Pore Diameter (nm) |
|---|---|---|---|
| αMnO2-NR | 29.103 | 0.192 | 5.428 |
| αMnO2-NR@TiO2 | 32.985 | 0.207 | 4.186 |
| Kinetic Models | αMnO2-NR without SO2 | αMnO2-NR@TiO2 without SO2 | αMnO2-NR with SO2 | αMnO2-NR@TiO2 with SO2 |
|---|---|---|---|---|
| Pseudo-first (R2) | 0.944 | 0.938 | 0.954 | 0.941 |
| Pseudo-second (R2) | 0.991 | 0.995 | 0.997 | 0.992 |
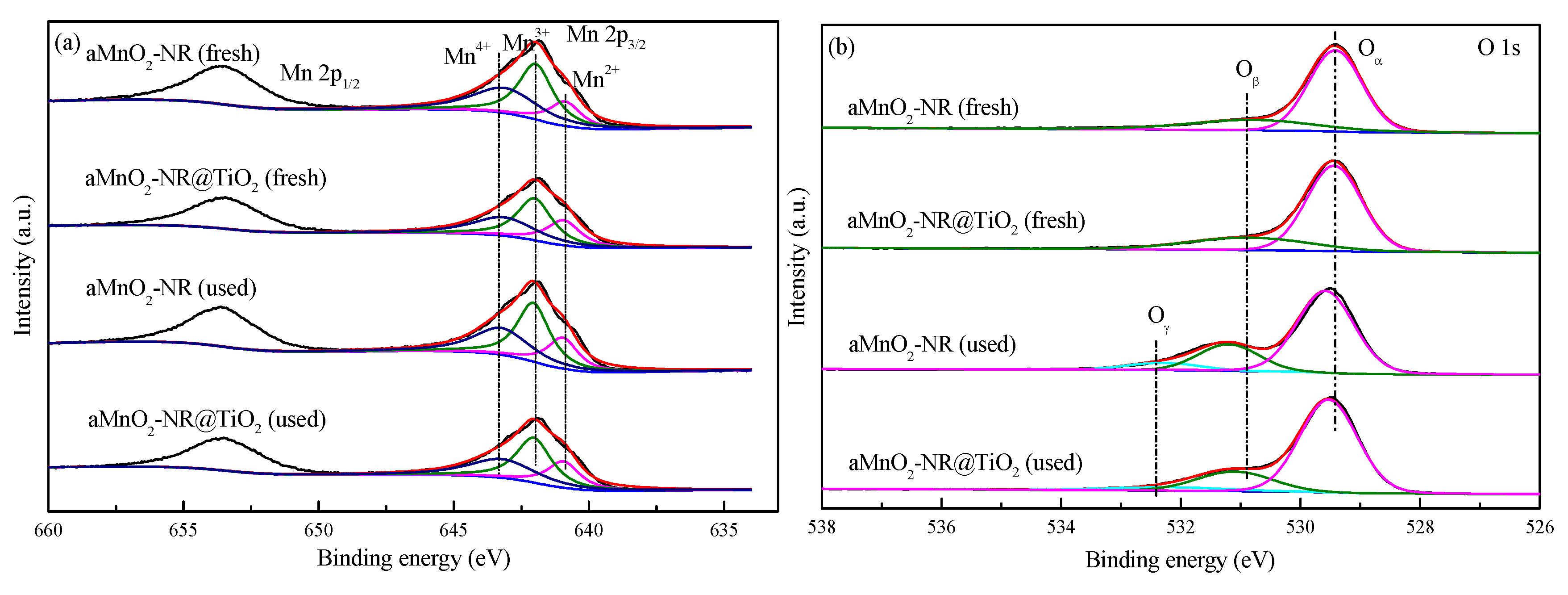
| Samples | S | Mn4+/Mn | Oβ/O |
|---|---|---|---|
| αMnO2-NR (fresh) | 3.17 | 37.8 | 26.0 |
| αMnO2-NR@TiO2 (fresh) | 2.27 | 33.4 | 24.7 |
| αMnO2-NR (used) | 4.97 | 34.0 | 22.8 |
| αMnO2-NR@TiO2 (used) | 2.66 | 33.0 | 20.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Han, X.; Li, C.; Song, X.; Zhu, H.; Bao, J.; Zhang, N.; He, G. Promoting Effect of the Core-Shell Structure of MnO2@TiO2 Nanorods on SO2 Resistance in Hg0 Removal Process. Catalysts 2020, 10, 72. https://doi.org/10.3390/catal10010072
Zhang X, Han X, Li C, Song X, Zhu H, Bao J, Zhang N, He G. Promoting Effect of the Core-Shell Structure of MnO2@TiO2 Nanorods on SO2 Resistance in Hg0 Removal Process. Catalysts. 2020; 10(1):72. https://doi.org/10.3390/catal10010072
Chicago/Turabian StyleZhang, Xiaopeng, Xiangkai Han, Chengfeng Li, Xinxin Song, Hongda Zhu, Junjiang Bao, Ning Zhang, and Gaohong He. 2020. "Promoting Effect of the Core-Shell Structure of MnO2@TiO2 Nanorods on SO2 Resistance in Hg0 Removal Process" Catalysts 10, no. 1: 72. https://doi.org/10.3390/catal10010072
APA StyleZhang, X., Han, X., Li, C., Song, X., Zhu, H., Bao, J., Zhang, N., & He, G. (2020). Promoting Effect of the Core-Shell Structure of MnO2@TiO2 Nanorods on SO2 Resistance in Hg0 Removal Process. Catalysts, 10(1), 72. https://doi.org/10.3390/catal10010072