
EMT and Treatment Resistance in Pancreatic Cancer


{kind=link}
Abstract
:1. Introduction
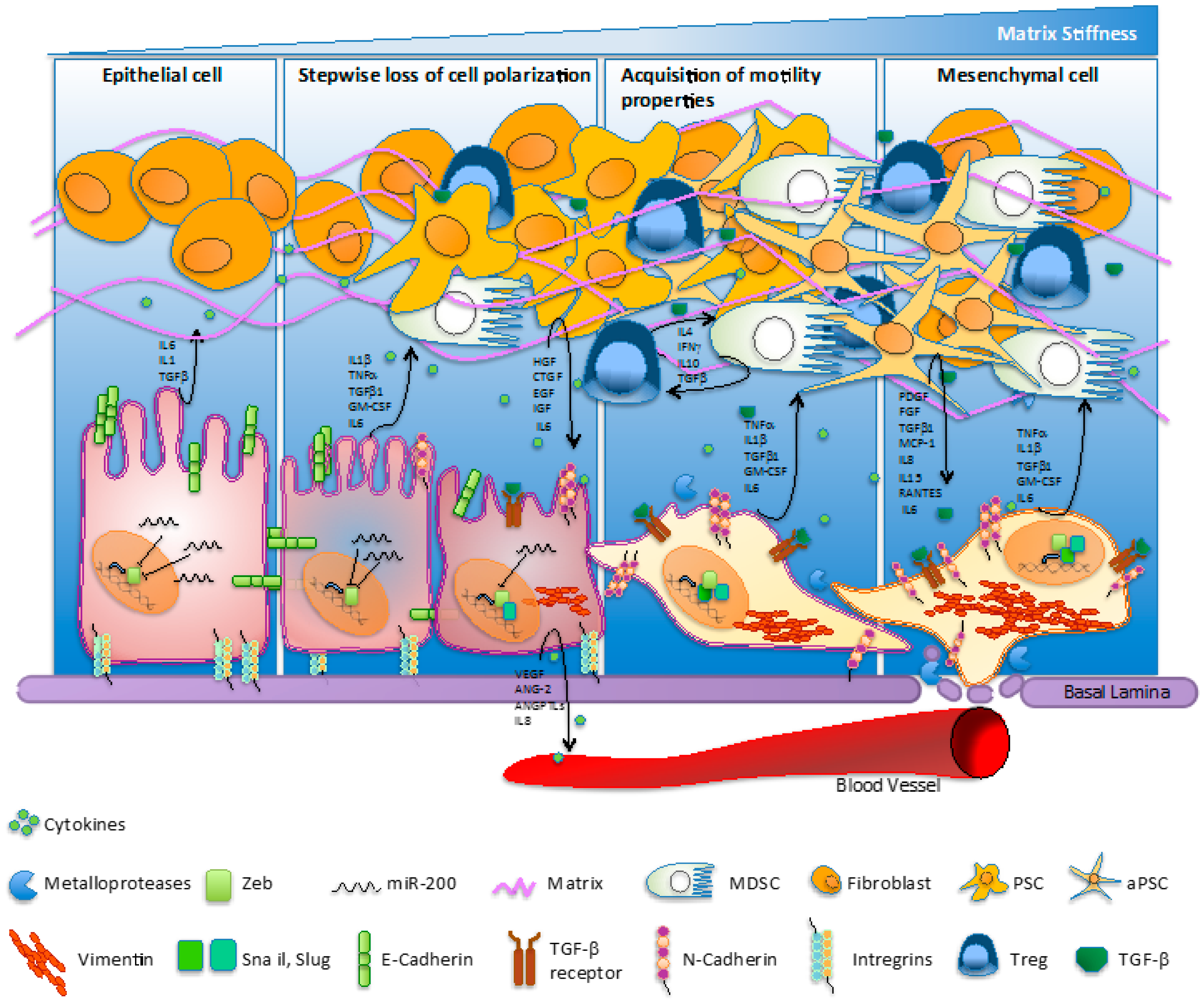
2. EMT and Cancer Progression
3. KRAS-Addiction of Pancreatic Cancer
4. Markers of EMT in Advanced Pancreatic Neoplasia
5. EMT and Metastasis
6. Molecular Mechanisms of EMT and PC Treatment Resistance
7. Role of Desmoplasia in Pancreatic Cancer
8. Inflammation Sustains EMT in Pancreatic Cancer
9. Microbiota, EMT, and Treatment Resistance
10. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Budillon, A. Pancreatic cancer: Between bench and bedside. Curr. Drug Targets 2012, 13, 729–730. [Google Scholar] [CrossRef] [PubMed]
- Tamburrino, A.; Piro, G.; Carbone, C.; Tortora, G.; Melisi, D. Mechanisms of resistance to chemotherapeutic and anti-angiogenic drugs as novel targets for pancreatic cancer therapy. Front. Pharmacol. 2013, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Calvetti, L.; Frizziero, M.; Tortora, G. Pancreatic cancer: Systemic combination therapies for a heterogeneous disease. Curr. Pharm. Des. 2014, 20, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, V.; Melisi, D.; Bria, E.; Cuppone, F.; Ciuffreda, L.; Pino, M.S.; Gelibter, A.; Tortora, G.; Cognetti, F.; Milella, M. Emerging pathways and future targets for the molecular therapy of pancreatic cancer. Expert Opin. Ther. Targets 2011, 15, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Maitra, A.; Yeo, C.J. Genetics and pathology of pancreatic cancer. HPB (Oxford) 2006, 8, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. Emt and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Groger, C.J.; Grubinger, M.; Waldhor, T.; Vierlinger, K.; Mikulits, W. Meta-analysis of gene expression signatures defining the epithelial to mesenchymal transition during cancer progression. PLoS ONE 2012, 7, e51136. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Sun, H.; Zhang, W.; Zhang, M.; Yang, X.; Kuang, R.; Zheng, H. Meta-analysis of emt datasets reveals different types of emt. PLoS ONE 2016, 11, e0156839. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Yamamoto, S.; Burnette, B.L.; Huang, D.; Gao, K.; Jamshidi, N.; Kuo, M.D. Transcriptome profiling reveals novel gene expression signatures and regulating transcription factors of tgfbeta-induced epithelial-to-mesenchymal transition. Cancer Med. 2016, 5, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. Ly2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Doi, R.; Toyoda, E.; Tsuji, S.; Wada, M.; Koizumi, M.; Tulachan, S.S.; Ito, D.; Kami, K.; Mori, T.; et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 2004, 10, 4125–4133. [Google Scholar] [CrossRef] [PubMed]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/akt pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor zeb1 (deltaef1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, M.A. The snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. Tgf-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Bonomi, S.; Gallo, S.; Ghigna, C. Making alternative splicing decisions during epithelial-to-mesenchymal transition (emt). Cell. Mol. Life Sci. 2012, 69, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Pichler, M.; Neureiter, D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol. Clin. Oncol. 2013, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Guerra, C.; Barbacid, M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential role for oncogenic ras in tumour maintenance. Nature 1999, 400, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic kras signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “k-ras addiction” reveals regulators of emt and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.N.; Gallick, G.E. Src, chemoresistance and epithelial to mesenchymal transition: Are they related? Anticancer Drugs 2007, 18, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Corbo, V.; Ponz-Sarvise, M.; Tuveson, D.A. The ras and yap1 dance, who is leading? EMBO J. 2014, 33, 2437–2438. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of yap/taz in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S. Role of yap/taz in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- Piro, G.; Carbone, C.; Cataldo, I.; Di Nicolantonio, F.; Giacopuzzi, S.; Aprile, G.; Simionato, F.; Boschi, F.; Zanotto, M.; Mina, M.M.; et al. An fgfr3 autocrine loop sustains acquired resistance to trastuzumab in gastric cancer patients. Clin. Cancer Res. 2016, 22, 6164–6175. [Google Scholar] [CrossRef] [PubMed]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef] [PubMed]
- Cates, J.M.; Byrd, R.H.; Fohn, L.E.; Tatsas, A.D.; Washington, M.K.; Black, C.C. Epithelial-mesenchymal transition markers in pancreatic ductal adenocarcinoma. Pancreas 2009, 38, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Filios, S.R.; Xu, G.; Chen, J.; Hong, K.; Jing, G.; Shalev, A. MicroRNA-200 is induced by thioredoxin-interacting protein and regulates zeb1 protein signaling and beta cell apoptosis. J. Biol. Chem. 2014, 289, 36275–36283. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Helm, J.; Coppola, D.; Kim, D.; Malafa, M.; Kim, S.J. MicroRNAs in pancreatic ductal adenocarcinoma. World J. Gastroenterol. 2011, 17, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Abou Tayoun, A.N.; Abo, K.M.; Pipas, J.M.; Gordon, S.R.; Gardner, T.B.; Barth, R.J., Jr.; Suriawinata, A.A.; Tsongalis, G.J. MicroRNAs as diagnostic markers for pancreatic ductal adenocarcinoma and its precursor, pancreatic intraepithelial neoplasm. Cancer Genet. 2013, 206, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, D.; Dehlendorff, C.; Boisen, M.K.; Hasselby, J.P.; Schultz, N.A.; Werner, J.; Immervoll, H.; Molven, A.; Hansen, C.P.; Johansen, J.S. Tissue microrna profiles as diagnostic and prognostic biomarkers in patients with resectable pancreatic ductal adenocarcinoma and periampullary cancers. Biomark Res. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Kadera, B.E.; Li, L.; Toste, P.A.; Wu, N.; Adams, C.; Dawson, D.W.; Donahue, T.R. MicroRNA-21 in pancreatic ductal adenocarcinoma tumor-associated fibroblasts promotes metastasis. PLoS ONE 2013, 8, e71978. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Carvajo, M.; Bauden, M.; Andersson, R. Pancreatic cancer stroma: Controversies and current insights. Scand. J. Gastroenterol. 2017, 52, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, B.; Liu, F.; Zhang, M.; Wang, Q.; Liu, Y.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (emt) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Dando, I.; Biondani, G.; Brandi, J.; Costanzo, C.; Zoratti, E.; Fassan, M.; Boschi, F.; Melisi, D.; Cecconi, D.; et al. Pancreatic ductal adenocarcinoma cell lines display a plastic ability to bidirectionally convert into cancer stem cells. Int. J. Oncol. 2015, 46, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Haeno, H.; Gonen, M.; Davis, M.B.; Herman, J.M.; Iacobuzio-Donahue, C.A.; Michor, F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 2012, 148, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Batra, S.K. Pancreatic cancer metastasis: Are we being pre-emted? Curr. Pharm. Des. 2015, 21, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ting, D.T.; Stott, S.L.; Wittner, B.S.; Ozsolak, F.; Paul, S.; Ciciliano, J.C.; Smas, M.E.; Winokur, D.; Gilman, A.J.; et al. RNA sequencing of pancreatic circulating tumour cells implicates wnt signalling in metastasis. Nature 2012, 487, 510–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Su, H.T.; Weng, C.C.; Hsiao, P.J.; Chen, L.H.; Kuo, T.L.; Chen, Y.W.; Kuo, K.K.; Cheng, K.H. Stem cell marker nestin is critical for tgf-beta1-mediated tumor progression in pancreatic cancer. Mol. Cancer Res. 2013, 11, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Carriere, C.; Seeley, E.S.; Goetze, T.; Longnecker, D.S.; Korc, M. The nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 4437–4442. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, A.; Raanan, C.; Polin, N.; Melzer, E.; Givol, D.; Schreiber, L. Modulation of c-kit expression in pancreatic adenocarcinoma: A novel stem cell marker responsible for the progression of the disease. Acta Histochem. 2014, 116, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, V.; Sperduti, I.; Vari, S.; Bria, E.; Melisi, D.; Garufi, C.; Nuzzo, C.; Scarpa, A.; Tortora, G.; Cognetti, F.; et al. Metastatic pancreatic cancer: Is there a light at the end of the tunnel? World J. Gastroenterol. 2015, 21, 4788–4801. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Melisi, D. Nf-kappab as a target for pancreatic cancer therapy. Expert Opin. Ther. Targets 2012, 16, S1–S10. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Chiao, P.J. Nf-kappa b as a target for cancer therapy. Expert Opin. Ther. Targets 2007, 11, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Piro, G.; Giacopuzzi, S.; Bencivenga, M.; Carbone, C.; Verlato, G.; Frizziero, M.; Zanotto, M.; Mina, M.M.; Merz, V.; Santoro, R.; et al. Tak1-regulated expression of BIRC3 predicts resistance to preoperative chemoradiotherapy in oesophageal adenocarcinoma patients. Br. J. Cancer 2015, 113, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to egfr and pi3k inhibitors and identifies axl as a therapeutic target for overcoming egfr inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Cardone, R.A.; Greco, M.R.; Zeeberg, K.; Zaccagnino, A.; Saccomano, M.; Bellizzi, A.; Bruns, P.; Menga, M.; Pilarsky, C.; Schwab, A.; et al. A novel nhe1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 2015, 17, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A. Targeted therapies for pancreatic cancer. Gastrointest. Cancer Res. 2008, 2, S16–S19. [Google Scholar] [PubMed]
- Tortora, G.; Melisi, D.; Ciardiello, F. Angiogenesis: A target for cancer therapy. Curr. Pharm. Des. 2004, 10, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Piro, G.; Fassan, M.; Tamburrino, A.; Mina, M.M.; Zanotto, M.; Chiao, P.J.; Bassi, C.; Scarpa, A.; Tortora, G.; et al. An angiopoietin-like protein 2 autocrine signaling promotes emt during pancreatic ductal carcinogenesis. Oncotarget 2015, 6, 13822–13834. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Moccia, T.; Zhu, C.; Paradiso, G.; Budillon, A.; Chiao, P.J.; Abbruzzese, J.L.; Melisi, D. Anti-vegf treatment-resistant pancreatic cancers secrete proinflammatory factors that contribute to malignant progression by inducing an emt cell phenotype. Clin. Cancer Res. 2011, 17, 5822–5832. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Tamburrino, A.; Piro, G.; Boschi, F.; Cataldo, I.; Zanotto, M.; Mina, M.M.; Zanini, S.; Sbarbati, A.; Scarpa, A.; et al. Combined inhibition of il1, cxcr1/2, and tgfbeta signaling pathways modulates in vivo resistance to anti-vegf treatment. Anticancer Drugs 2016, 27, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Piro, G.; Simionato, F.; Ligorio, F.; Cremolini, C.; Loupakis, F.; Ali, G.; Rossini, D.; Merz, V.; Santoro, R.; et al. Homeobox b9 mediates resistance to anti-vegf therapy in colorectal cancer patients. Clin. Cancer Res. 2017, 15, 4312–4322. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Li, L.; Li, Z.; Xie, K. Targeted destruction of the orchestration of the pancreatic stroma and tumor cells in pancreatic cancer cases: Molecular basis for therapeutic implications. Cytokine Growth Factor Rev. 2012, 23, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells and pancreatic cancer cells: An unholy alliance. Cancer Res. 2008, 68, 7707–7710. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Thakar, D.; Weaver, V.M. Force-dependent breaching of the basement membrane. Matrix Biol. 2017, 57–58, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, J.; Chen, X.; Duan, W.; Ma, Q.; Li, X. Disrupting the balance between tumor epithelia and stroma is a possible therapeutic approach for pancreatic cancer. Med. Sci. Monit. 2014, 20, 2002–2006. [Google Scholar] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, U.K.; Moore, T.T.; Joo, H.G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory t cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Nummer, D.; Suri-Payer, E.; Schmitz-Winnenthal, H.; Bonertz, A.; Galindo, L.; Antolovich, D.; Koch, M.; Buchler, M.; Weitz, J.; Schirrmacher, V.; et al. Role of tumor endothelium in cd4+ cd25+ regulatory t cell infiltration of human pancreatic carcinoma. J. Natl. Cancer Inst. 2007, 99, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through tlr4/il-10 signaling pathway. Lab. Invest. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Jiang, Y.; Pillarisetty, V.G. Role of immune cells in pancreatic cancer from bench to clinical application: An updated review. Medicine 2016, 95, e5541. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. Emt and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Von Bernstorff, W.; Spanjaard, R.A.; Chan, A.K.; Lockhart, D.C.; Sadanaga, N.; Wood, I.; Peiper, M.; Goedegebuure, P.S.; Eberlein, T.J. Pancreatic cancer cells can evade immune surveillance via nonfunctional fas (apo-1/cd95) receptors and aberrant expression of functional fas ligand. Surgery 1999, 125, 73–84. [Google Scholar] [CrossRef]
- Trapani, J.A. The dual adverse effects of tgf-beta secretion on tumor progression. Cancer Cell 2005, 8, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. Pd-1/pd-l1 blockade together with vaccine therapy facilitates effector t-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Andersson, R.; Bauden, M.; Hammes, S.; Holdenrieder, S.; Ansari, D. Immune checkpoint therapy for pancreatic cancer. World J. Gastroenterol. 2016, 22, 9457–9476. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; O’Neill, E.; Gordon-Weeks, A.; Mukherjee, S.; McKenna, W.G.; Muschel, R.J. Pancreatic ductal adenocarcinoma: From genetics to biology to radiobiology to oncoimmunology and all the way back to the clinic. Biochim. Biophys. Acta 2015, 1855, 61–82. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of ctla-4 on both effector and regulatory t cell compartments contributes to the antitumor activity of anti-ctla-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (anti-ctla-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Myint, Z.W.; Goel, G. Role of modern immunotherapy in gastrointestinal malignancies: A review of current clinical progress. J. Hematol. Oncol. 2017, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. Pd-l1 regulates the development, maintenance, and function of induced regulatory t cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of pd-l1 on tumor cells in the escape from host immune system and tumor immunotherapy by pd-l1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the pd-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Izard, J. Microbiota, oral microbiome, and pancreatic cancer. Cancer J. 2014, 20, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Ertz-Archambault, N.; Keim, P.; Von Hoff, D. Microbiome and pancreatic cancer: A comprehensive topic review of literature. World J. Gastroenterol. 2017, 23, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Said, N.A.; Williams, E.D. Growth factors in induction of epithelial-mesenchymal transition and metastasis. Cells Tissues Organs 2011, 193, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Nakagawa, S.; Sawayama, H.; Ishimoto, T.; Imai, K.; Iwatsuki, M.; Hashimoto, D.; Baba, Y.; Yamashita, Y.I.; Yoshida, N.; et al. The microbiome and hepatobiliary-pancreatic cancers. Cancer Lett. 2017, 402, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by ctla-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-pd-l1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G.; Stebbing, J.; Adamson, D.J.; Arif, S.S.; Bidoli, P.; Chang, D.; Cheeseman, S.; Diaz-Beveridge, R.; Fernandez-Martos, C.; Glynne-Jones, R.; et al. Randomised, open-label, phase ii study of gemcitabine with and without imm-101 for advanced pancreatic cancer. Br. J. Cancer 2016, 115, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with gvax pancreas prime and listeria monocytogenes-expressing mesothelin (crs-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaianigo, N.; Melisi, D.; Carbone, C. EMT and Treatment Resistance in Pancreatic Cancer. Cancers 2017, 9, 122. https://doi.org/10.3390/cancers9090122
Gaianigo N, Melisi D, Carbone C. EMT and Treatment Resistance in Pancreatic Cancer. Cancers. 2017; 9(9):122. https://doi.org/10.3390/cancers9090122
Chicago/Turabian StyleGaianigo, Nicola, Davide Melisi, and Carmine Carbone. 2017. "EMT and Treatment Resistance in Pancreatic Cancer" Cancers 9, no. 9: 122. https://doi.org/10.3390/cancers9090122