
Targeting Platelets for the Treatment of Cancer



Abstract
1. Introduction
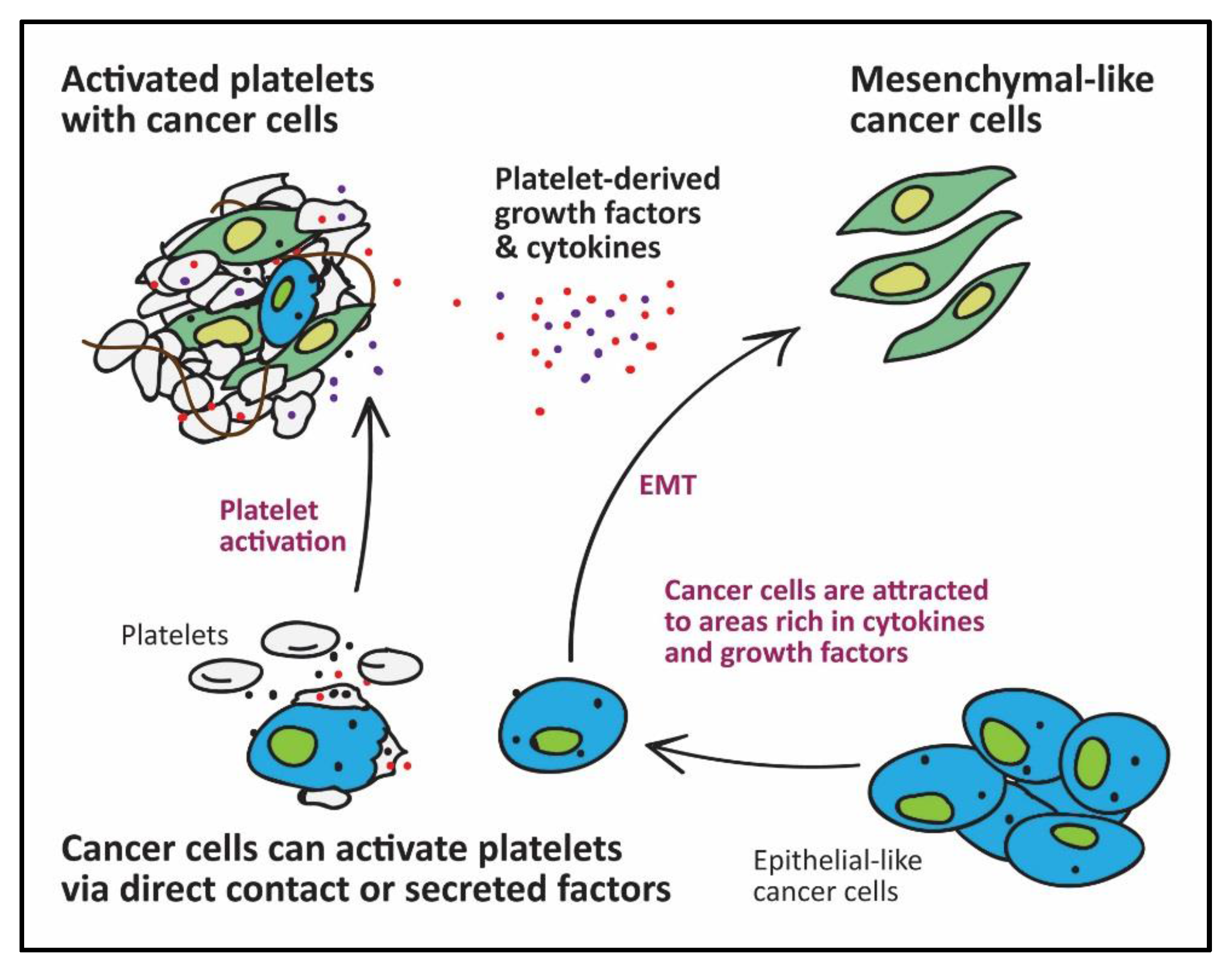
2. The Role of Platelets in Cancer Metastasis
3. The Role of Platelets in Tumour Angiogenesis
4. The Role of Platelets in Tumour Growth
5. The Role of Platelets in Chemotherapy Resistance
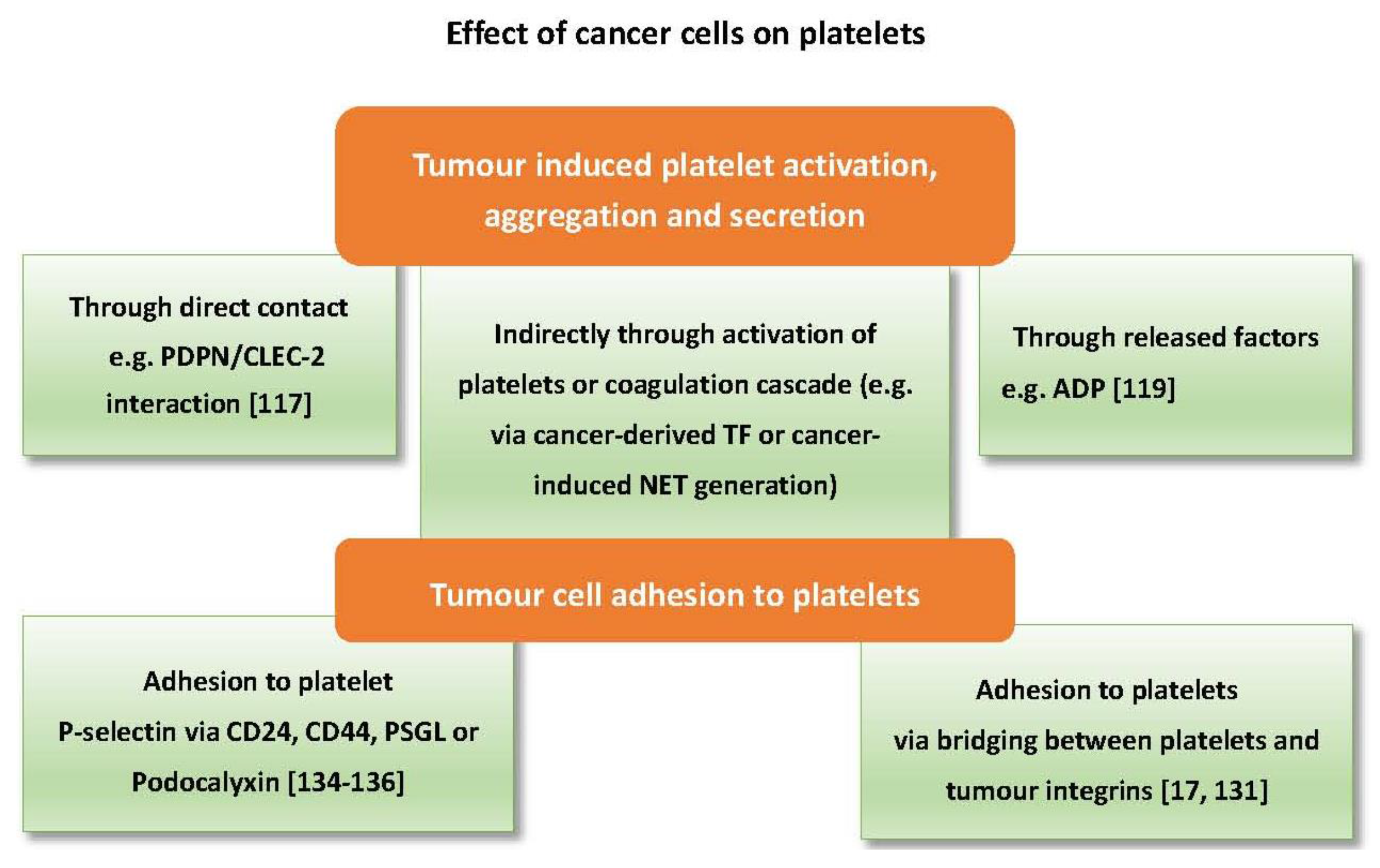
6. The Effects of Cancer Cells on Platelets
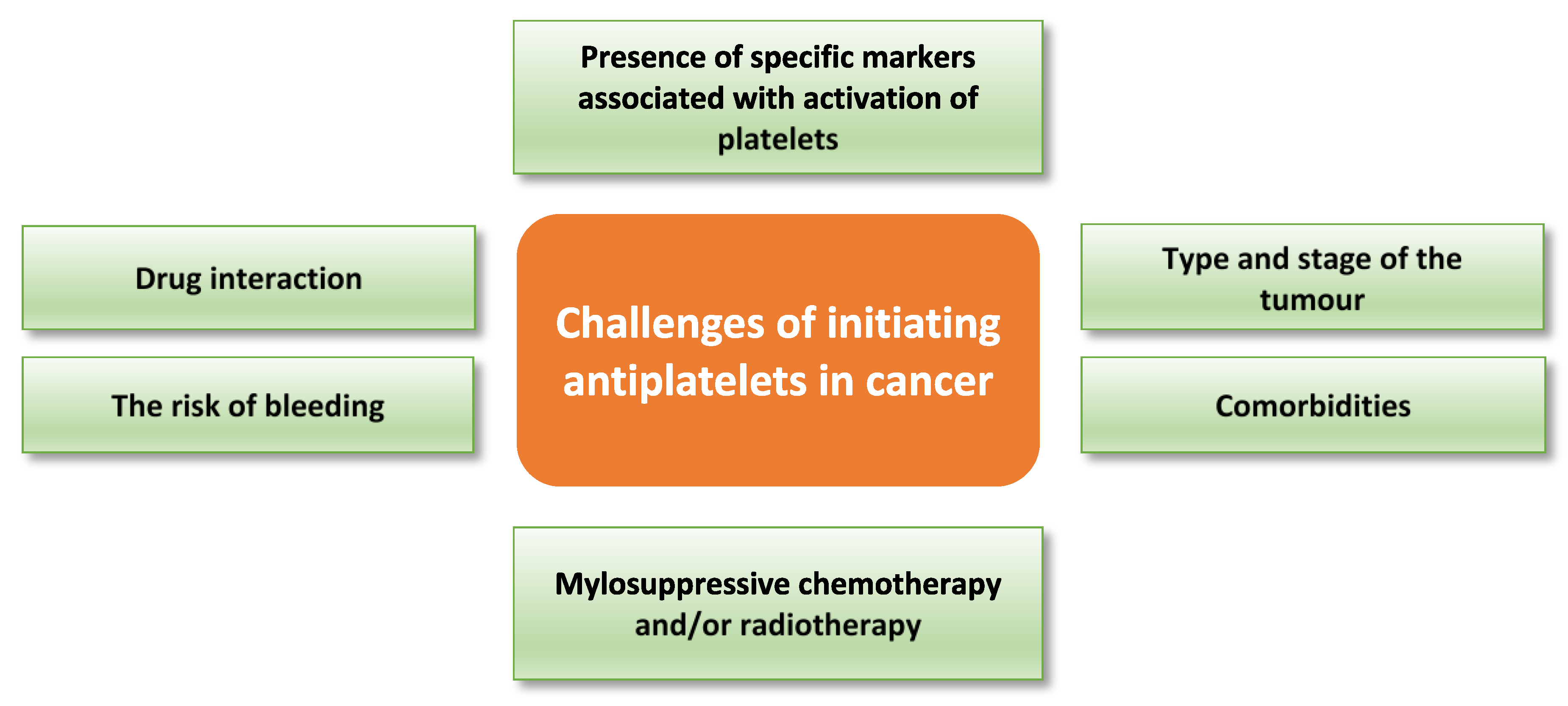
7. Challenges to Antiplatelet Therapeutic Approaches in Cancer
8. Clinical and Preclinical Use of Antiplatelet Therapies in Cancer
8.1. Aspirin in Cancer
8.1.1. Preclinical Studies
8.1.2. Clinical Studies
8.2. Other Antiplatelet Strategies in Cancer Therapy
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Erpenbeck, L.; Schon, M.P. Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Berger, S. Platelet function: A review. I. normal function. Can. Med. Assoc. J. 1970, 102, 1271–1274. [Google Scholar] [PubMed]
- Koenen, R.R. The prowess of platelets in immunity and inflammation. Thromb. Haemost. 2016, 116, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Gasic, G.J.; Gasic, T.B.; Galanti, N.; Johnson, T.; Murphy, S. Platelet-tumor-cell interactions in mice. The role of platelets in the spread of malignant disease. Int. J. Cancer 1973, 11, 704–718. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; Pacchiarini, L.; Ricetti, M.M.; Dionigi, P.; Jemos, V.; Zucchella, M.; Fratino, P. Mechanisms of platelet activation by cultured human cancer cells and cells freshly isolated from tumor tissues. Invasion Metastasis 1989, 9, 298–309. [Google Scholar] [PubMed]
- Boukerche, H.; Berthiervergnes, O.; Penin, F.; Tabone, E.; Lizard, G.; Bailly, M.; Mcgregor, J.L. Human-melanoma cell-lines differ in their capacity to release adp and aggregate platelets. Br. J. Haematol. 1994, 87, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Serre, C.M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Investig. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yan, X.; Pan, Y.; Wang, Y.; Wang, N.; Li, L.; Liu, Y.; Chen, X.; Zhang, C.Y.; Gu, H.; et al. MicroRNA-223 delivered by platelet-derived microvesicles promotes lung cancer cell invasion via targeting tumor suppressor EPB41L3. Mol. Cancer 2015, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Weiler, H. A platelet cloak for tumor cells. Blood 2005, 105, 5–6. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates nkg2d thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Felding-Habermann, B.; Habermann, R.; Saldivar, E.; Ruggeri, Z.M. Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J. Biol. Chem. 1996, 271, 5892–5900. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.; Cooke, N.; Kenny, D. Living in shear: Platelets protect cancer cells from shear induced damage. Clin. Exp. Metastasis 2014, 31, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Cai, Y.; Zhou, X.D.; Yang, H.P. activated platelets interact with lung cancer cells through p-selectin glycoprotein ligand-1. Pathol. Oncol. Res. 2012, 18, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kaneko, M.; Sata, M.; Fujita, N.; Tsuruo, T.; Osawa, M. Enhanced expression of Aggrus (T1alpha/podoplanin), a platelet-aggregation-inducing factor in lung squamous cell carcinoma. Tumour Biol. 2005, 26, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Da Prada, M.; Picotti, G.B. Content and subcellular localization of catecholamines and 5-hydroxytryptamine in human and animal blood platelets: Monoamine distribution between platelets and plasma. Br. J. Pharmacol. 1979, 65, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Nagy, J.A.; Pal, S.; Vasile, E.; Eckelhoefer, I.A.; Bliss, V.S.; Manseau, E.J.; Dasgupta, P.S.; Dvorak, H.F.; Mukhopadhyay, D. The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat. Med. 2001, 7, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Zamani, A.; Qu, Z. Serotonin activates angiogenic phosphorylation signaling in human endothelial cells. FEBS Lett. 2012, 586, 2360–2365. [Google Scholar] [CrossRef] [PubMed]
- Battinelli, E.M.; Markens, B.A.; Italiano, J.E., Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: Modulation of physiologic and pathologic angiogenesis. Blood 2011, 118, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.G.; Metharom, P.; Berndt, M.C. The functional role of platelets in the regulation of angiogenesis. Platelets 2015, 26, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [PubMed]
- Radziwon-Balicka, A.; Medina, C.; O’Driscoll, L.; Treumann, A.; Bazou, D.; Inkielewicz-Stepniak, I.; Radomski, A.; Jow, H.; Radomski, M.W. Platelets increase survival of adenocarcinoma cells challenged with anticancer drugs: Mechanisms and implications for chemoresistance. Br. J. Pharmacol. 2012, 167, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lan, X.; Liu, M.; Zhou, B.; Wang, B.; Chen, P. Direct TGF-beta 1 signaling between activated platelets and pancreatic cancer cells primes cisplatin insensitivity. Cell Biol. Int. 2013, 37, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Rickelt, S.; Newton, A.P.; Garris, C.; Nunes, E.; Evavold, C.; Pfirschke, C.; Engblom, C.; Mino-Kenudson, M.; Hynes, R.O.; et al. PF4 Promotes Platelet Production and Lung Cancer Growth. Cell Rep. 2016, 17, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Fine, R.L. Pancreatic cancer and thromboembolic disease. Lancet Oncol. 2004, 5, 655–663. [Google Scholar] [CrossRef]
- Lyman, G.H.; Khorana, A.A. Cancer, clots and consensus: New understanding of an old problem. J. Clin. Oncol. 2009, 27, 4821–4826. [Google Scholar] [CrossRef] [PubMed]
- Pelosof, L.C.; Gerber, D.E. Paraneoplastic Syndromes: An Approach to Diagnosis and Treatment. Mayo Clin. Proc. 2010, 85, 837–854. [Google Scholar] [CrossRef] [PubMed]
- Karolak, L.; Chandra, A.; Khan, W.; Marks, B.; Petros, W.P.; Peters, W.P.; Greenberg, C.S.; Hannun, Y.A. High-dose chemotherapy-induced platelet defect: Inhibition of platelet signal transduction pathways. Mol. Pharmacol. 1993, 43, 37–44. [Google Scholar] [PubMed]
- Tajima, H.; Ohta, T.; Miyashita, T.; Nakanuma, S.; Matoba, M.; Miyata, T.; Sakai, S.; Okamoto, K.; Makino, I.; Kinoshita, J.; et al. Oxaliplatin-based chemotherapy induces extravasated platelet aggregation in the liver. Mol. Clin. Oncol. 2015, 3, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kawada, M.; Inoue, H.; Arakawa, M.; Ikeda, D. Transforming growth factor-beta1 modulates tumor-stromal cell interactions of prostate cancer through insulin-like growth factor-I. Anticancer Res. 2008, 28, 721–730. [Google Scholar] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGF beta in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.T.; Zhang, S.Q.; Miao, L.L.; Wang, J.B.; Jin, Z.J.; Gu, B.; Duan, Z.H.; Zhao, Z.L.; Ma, S.M.; Zhang, W.J.; et al. Activation of platelet protease-activated receptor-1 induces epithelial-mesenchymal transition and chemotaxis of colon cancer cell line SW620. Oncol. Rep. 2015, 33, 2681–2688. [Google Scholar] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Crowley, D.J.; Smyth, P.A.; O’Toole, S.A.; Egan, K.; Kenny, D.; Sheils, O.M.; O’Leary, J.J. The epithelial mesenchymal transition (emt) profile of platelet cloaked cancer cells. Lab. Invest. 2011, 91, 441A. [Google Scholar]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [PubMed]
- Orellana, R.; Kato, S.; Erices, R.; Bravo, M.L.; Gonzalez, P.; Oliva, B.; Cubillos, S.; Valdivia, A.; Ibanez, C.; Branes, J.; et al. Platelets enhance tissue factor protein and metastasis initiating cell markers, and act as chemoattractants increasing the migration of ovarian cancer cells. BMC Cancer 2015, 15, 290. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.W.; Clair, T.; Kim, Y.S.; McMarlin, A.; Schiffmann, E.; Liotta, L.A.; Stracke, M.L. Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor. Cancer Res. 2001, 61, 6938–6944. [Google Scholar] [PubMed]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of Autotaxin and Lysophosphatidic Acid Receptors Increases Mammary Tumorigenesis, Invasion, and Metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Wannecq, E.; Descotes, F.; Jansen, S.; Deux, B.; Ribeiro, J.; Serre, C.M.; Gres, S.; Bendriss-Vermare, N.; Bollen, M.; et al. Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. Plos ONE 2010, 5, e9741. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.; et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Lee, S.-C.; David, M.; Bordet, J.-C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alpha(V)beta(3) controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Radad, K.; Gille, G.; Rausch, W.D. Short review on dopamine agonists: Insight into clinical and research studies relevant to Parkinson’s disease. Pharmacol. Rep. 2005, 57, 701–712. [Google Scholar] [PubMed]
- Bonhomme, N.; Esposito, E. Involvement of serotonin and dopamine in the mechanism of action of novel antidepressant drugs: A review. J. Clin. Psychopharmacol. 1998, 18, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Zadeh, L.F.; Moses, L.; Gwaltney-Brant, S.M. Serotonin: A review. J. Vet. Pharmacol. Ther. 2008, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Smith, M.; Lu, C.; Shahzad, M.M.; Pena, G.N.; Allen, J.K.; Stone, R.L.; Mangala, L.S.; Han, H.D.; Kim, H.S.; Farley, D.; et al. Dopamine blocks stress-mediated ovarian carcinoma growth. Clin. Cancer Res. 2011, 17, 3649–3659. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, D.; Sarkar, C.; Mitra, R.B.; Banerjee, S.; Dasgupta, P.S.; Basu, S. Depleted dopamine in gastric cancer tissues: Dopamine treatment retards growth of gastric cancer by inhibiting angiogenesis. Clin. Cancer Res. 2004, 10, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Chakroborty, D.; Chowdhury, U.R.; Dasgupta, P.S.; Basu, S. Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 2008, 14, 2502–2510. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Sinha, S.; Yang, S.P.; Patra, C.; Dutta, S.; Wang, E.; Mukhopadhyay, D. The neurotransmitter dopamine modulates vascular permeability in the endothelium. J. Mol. Signal 2008, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, E.J.; Shabbir, M.A.; Mikhailidis, D.P.; Mumtaz, F.H.; Thompson, C.S. The effect of serotonin and serotonin antagonists on bladder cancer cell proliferation. BJU Int. 2006, 97, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, E.J.; Shabbir, M.; Mikhailidis, D.P.; Thompson, C.S.; Mumtaz, F.H. The role of serotonin (5-hydroxytryptamine1A and 1B) receptors in prostate cancer cell proliferation. J. Urol. 2006, 176, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Soll, C.; Jang, J.H.; Riener, M.O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.A. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology 2010, 51, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Mohle, R.; Green, D.; Moore, M.A.; Nachman, R.L.; Rafii, S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. USA 1997, 94, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E., Jr.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Pinedo, H.M.; Verheul, H.M.; D’Amato, R.J.; Folkman, J. Involvement of platelets in tumour angiogenesis? Lancet 1998, 352, 1775–1777. [Google Scholar] [CrossRef]
- Cross, M.J.; Dixelius, J.; Matsumoto, T.; Claesson-Welsh, L. VEGF-receptor signal transduction. Trends Biochem. Sci. 2003, 28, 488–494. [Google Scholar] [CrossRef]
- Battinelli, E.M.; Markens, B.A.; Kulenthirarajan, R.A.; Machlus, K.R.; Flaumenhaft, R.; Italiano, J.E., Jr. Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 2014, 123, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.; Lolkema, M.P.; Qian, D.Z.; Hilkes, Y.H.; Liapi, E.; Akkerman, J.W.; Pili, R.; Voest, E.E. Platelets take up the monoclonal antibody bevacizumab. Clin. Cancer Res. 2007, 13, 5341–5347. [Google Scholar] [CrossRef] [PubMed]
- Klement, G.L.; Yip, T.T.; Cassiola, F.; Kikuchi, L.; Cervi, D.; Podust, V.; Italiano, J.E.; Wheatley, E.; Abou-Slaybi, A.; Bender, E.; et al. Platelets actively sequester angiogenesis regulators. Blood 2009, 113, 2835–2842. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Fan, F.; Liu, Z.; Zhang, F.; Liu, Y.; Wei, Z.; Shen, C.; Cao, Y.; Wang, A.; Lu, Y. The angiogenic responses induced by release of angiogenic proteins from tumor cell-activated platelets are regulated by distinct molecular pathways. IUBMB Life 2015, 67, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Caine, G.J.; Lip, G.Y.; Blann, A.D. Platelet-derived VEGF, Flt-1, angiopoietin-1 and P-selectin in breast and prostate cancer: Further evidence for a role of platelets in tumour angiogenesis. Ann. Med. 2004, 36, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cao, F.L.; Wang, B.Z.; Mu, X.R.; Li, G.Y.; Wang, X.W. Expression of angiogenesis regulatory proteins and epithelial-mesenchymal transition factors in platelets of the breast cancer patients. Sci. World J. 2014, 2014, 878209. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, N.; Patruno, R.; Zito, F.A.; Di Summa, A.; Tinelli, A.; Troilo, S.; Misino, A.; Ruggieri, E.; Goffredo, V.; Gadaleta, C.D.; et al. Vascular endothelial growth factor concentrations from platelets correlate with tumor angiogenesis and grading in a spontaneous canine non-Hodgkin lymphoma model. Leuk Lymphoma 2010, 51, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.E.; Zurakowski, D.; Italiano, J.E., Jr.; Michel, L.V.; Fox, L.; Klement, G.L.; Folkman, J. Normal ranges of angiogenesis regulatory proteins in human platelets. Am. J. Hematol. 2010, 85, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Vavken, P.; Kevy, S.; Jacobson, M.; Zurakowski, D.; Murray, M.M. Platelet activation by collagen provides sustained release of anabolic cytokines. Am. J. Sports Med. 2011, 39, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Bambace, N.M.; Levis, J.E.; Holmes, C.E. The effect of P2Y-mediated platelet activation on the release of VEGF and endostatin from platelets. Platelets 2010, 21, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Ibele, G.M.; Kay, N.E.; Johnson, G.J.; Jacob, H.S. Human platelets exert cytotoxic effects on tumor cells. Blood 1985, 65, 1252–1255. [Google Scholar] [PubMed]
- Okada, M.; Sagawa, T.; Tominaga, A.; Kodama, T.; Hitsumoto, Y. Two mechanisms for platelet-mediated killing of tumour cells: One cyclo-oxygenase dependent and the other nitric oxide dependent. Immunology 1996, 89, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Sagawa, T.; Tominaga, A.; Kodama, T.; Okada, M. Cytotoxicity of unstimulated and thrombin-activated platelets to human tumour cells. Immunology 1993, 78, 650–656. [Google Scholar] [PubMed]
- Crist, S.A.; Elzey, B.D.; Ludwig, A.T.; Griffith, T.S.; Staack, J.B.; Lentz, S.R.; Ratliff, T.L. Expression of TNF-related apoptosis-inducing ligand (TRAIL) in megakaryocytes and platelets. Exp. Hematol. 2004, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Menezes, J.; Knafo, L.; Ahmad, A. Activated human platelets express Fas-L and induce apoptosis in Fas-positive tumor cells. J. Leukoc. Biol. 2001, 69, 123–128. [Google Scholar] [PubMed]
- Wang, Y.; Zhang, H. Platelet-induced inhibition of tumor cell growth. Thromb. Res. 2008, 123, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Bottsford-Miller, J.; Vasquez, H.G.; Stone, R.; Zand, B.; Kroll, M.H.; Sood, A.K.; Afshar-Kharghan, V. Platelets increase the proliferation of ovarian cancer cells. Blood 2012, 120, 4869–4872. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.-J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.L.; Nick, A.M.; McNeish, I.A.; Balkwill, F.; Han, H.D.; Bottsford-Miller, J.; Rupaimoole, R.; Armaiz-Pena, G.N.; Pecot, C.V.; Coward, J.; et al. Paraneoplastic thrombocytosis in ovarian cancer. N. Engl. J. Med. 2012, 366, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Gungor, T.; Kanat-Pektas, M.; Sucak, A.; Mollamahmutoglu, L. The role of thrombocytosis in prognostic evaluation of epithelial ovarian tumors. Arch. Gynecol. Obstet. 2009, 279, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, S.W.; Nam, E.J.; Yim, G.W.; Kim, S.; Kim, Y.T. The impact of pretreatment thrombocytosis and persistent thrombocytosis after adjuvant chemotherapy in patients with advanced epithelial ovarian cancer. Gynecol. Oncol. 2014, 122, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Li, A.J.; Madden, A.C.; Cass, I.; Leuchter, R.S.; Lagasse, L.D.; Karlan, B.Y. The prognostic significance of thrombocytosis in epithelial ovarian carcinoma. Gynecol. Oncol. 2004, 92, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wei, B.; Zhou, W.; Yang, Y.; Li, B.; Guo, S.; Li, J.; Ye, J.; Li, J.; Zhang, Q.; et al. P-selectin-mediated platelet adhesion promotes tumor growth. Oncotarget 2015, 6, 6584–6596. [Google Scholar] [CrossRef] [PubMed]
- Carr, B.I.; Cavallini, A.; D’Alessandro, R.; Refolo, M.G.; Lippolis, C.; Mazzocca, A.; Messa, C. Platelet extracts induce growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Tucker, R.F.; Shipley, G.D.; Moses, H.L.; Holley, R.W. Growth inhibitor from BSC-1 cells closely related to platelet type beta transforming growth factor. Science 1984, 226, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Anzano, M.A.; Wakefield, L.M.; Roche, N.S.; Stern, D.F.; Sporn, M.B. Type beta transforming growth factor: A bifunctional regulator of cellular growth. Proc. Natl. Acad. Sci. USA 1985, 82, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A.; Bertino, J.; Cleary, J.; Ortiz, T.; Lane, A.; Supko, J.G.; Ryan, D. Cytotoxic Agents. In Goodman and Gilman’s The Pharmacological Basis of Therapeutics, Twelfth Edition (Chapter 61 Cytotoxic Agents), Twelfth edition; Brunton, L.L., Chabner, B.A., Knollmann, B.C., Eds.; The McGraw-Hill Companies: New York, NY, USA, 2010; pp. 1677–1730. [Google Scholar]
- Tonato, M.; Mosconi, A.M.; Martin, C. Safety profile of gemcitabine. Anticancer Drugs 1995, 6, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Velez, P.; Izquierdo, I.; Rosa, I.; Garcia, A. A 2D-DIGE-based proteomic analysis reveals differences in the platelet releasate composition when comparing thrombin and collagen stimulations. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Slabuszewska-Jozwiak, A.; Dmoch-Gajzlerska, E.; Kozakiewicz, B.; Jakiel, G. The prognostic significance of thrombocytosis in ovarian cancer. Ann. Agric. Environ. Med. 2015, 22, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Choi, G.S.; Park, J.S.; Park, S.; Kawai, K.; Watanabe, T. Clinical significance of thrombocytosis before preoperative chemoradiotherapy in rectal cancer: Predicting pathologic tumor response and oncologic outcome. Ann. Surg. Oncol. 2015, 22, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Wang, Z.; Sarkar, S.H.; Li, Y.; Banerjee, S.; Saliganan, A.; Kim, H.R.; Cher, M.L.; Sarkar, F.H. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells 2008, 26, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, X. Platelets are associated with xenograft tumor growth and the clinical malignancy of ovarian cancer through an angiogenesis-dependent mechanism. Mol. Med. Rep. 2015, 11, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Mitrugno, A.; Sylman, J.L.; Ngo, A.T.; Pang, J.; Sears, R.C.; Williams, C.; McCarty, O.J. Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: Implications for the oncoprotein c-MYC. Am. J. Physiol. Cell Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, K.; Agarwal, K.; Kumar, N. Platelet concentrates: Regenerating the lost tissues. J. Pharm. Bioallied Sci. 2013, 5, 329–330. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, R.; Refolo, M.G.; Lippolis, C.; Giannuzzi, G.; Carella, N.; Messa, C.; Cavallini, A.; Carr, B.I. Antagonism of sorafenib and regorafenib actions by platelet factors in hepatocellular carcinoma cell lines. BMC Cancer 2014, 14, 351. [Google Scholar] [CrossRef] [PubMed]
- Bottsford-Miller, J.; Choi, H.J.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Haemmerle, M.; Nick, A.M.; Pradeep, S.; Zand, B.; Previs, R.A.; et al. Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clin. Cancer Res. 2015, 21, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Miyashita, T.; Inokuchi, M.; Hayashi, H.; Oyama, K.; Tajima, H.; Takamura, H.; Ninomiya, I.; Ahmed, A.K.; Harman, J.W.; et al. Platelets surrounding primary tumor cells are related to chemoresistance. Oncol. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Tsukasa, K.; Ding, Q.; Yoshimitsu, M.; Miyazaki, Y.; Matsubara, S.; Takao, S. Slug contributes to gemcitabine resistance through epithelial-mesenchymal transition in CD133(+) pancreatic cancer cells. Hum. Cell 2015, 28, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Y.; Abbatiello, T.C.; Wu, W.L.; Kim, J.R.; Sarkissyan, M.; Sarkissyan, S.; Chung, S.S.; Elshimali, Y.; Vadgama, J.V. Slug contributes to cancer progression by direct regulation of ERalpha signaling pathway. Int. J. Oncol. 2015, 46, 1461–1472. [Google Scholar] [PubMed]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, J.M.; Gordon, S.; Zucker, S. Identification of tissue factor in two human pancreatic cancer cell lines. Cancer Res. 1989, 49, 5443–5447. [Google Scholar] [PubMed]
- Heinmoller, E.; Schropp, T.; Kisker, O.; Simon, B.; Seitz, R.; Weinel, R.J. Tumor cell-induced platelet aggregation in vitro by human pancreatic cancer cell lines. Scand. J. Gastroenterol. 1995, 30, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Zucchella, M.; Dezza, L.; Pacchiarini, L.; Meloni, F.; Tacconi, F.; Bonomi, E.; Grignani, G.; Notario, A. Human tumor cells cultured “in vitro” activate platelet function by producing ADP or thrombin. Haematologica 1989, 74, 541–545. [Google Scholar] [PubMed]
- Mitrugno, A.; Williams, D.; Kerrigan, S.W.; Moran, N. A novel and essential role for Fc gamma RIIa in cancer cell-induced platelet activation. Blood 2014, 123, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, N.; Ishii, G.; Kojima, M.; Sanada, M.; Fujii, S.; Ochiai, A. Podoplanin, a novel marker of tumor-initiating cells in human squamous cell carcinoma A431. Biochem. Biophys. Res. Commun. 2008, 373, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Sato, S.; Oh-hara, T.; Takami, M.; Koike, S.; Mishima, Y.; Hatake, K.; Fujita, N. Platelets Promote Tumor Growth and Metastasis via Direct Interaction between Aggrus/Podoplanin and CLEC-2. PLoS ONE 2013, 8, e73609. [Google Scholar] [CrossRef] [PubMed]
- Murugappa, S.; Kunapuli, S.P. The role of ADP receptors in platelet function. Front. Biosci. 2006, 11, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; Jamieson, G.A. Platelets in tumor-metastasis-generation of adenosine-diphosphate by tumor-cells is specific but unrelated to metastatic potential. Blood 1988, 71, 844–849. [Google Scholar] [PubMed]
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Geddings, J.E.; Hisada, Y.; Boulaftali, Y.; Getz, T.M.; Whelihan, M.; Fuentes, R.; Dee, R.; Cooley, B.C.; Key, N.S.; Wolberg, A.S.; et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J. Thromb. Haemost 2016, 14, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Gerotziafas, G.T.; Galea, V.; Mbemba, E.; Khaterchi, A.; Sassi, M.; Baccouche, H.; Prengel, C.; van Dreden, P.; Hatmi, M.; Bernaudin, J.F.; et al. Tissue factor over-expression by human pancreatic cancer cells BXPC3 is related to higher prothrombotic potential as compared to breast cancer cells MCF7. Thromb. Res. 2012, 129, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Delluc, A.; Rousseau, A.; Delluc, C.; Le Moigne, E.; Le Gal, G.; Mottier, D.; van Dreden, P.; Lacut, K. Venous thromboembolism in patients with pancreatic cancer: Implications of circulating tissue factor. Blood Coagul. Fibrinolysis 2011, 22, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.N.; Afshar-Kharghan, V.; Sood, A.K. Platelet effects on ovarian cancer. Semin. Oncol. 2014, 41, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [PubMed]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic Cancer-Induced Neutrophil Extracellular Traps: A Potential Contributor to Cancer-Associated Thrombosis. Int. J. Mol. Med. Sci. 2017, 18, 487. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.C.; Weitz, J.I.; Liaw, P.C. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Felding-Habermann, B.; O’Toole, T.E.; Smith, J.W.; Fransvea, E.; Ruggeri, Z.M.; Ginsberg, M.H.; Hughes, P.E.; Pampori, N.; Shattil, S.J.; Saven, A.; et al. Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Quang, P.; Azab, F.; Pitsillides, C.; Thompson, B.; Chonghaile, T.; Patton, J.T.; Maiso, P.; Monrose, V.; Sacco, A.; et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood 2012, 119, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Dimitroff, C.J.; Descheny, L.; Trujillo, N.; Kim, R.; Nguyen, V.; Huang, W.; Pienta, K.J.; Kutok, J.L.; Rubin, M.A. Identification of leukocyte E-selectin ligands, P-selectin glycoprotein ligand-1 and E-selectin ligand-1, on human metastatic prostate tumor cells. Cancer Res. 2005, 65, 5750–5760. [Google Scholar] [CrossRef] [PubMed]
- Aigner, S.; Ramos, C.L.; Hafezi-Moghadam, A.; Lawrence, M.B.; Friederichs, J.; Altevogt, P.; Ley, K. CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J. 1998, 12, 1241–1251. [Google Scholar] [PubMed]
- Alves, C.S.; Burdick, M.M.; Thomas, S.N.; Pawar, P.; Konstantopoulos, K. The dual role of CD44 as a functional P-selectin ligand and fibrin receptor in colon carcinoma cell adhesion. Am. J. Physiol. Cell Physiol. 2008, 294, C907–C916. [Google Scholar] [CrossRef] [PubMed]
- Theoret, J.F.; Yacoub, D.; Hachem, A.; Gillis, M.A.; Merhi, Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb. Res. 2011, 128, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Larrucea, S.; Butta, N.; Rodriguez, R.B.; Alonso-Martin, S.; Arias-Salgado, E.G.; Ayuso, M.S.; Parrilla, R. Podocalyxin enhances the adherence of cells to platelets. Cell. Mol. Life Sci. 2007, 64, 2965–2974. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.T.; Minton, A.R.; Hayes, C.S.; Goss, A.; van Ryn, J.; Gilmour, S.K. Thrombin inhibition and cyclophosphamide synergistically block tumor progression and metastasis. Cancer Biol. Ther. 2015, 16, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Liebman, H.A. Thrombocytopenia in cancer patients. Thromb. Res. 2014, 133, S63–S69. [Google Scholar] [CrossRef]
- Baaten, C.C.; Moenen, F.C.; Henskens, Y.M.; Swieringa, F.; Wetzels, R.; van Oerle, R.; Ten Cate, H.; Beckers, E.A.; Heemskerk, J.W.; van der Meijden, P.E. OC-08—Multiple functional defects in platelets from thrombocytopenic cancer patients undergoing chemotherapy. Thromb. Res. 2016, 140, S171. [Google Scholar] [CrossRef]
- Gasic, G.; Gasic, T. Removal of sialic acid from the cell coat in tumor cells and vascular endothelium, and its effects on metastasis. Proc. Natl. Acad. Sci. USA 1962, 48, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Pacchiarini, L.; Serra, L.; Grignani, G.; Gamba, G.; Gorini, M. In vitro effect of culture fluids from neoplastic tissues on platelet aggregation. II. Experimental tumors. Boll. Soc. Ital. Biol. Sper. 1982, 58, 854–859. [Google Scholar] [PubMed]
- Bradley, C.J.; Dauer, R.J.; Thurlow, P.J.; Connellan, J.M. Characterization of platelet aggregation induced by the human carcinosarcoma Colo 526: Role of platelet activation, tumor cell cytoskeleton and tumor cell plasma membrane. Pathology 1997, 29, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.C.; Purushotham, A.D.; George, W.D. Inhibition of pulmonary tumor seeding by antiplatelet and fibrinolytic therapy in an animal experimental model. J. Surg. Oncol. 1994, 55, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Cylooxygenase-dependent Formation of the Isoprostane, 8-Epi Prostaglandin F2α. J. Biol. Chem. 1995, 270, 9800–9808. [Google Scholar] [CrossRef] [PubMed]
- Rinder, C.S.; Student, L.A.; Bonan, J.L.; Rinder, H.M.; Smith, B.R. Aspirin does not inhibit adenosine diphosphate-induced platelet alpha-granule release. Blood 1993, 82, 505–512. [Google Scholar] [PubMed]
- Chung, A.W.; Jurasz, P.; Hollenberg, M.D.; Radomski, M.W. Mechanisms of action of proteinase-activated receptor agonists on human platelets. Br. J. Pharmacol. 2002, 135, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Barstad, R.M.; Orvim, U.; Hamers, M.J.; Tjonnfjord, G.E.; Brosstad, F.R.; Sakariassen, K.S. Reduced effect of aspirin on thrombus formation at high shear and disturbed laminar blood flow. Thromb. Haemost. 1996, 75, 827–832. [Google Scholar] [PubMed]
- Kune, G.A.; Kune, S.; Watson, L.F. colorectal-cancer risk, chronic illnesses, operations, and medications - case control results from the melbourne colorectal-cancer study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar] [CrossRef] [PubMed]
- Benamouzig, R.; Deyra, J.; Martin, A.; Girard, B.; Jullian, E.; Piednoir, B.; Couturier, D.; Coste, T.; Little, J.; Chaussade, S.; et al. Daily soluble aspirin and prevention of colorectal adenoma recurrence: One-year results of the APACC trial. Gastroenterology 2003, 125, 328–336. [Google Scholar] [CrossRef]
- Baron, J.A.; Cole, B.F.; Sandler, R.S.; Haile, R.W.; Ahnen, D.; Bresalier, R.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.; Burke, C.A.; et al. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med. 2003, 348, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Wakabayashi, K.; Suzuki, S.; Mutoh, M.; Hirata, K.; Nakamura, T.; Takeyama, I.; Kawano, A.; Gondo, N.; Abe, T.; et al. Preventive effects of low-dose aspirin on colorectal adenoma growth in patients with familial adenomatous polyposis: Double-blind, randomized clinical trial. Cancer Med. 2013, 2, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin use and survival after diagnosis of colorectal cancer. JAMA 2009, 302, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef]
- Cao, Y.; Nishihara, R.; Wu, K.; Wang, M.; Ogino, S.; Willett, W.C.; Spiegelman, D.; Fuchs, C.S.; Giovannucci, E.L.; Chan, A.T. Population-wide Impact of Long-term Use of Aspirin and the Risk for Cancer. JAMA Oncol. 2016, 2, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Frouws, M.A.; Bastiaannet, E.; Langley, R.E.; Chia, W.K.; van Herk-Sukel, M.P.; Lemmens, V.E.; Putter, H.; Hartgrink, H.H.; Bonsing, B.A.; van de Velde, C.J.; et al. Effect of low-dose aspirin use on survival of patients with gastrointestinal malignancies; an observational study. Br. J. Cancer 2017, 116, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Risch, H.A.; Lu, L.; Streicher, S.A.; Wang, J.; Zhang, W.; Ni, Q.; Kidd, M.S.; Yu, H.; Gao, Y.T. Aspirin Use and Reduced Risk of Pancreatic Cancer. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.E.; Huang, J.C.; Pace, T.R.; Howard, A.B.; Muss, H.B. Tamoxifen and aromatase inhibitors differentially affect vascular endothelial growth factor and endostatin levels in women with breast cancer. Clin. Cancer Res. 2008, 14, 3070–3076. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.M.; Sullivan, F.M.; Thompson, A.M.; McCowan, C. Aspirin use and survival after the diagnosis of breast cancer: A population-based cohort study. Br. J. Cancer 2014, 111, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.D.; Chen, W.Y.; Li, L.; Hertzmark, E.; Spiegelman, D.; Hankinson, S.E. Aspirin Intake and Survival After Breast Cancer. J. Clin. Oncol. 2010, 28, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, X.; Chen, L.; Ma, T.; Tang, J.; Zhao, J. Association between aspirin use and mortality in breast cancer patients: A meta-analysis of observational studies. Breast Cancer Res. Treat. 2015, 150, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.J.; Cooper, J.A.; Hughes, C.M.; Powe, D.G.; Cardwell, C.R. Post-diagnostic prescriptions for low-dose aspirin and breast cancer-specific survival: A nested case-control study in a breast cancer cohort from the UK Clinical Practice Research Datalink. Breast Cancer Res. 2014, 16, R34. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.D.; Olsson, H.; Pawitan, Y.; Holm, J.; Lundholm, C.; Andersson, T.M.L.; Adami, H.-O.; Askling, J.; Smedby, K.E. Aspirin intake and breast cancer survival—A nation-wide study using prospectively recorded data in Sweden. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Roop, R.P.; Naughton, M.J.; Van Poznak, C.; Schneider, J.G.; Lammers, P.E.; Pluard, T.J.; Johnson, F.; Eby, C.S.; Weilbaecher, K.N. A randomized phase II trial investigating the effect of platelet function inhibition on circulating tumor cells in patients with metastatic breast cancer. Clin. Breast Cancer 2013, 13, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, S.; Wallace, K.; Day, T.A.; Alberg, A.J.; Li, Z. Lower circulating platelet counts and antiplatelet therapy independently predict better outcomes in patients with head and neck squamous cell carcinoma. J. Hematol. Oncol. 2014, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Furlan, C.; Steffan, A.; Polesel, J.; Trovo, M.; Gobitti, C.; Vaccher, E.; Serraino, D.; Barzan, L.; Franchin, G. Lower platelet counts and antiplatelet therapy independently predict better outcomes in patients with head and neck squamous cell carcinoma: A retrospective analysis. Biomark Res. 2015, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, O.; Bhatt, D.L. The evolution of antiplatelet therapy in cardiovascular disease. Nat. Rev. Cardiol. 2011, 8, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Metharom, P.; Berndt, M.C.; Baker, R.I.; Andrews, R.K. Current state and novel approaches of antiplatelet therapy. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.Y.; Levine, M.N. Venous Thromboembolism and Cancer: Risks and Outcomes. Circulation 2003, 107, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Cronin-Fenton, D.P.; Søndergaard, F.; Pedersen, L.A.; Fryzek, J.P.; Cetin, K.; Acquavella, J.; Baron, J.A.; Sørensen, H.T. Hospitalisation for venous thromboembolism in cancer patients and the general population: A population-based cohort study in Denmark, 1997–2006. Br. J. Cancer 2010, 103, 947. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.J.; Card, T.R.; West, J.; Crooks, C.; Grainge, M.J. Incidence of venous thromboembolism in patients with cancer—A cohort study using linked United Kingdom databases. Eur. J. Cancer 2013, 49, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Horsted, F.; West, J.; Grainge, M.J. Risk of venous thromboembolism in patients with cancer: A systematic review and meta-analysis. PLoS Med. 2012, 9, e1001275. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, Y.W.; Osanto, S.; Reitsma, P.H.; Versteeg, H.H. The relationship between tissue factor and cancer progression: Insights from bench and bedside. Blood 2012, 119, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Connolly, G.C.; Phipps, R.P.; Francis, C.W. Platelets and cancer-associated thrombosis. Semin. Oncol. 2014, 41, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Mezouar, S.; Frere, C.; Darbousset, R.; Mege, D.; Crescence, L.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Role of platelets in cancer and cancer-associated thrombosis: Experimental and clinical evidences. Thromb. Res. 2016, 139, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Jenne, C.N. Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Tzanakakis, G.N.; Agarwal, K.C.; Vezeridis, M.P. Prevention of human pancreatic cancer cell-induced hepatic metastasis in nude mice by dipyridamole and its analog RA-233. Cancer 1993, 71, 2466–2471. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef] [PubMed]
- Hicks, B.M.; Murray, L.J.; Hughes, C.; Cardwell, C.R. Clopidogrel use and cancer-specific mortality: A population-based cohort study of colorectal, breast and prostate cancer patients. Pharmacoepidemiol. Drug Saf. 2015, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Mezouar, S.; Darbousset, R.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int. J. Cancer 2015, 136, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Sitia, G.; Aiolfi, R.; Di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar] [CrossRef] [PubMed]
- Leader, A.; Zelikson-Saporta, R.; Rozovski, U.; Pereg, D.; Raanani, P.; Spectre, G.; Lishner, M.; Hermoni, D. Clopidogrel treatment is associated with a decrease in cancer incidence. Blood 2015, 126, 1124. [Google Scholar]
- Gebremeskel, S.; LeVatte, T.; Liwski, R.S.; Johnston, B.; Bezuhly, M. The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int. J. Cancer 2015, 136, 234–240. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Angiogenic Factor | Normal Physiological Level in 106 Platelets (Median (Range)) |
|---|---|
| VEGF | 0.68 (0.02–1.47) pg [75], 0.9 (0.1–2.3) pg [73] |
| PDGF | 21 (12–33) pg [75], 19.1 (9.3–48.9) pg [73] |
| PF4 | 10 (2.4–22) ng [75], 10.2 (4.2–20.5) ng [73] |
| TSP-1 | 27 (7–54) ng [75] |
| bFGF | 0.42 (0.15–0.75) pg [75] |
| Drug | Side Effects |
|---|---|
| Alkylating Agents | |
| Cyclophosphamide | Lesser effect on peripheral platelet count compared to other alkylating agents. |
| Ifosfamide | Greater suppression of platelet count than cyclophosphamide |
| Carmustine | Delayed and prolonged suppression of platelet count, reaching a nadir at 4–6 weeks after administration, with slow reversal |
| Busulfan | Prolonged and cumulative effect lasting months or years |
| Thiotepa | Delayed effect compared to cyclophosphamide with platelet nadir at 3 weeks |
| Streptozocin | Suppression of platelet count in 20% of patients |
| Dacarbazine | Mild suppression of platelet count which is reversible within 1.2 weeks |
| Temozolomide | Similar to dacarbazine |
| Procarbazine | Suppression of platelet count after one week of initiating treatment and reversed within two weeks off treatment |
| Platinum analogues | |
| Cisplatin | Transient thrombocytopenia |
| Antimetabolites | |
| Methotrexate | Effect on platelets is completely reversed within two weeks. However, prolonged suppression may occur in patients with compromised renal function. |
| 5-Florouracil | Thrombocytopenia, less often with infusion compared to bolus regimen |
| Cytarabine | Potent myelosuppression with severe thrombocytopenia |
| Gemcitabine | Mild haematological toxicities [95]. Myelosuppression is more prominent with longer duration infusion. |
| 6-mercaptopurine | Gradual thrombocytopenia |
| Cladribine | Cumulative thrombocytopenia with repeated administration. |
| Others | |
| Topotecan | Neutropenia with or without thrombocytopenia. |
| Etoposide | Infrequent thrombocytopenia, which is usually, not severe. |
| Bleomycin | Minor myelosuppression |
| Mitomycin | Marked thrombocytopenia |
| Hydroxyurea | Occasional thrombocytopenia |
| Vorinostat | Thrombocytopenia is more prominent with intravenous administration. |
| Platelets Decrease Tumour Growth | Platelets Enhance Tumour Growth |
|---|---|
| In in vitro experiments, platelets showed a cytotoxic effect on cancer cells (Malme, a melanoma cell line, and 786, a renal carcinoma cwll line). The platelet effect was abrogated by aspirin [78] | Platelet-derived TGFβ1 enhances ovarian cancer growth in vitro and in vivo [84]. |
| Platelets kill tumour cells (LU99A, a lung cell line, and K562, a chronic myeloid leukaemia cell line) via cyclooxygenase or nitric oxide-dependent pathways [79]. | Platelets promoted proliferation of cancer cells (PLC/PRF/5, Hep3B and HepG2 cells hepatocellular carcinoma cell lines) in vitro via activation of the MAPK pathway [91]. |
| Platelets kill tumour cells via activation of an apoptosis pathway in cancer cells (CEM, leukaemia cell line) through interaction between platelet-derived FAS-L and FAS receptor on the cancer cell [82]. | Platelets enhance the growth of an SKOV3 human ovarian cancer xenograft [101]. |
| Platelets prevented murine cancer cell growth (EG7 (H-2b), L1210, YAC-1 (H-2a) lymphoma cell lines, B16 H-2b, a melanoma cell line, and RM1 (H-2b), a prostate cancer cell line) by inducing cell cycle arrest rather than activating apoptosis [83]. | Deposition of platelets in a solid tumour, as well as tumour growth (pancreatic islet insulinoma), was significantly reduced in P-selectin deficient mice [90]. |
| In a genetically modified lung cancer mouse model, PF4 enhanced platelet production and accumulation in the lung, which accelerated cancer progression [30]. | |
| Platelets enhance the proliferation of colon and pancreatic cancer cells by upregulating the oncoprotein c-MYC [102]. |
| Antiplatelet drugs | Study outcome | References |
|---|---|---|
| Dipyridamole and RA-233 | In a pancreatic cancer mouse model, the combination of dipyridamole and RA-233 (cAMP-PDE inhibitor) reduced hepatic metastasis | [179] |
| Prasugrel | In the TRITON-TIMI 38 double-blinded randomised multicentre clinical trial of more than 13000 individuals assessing prasugrel versus clopidogrel in patients with acute coronary syndrome, prasugrel was associated with an increased incidence of gastrointestinal cancer. The exact mechanism is not entirely understood. | [180] |
| Clopidogrel | In prostate, breast and colorectal cancer patients, there was no increased risk of cancer-specific mortality among clopidogrel users. This study was in response to TRITON-TIMI 38 In a pancreatic cancer mouse model, clopidogrel reduced tumour growth, metastasis and thrombosis associated with cancer cell microparticle-derived tissue factor In a lung adenocarcinoma mouse model, clopidogrel reduced cancer growth and progression. [181] | [30,181,182] |
| Aspirin /Clopidogrel | In HBV transgenic mice, aspirin /clopidogrel delayed or prevented the development of hepatocellular carcinoma and improved the overall survival. | [183] |
| Clopidogrel with or without aspirin | In a large retrospective study involving 184,781 patients, use of clopidogrel with or without aspirin was associated with lower incidence of cancer | [184] |
| Ticagrelor | In melanoma and breast cancer mouse models, ticagrelor significantly reduced cancer metastasis and improved survival. | [185] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elaskalani, O.; Berndt, M.C.; Falasca, M.; Metharom, P. Targeting Platelets for the Treatment of Cancer. Cancers 2017, 9, 94. https://doi.org/10.3390/cancers9070094
Elaskalani O, Berndt MC, Falasca M, Metharom P. Targeting Platelets for the Treatment of Cancer. Cancers. 2017; 9(7):94. https://doi.org/10.3390/cancers9070094
Chicago/Turabian StyleElaskalani, Omar, Michael C. Berndt, Marco Falasca, and Pat Metharom. 2017. "Targeting Platelets for the Treatment of Cancer" Cancers 9, no. 7: 94. https://doi.org/10.3390/cancers9070094
APA StyleElaskalani, O., Berndt, M. C., Falasca, M., & Metharom, P. (2017). Targeting Platelets for the Treatment of Cancer. Cancers, 9(7), 94. https://doi.org/10.3390/cancers9070094