
Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy

Abstract
:1. Introduction
2. Regional Delivery of CAR-T Cells in Clinical Practice
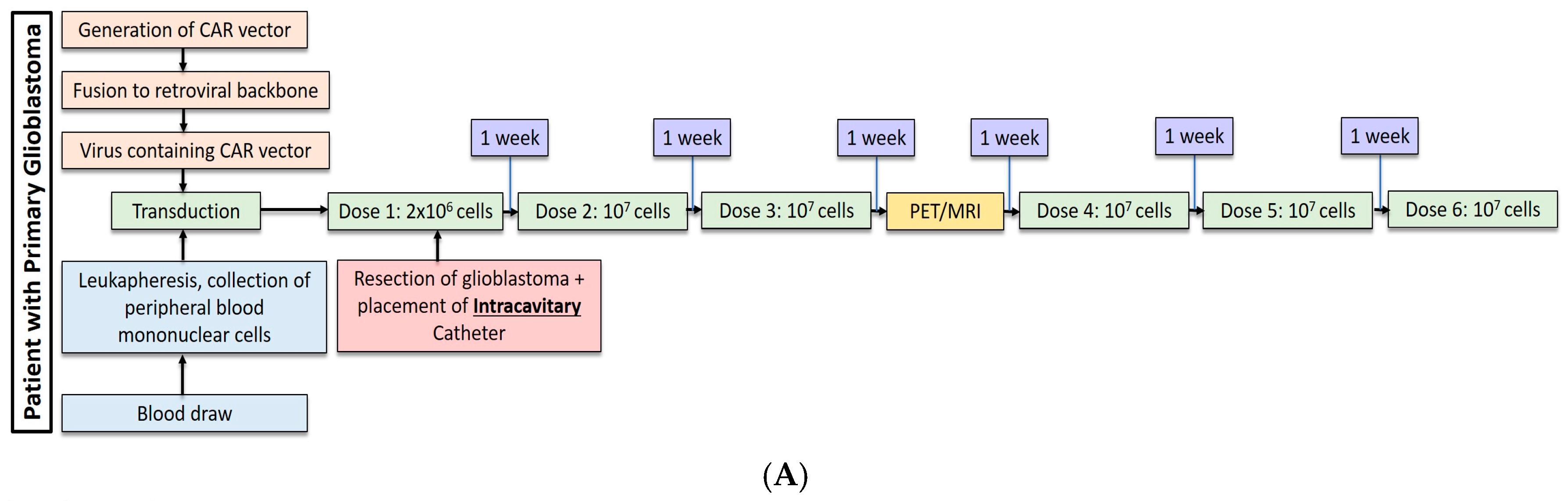
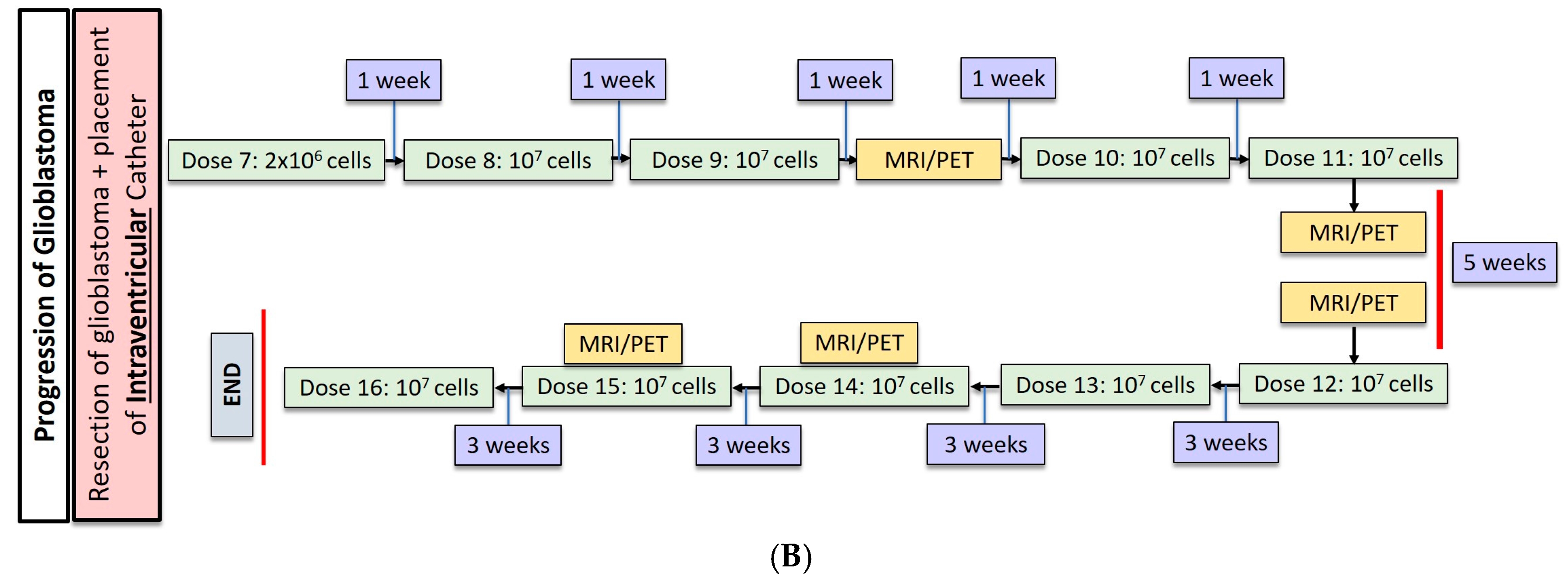
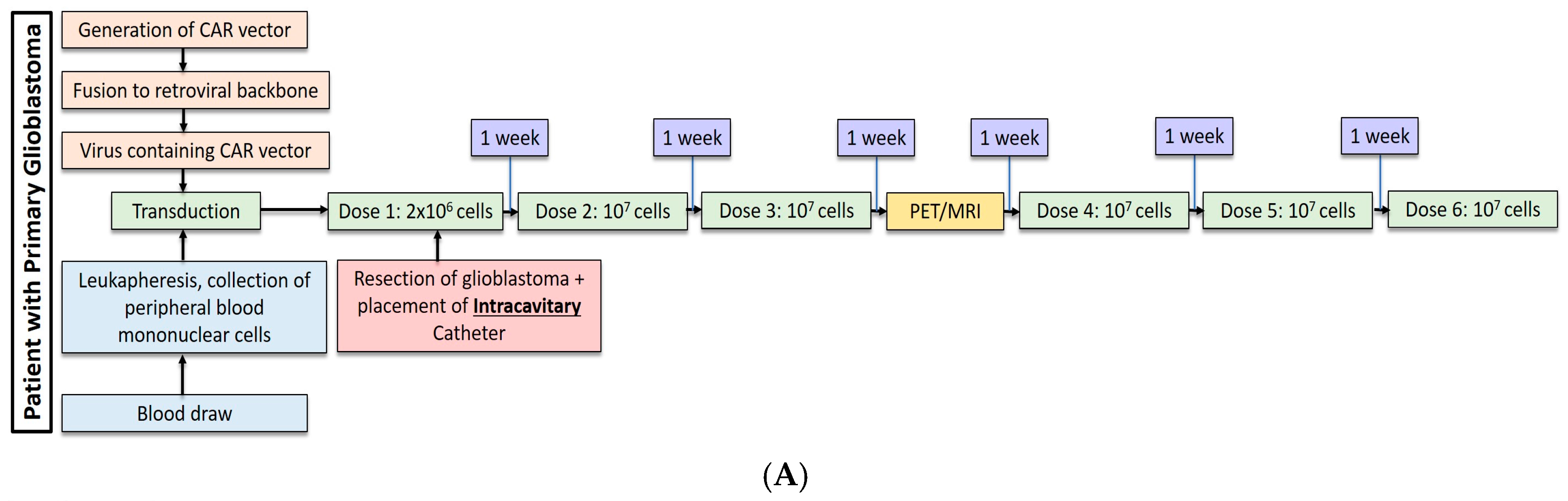
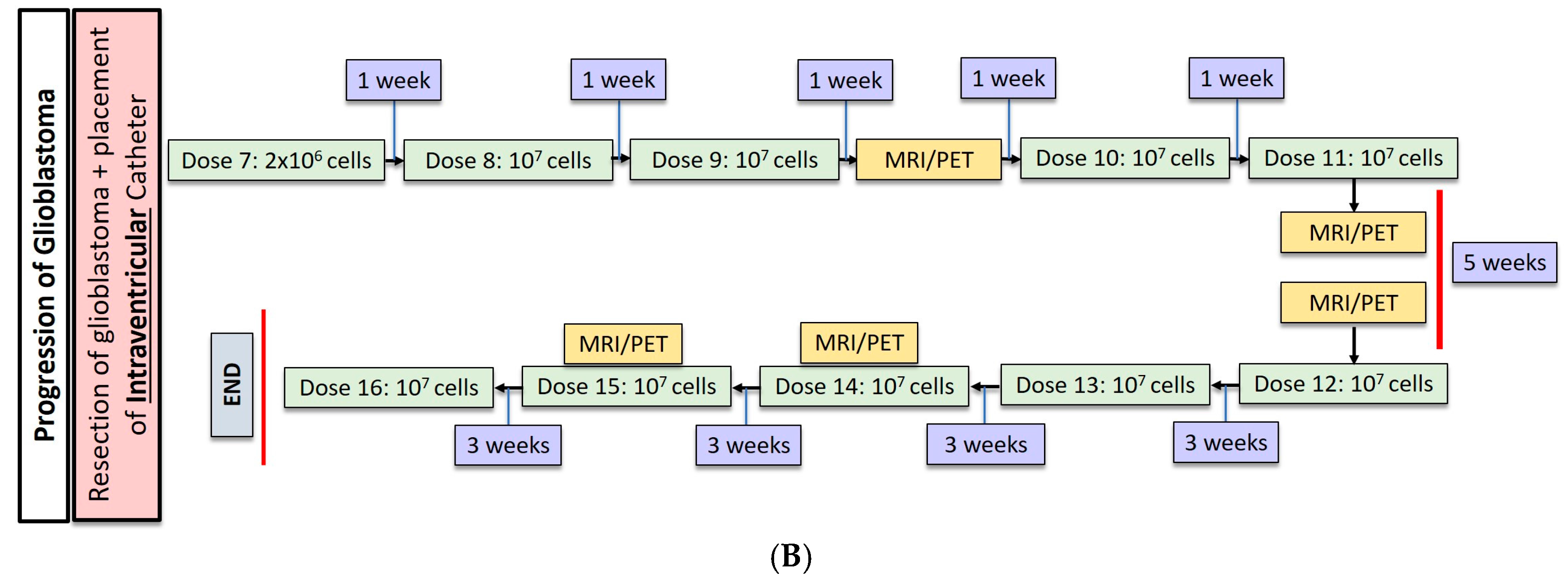
2.1. Gliobastoma
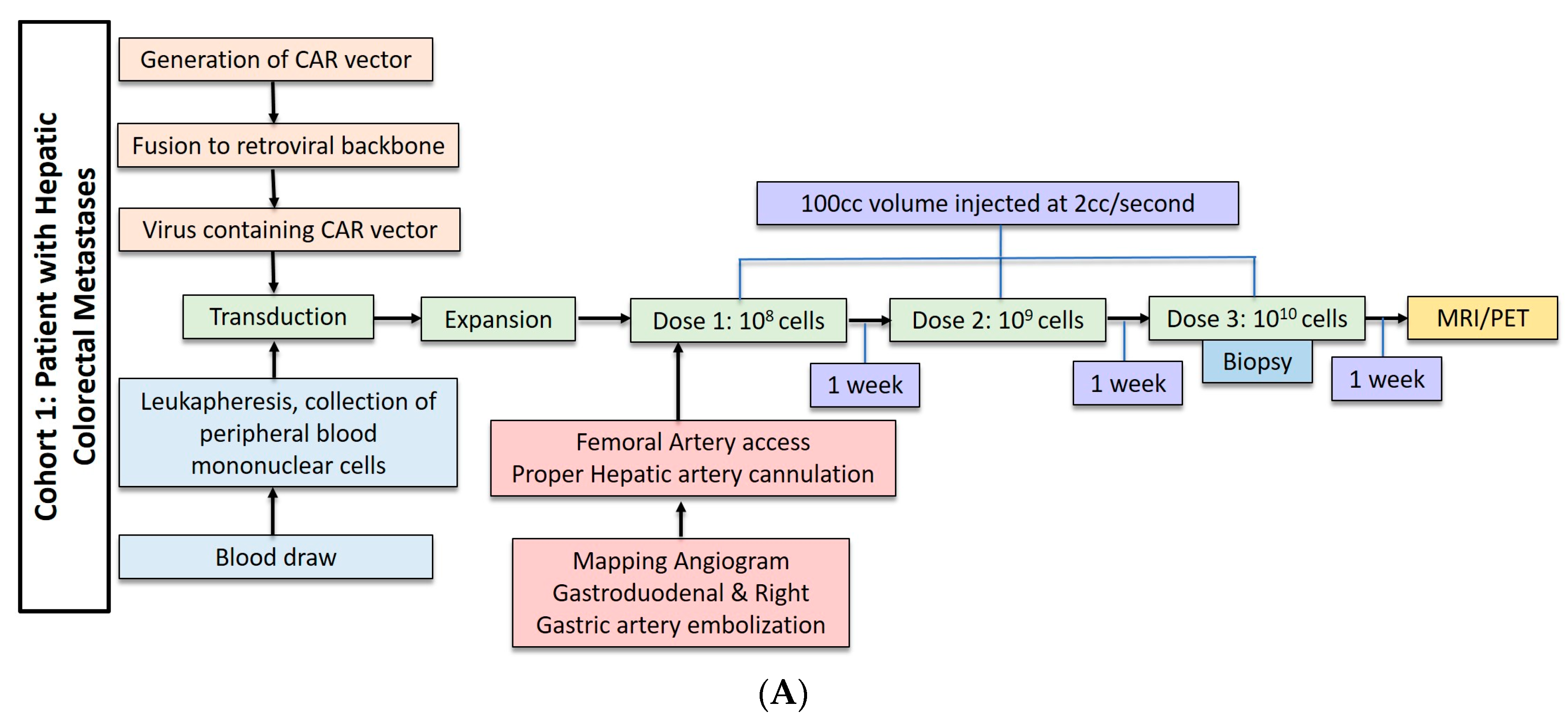
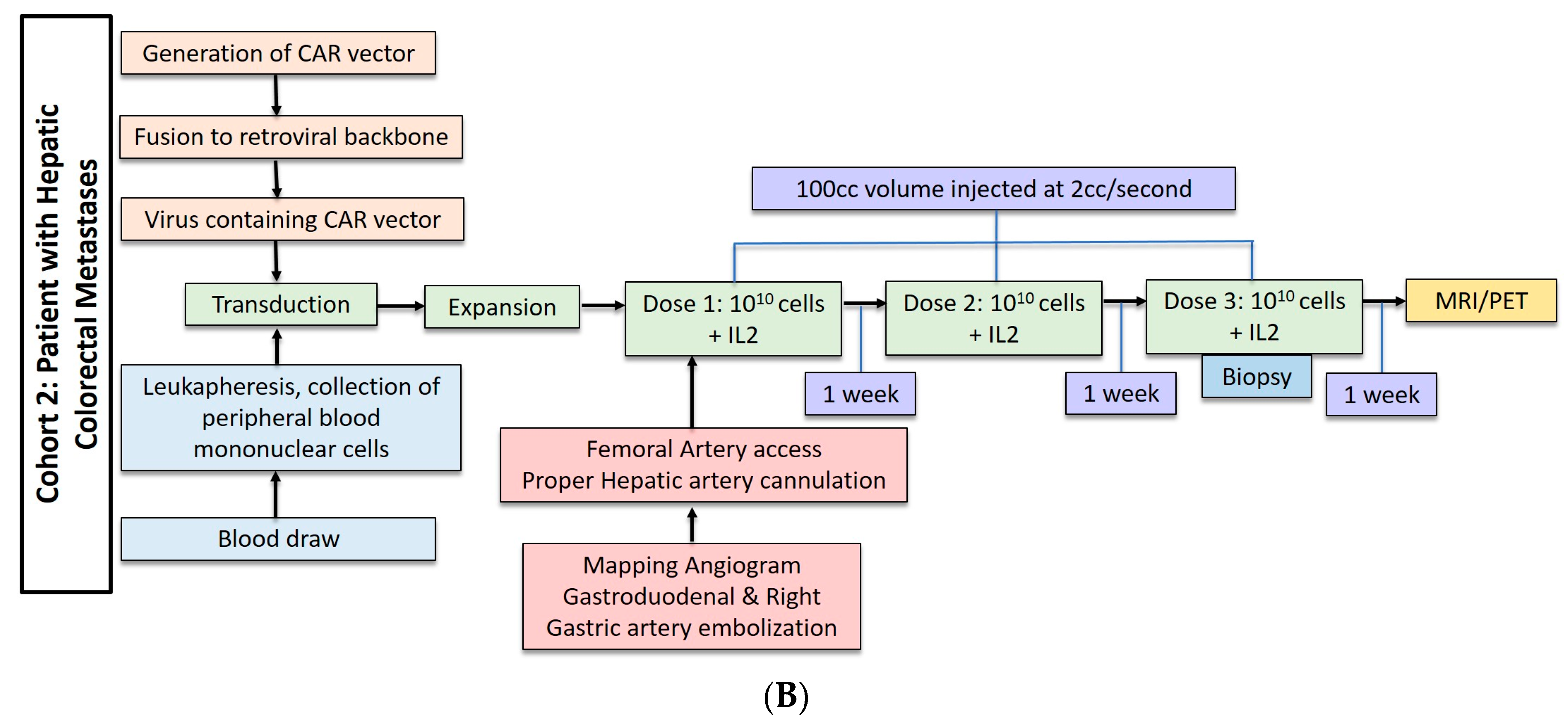
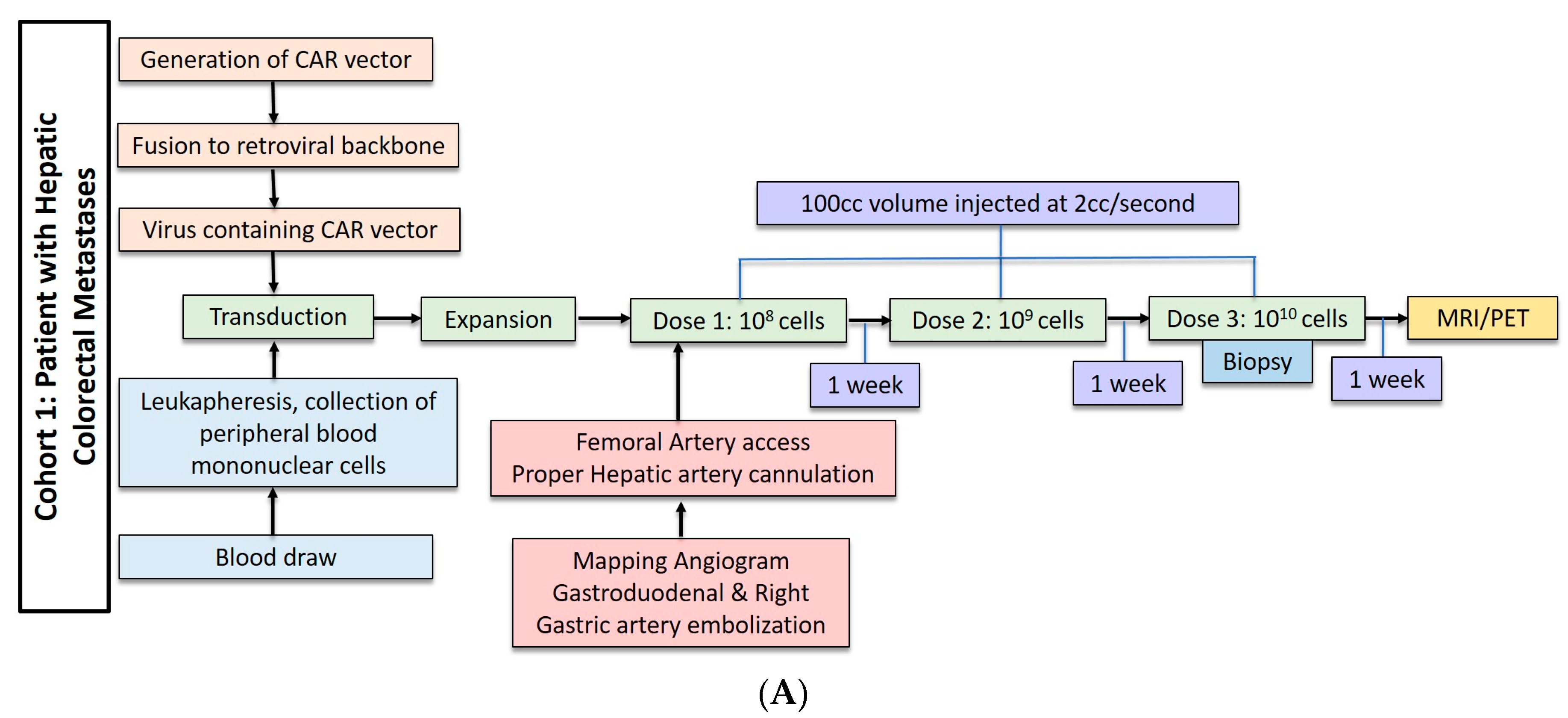
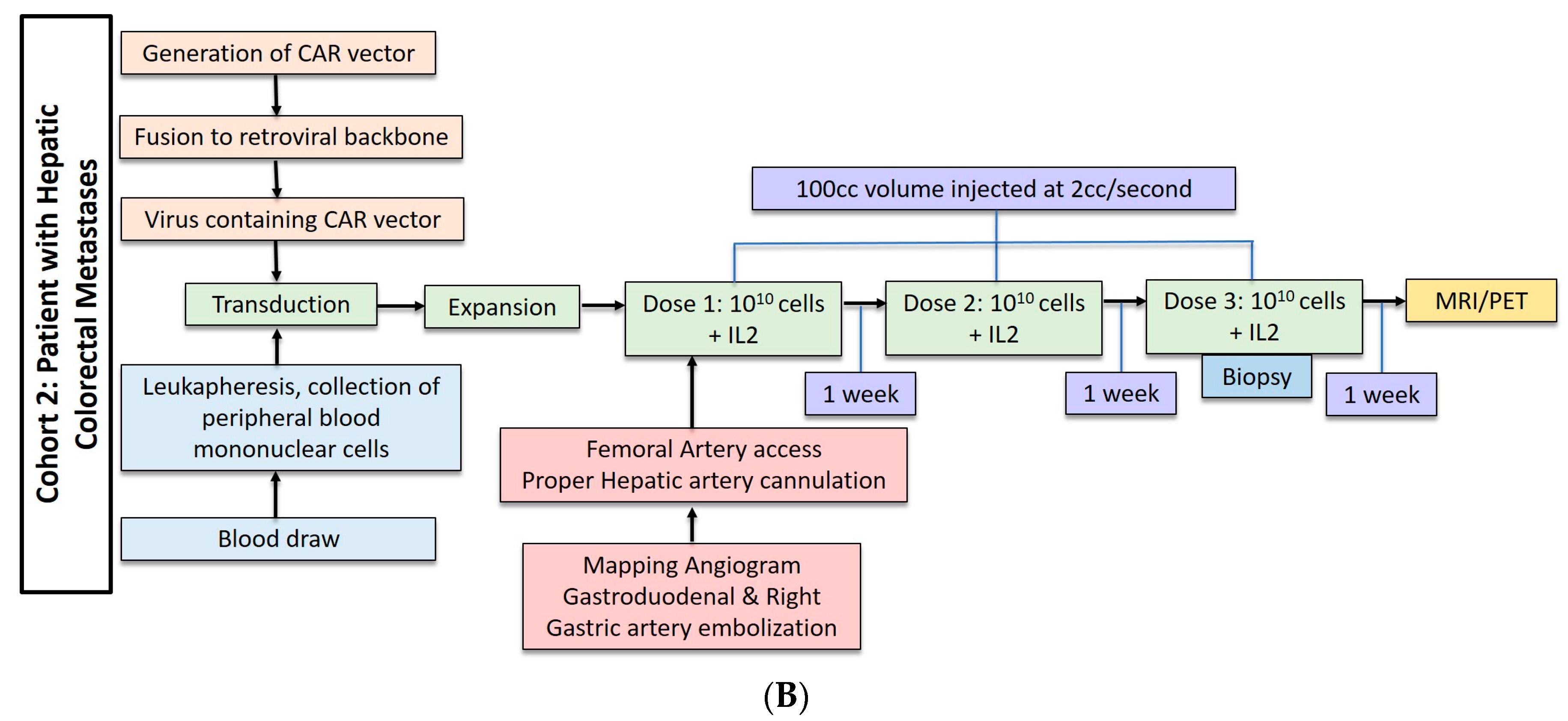
2.2. Hepatic Metastases of Colorectal Adenocarcinoma
3. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Newick, K.; Brien, S.O.; Moon, E.; Albelda, S.M. CAR-T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; Moon, E.; Albelda, S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Oncolytics 2016, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Xu, H. A new insight in chimeric antigen receptor-engineered T cells for cancer immunotherapy. J. Hematol. Oncol. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Vecchio, M.D.; Creighton, C.J.; Ittmann, M.T.; et al. T Lymphocytes Redirected against the Chondroitin Sulfate Proteoglycan-4 Control the Growth of Multiple Solid Tumors both In Vitro and In Vivo. Clin. Cancer Res. 2015, 20, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer- Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Prapa, M.; Caldrer, S.; Spano, C.; Bestagno, M.; Golinelli, G.; Grisendi, G.; Petrachi, T.; Conte, P.; Horwitz, E.M.; Paolucci, P.; et al. A novel anti-GD2/4–1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, Y. Increasing the safety and ef fi cacy of chimeric antigen receptor T cell therapy. Protein Cell 2017. [Google Scholar] [CrossRef]
- Maus, M.V.; June, C.H. CCR FOCUS Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin. Cancer Res. 2016, 22, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Cherkassky, L.; Villena-vargas, J.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M.; et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Julie, R.; Chang, W.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and safety of IL13Ra2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2016, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; Kurien, A.; Priceman, S.J.; Wang, X.; et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.B.; Suryadevara, M.C.; Gedeon, C.P.; Herndon, E.J.; Sanchez-Perez, L.; Sampson, H.J. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J. Clin. Neurosci. 2015, 21, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.N.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Burga, R.A.; Mccormack, E.; Wang, L.J.; Mooring, W.; Point, G.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor modified T cell therapy for CEA+ liver metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.; Point, R.G.; Cunetta, M.; Thorn, M.; Guha, P.; Espat, N.J.; Boutros, C.; Hanna, N.; Junghans, P.R.; et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016, 23, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, S.S.; Jensen, M.C.; Satyamurthy, N.; Paik, D.; Czernin, J.; Gambh, S.S. Non-invasive detection of therapeutic cytolytic T Cells in patients with [18-F]FHBG positron emission tomography in a glioma patient. Nat. Clin. Pract. Oncol. 2009, 6, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Paty, P.; Kemeny, N.; Tong, W.; Sullivan, D.; Fong, Y.; Jarnagin, W. A phase I/II study of hepatic arterial infusion (HAI) of floxuridine (FUDR) and dexamethasone (DEX) with systemic irinotecan (CPT-11) for unresectable hepatic metastases from colorectal cancer. Proc. Am. Soc. Clin. Oncol. 2000, 19, 260a. [Google Scholar] [CrossRef]
- Kemeny, N.; Jarnagin, W.; Paty, P.; Gönen, M.; Schwartz, L.; Morse, M.; Leonard, G.; D’Angelica, M.; DeMatteo, R.; Blumgart, L.; et al. Phase I trial of systemic oxaliplatin combination chemotherapy with hepatic arterial infusion in patients with unresectable liver metastases from colorectal cancer. J. Clin. Oncol. 2005, 23, 4888–4896. [Google Scholar] [CrossRef] [PubMed]
- Kemeny, N.E.; Huitzil Melendez, F.D.; Capanu, M.; Paty, P.B.; Fong, Y.; Schwartz, L.H.; Jarnagin, W.R.; Patel, D.; D’Angelica, M.; et al. Conversion to resectability using hepatic artery infusion plus systemic chemotherapy for the treatment of unresectable liver metastases from colorectal carcinoma. J. Clin. Oncol. 2009, 27, 3465–3471. [Google Scholar] [CrossRef] [PubMed]
- Corrigan-Curay, J.; Kiem, H.-P.; Baltimore, D.; O’Reilly, M.; Brentjens, R.J.; Cooper, L.; Forman, S.; Gottschalk, S.; Greenberg, P.; Junghans, R.; et al. T-Cell immunotherapy: Looking forward. Mol. Ther. 2014, 22, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Misiakos, E.P.; Nikolaos, P.; Kouraklis, G. Current treatment for colorectal liver metastases. World J. Gastroenterol. 2011, 17, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Guha, P.; Reha, J.; Katz, S.C. Immunosuppression in liver tumors: Opening the portal to effective immunotherapy. Nat. Publ. Gr. 2016. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Institutes of Health. Phase Ib Trial of CAR-T Hepatic Artery Infusions Followed by Selective Internal Radiation Therapy (SIRT) with Yttrium-90 Sir-Spheres® for CEA-Expressing Liver Metastases. ClinicalTrials.gov Identifier NCT02414269. Available online: http://clinicaltrials.gov/ (accessed on 8 May 2017).
- Klampatsa, A.; Achkova, D.Y.; Davies, D.M.; Parente-Pereira, A.C.; Woodman, N.; Rosekilly, J.; Osborne, G.; Thayaparan, T.; Bille, A.; Sheaf, M.; et al. Intracavitary “T4 immunotherapy” of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017, 393, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Van der Stegen, S.J.C.; Davies, D.M.; Wilkie, S.; Foster, J.; Sosabowski, J.K.; Burnet, J.; Whilding, L.M.; Petrovic, R.M.; Ghaem-Maghami, S.; Mather, S.; et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: Identifying a window of therapeutic opportunity? J. Immunol. 2013, 191, 4589–4598. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Institutes of Health. Malignant pleural disease treated with autologous T-cells genetically engineered to target the cancer-cell surface antigen mesothelin. Identifier NCT02414269. Available online: http://clinicaltrials.gov/ (accessed on 8 May 2017).
- Davies, D.; Foster, J. Flexible Targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012, 18, 1. [Google Scholar] [CrossRef]
- Van Schalkwyk, M.C.I.; Papa, S.E.; Jeannon, J.-P.; Guerrero Urbano, T.; Spicer, J.F.; Maher, J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum. Gene Ther. Clin. Dev. 2013, 24, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Globerson-Levin, A.; Waks, T.; Eshhar, Z. Elimination of progressive mammary cancer by repeated administrations of chimeric antigen receptor-modified T cells. Mol. Ther. 2014, 22, 1029–1038. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Cancer | Study Type | CAR | Stimulatory Signal | Inhibitory Signal | Additional Therapy | Delivery Method |
|---|---|---|---|---|---|---|---|
| Yaghoubi et al. [19] | Glioblastoma | Clinical Phase I | IL13Rα2 | - | - | Surgical Resection | Catheter infusion into resection cavity |
| Brown et al. [12] | Glioblastoma | Clinical Phase I | IL13Rα2 | - | - | Surgical Resection | Catheter infusion into resection cavity |
| Brown et al. [13] | Glioblastoma | Clinical Phase I | IL13Rα2 | CD137 | IgG4-Fc mutant A | Surgical Resection | Catheter infusion into resection cavity |
| Katz et al. [17] | Hepatic colorectal metastases | Clinical Phase I | CEA | IL2 | - | Chemotherapy | Hepatic artery infusion |
| Katz et al. [26] | Hepatic colorectal metastases | Clinical Phase I | CEA | IL2 | - | Chemotherapy + Yttrium spheres | Hepatic Artery Infusion |
| van Schalkwyk et al. [31] | Head and Neck | Clinical Phase I | T1E28z (ErbB) B | Cytokine receptor 4αβ C | - | Cyclophoshamide | Ultrasound guided intratumoral injection |
| Adusumilli et al. [29] | Pleural Mesothelioma | Clinical Phase I | M28z D | CD28 | - | - | Intrapleural |
| Katz et al. [18] | Peritoneal carcinomatosis | Pre-clinical | CEA | IL2 | Anti-PDL1 Anti-Gr1 Anti-GITR E | - | Intraperitoneal |
| Adusumilli et al. [11] | Pleural Mesothelioma | Pre-clinical | M28z | CD28 | - | Cyclophosphamide | Intrapleural |
| van der Stegen et al. [28] | Head and Neck | Pre-clinical | T1E28z (ErbB) | Cytokine receptor 4αβ C | - | - | Intraperitoneal |
| Davies et al. [30] | Head and Neck | Pre-clinical | T1E28z (ErbB) | Cytokine receptor 4αβ F | - | - | Intraperitoneal |
| Klampatsa et al. [27] | Mesothelioma | Pre-clinical | T1E28z (ErbB) | Cytokine receptor 4αβ | - | - | Intraperitoneal |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sridhar, P.; Petrocca, F. Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy. Cancers 2017, 9, 92. https://doi.org/10.3390/cancers9070092
Sridhar P, Petrocca F. Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy. Cancers. 2017; 9(7):92. https://doi.org/10.3390/cancers9070092
Chicago/Turabian StyleSridhar, Praveen, and Fabio Petrocca. 2017. "Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy" Cancers 9, no. 7: 92. https://doi.org/10.3390/cancers9070092
APA StyleSridhar, P., & Petrocca, F. (2017). Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy. Cancers, 9(7), 92. https://doi.org/10.3390/cancers9070092