
The Role of miRNAs in Angiogenesis, Invasion and Metabolism and Their Therapeutic Implications in Gliomas

Abstract
1. Introduction
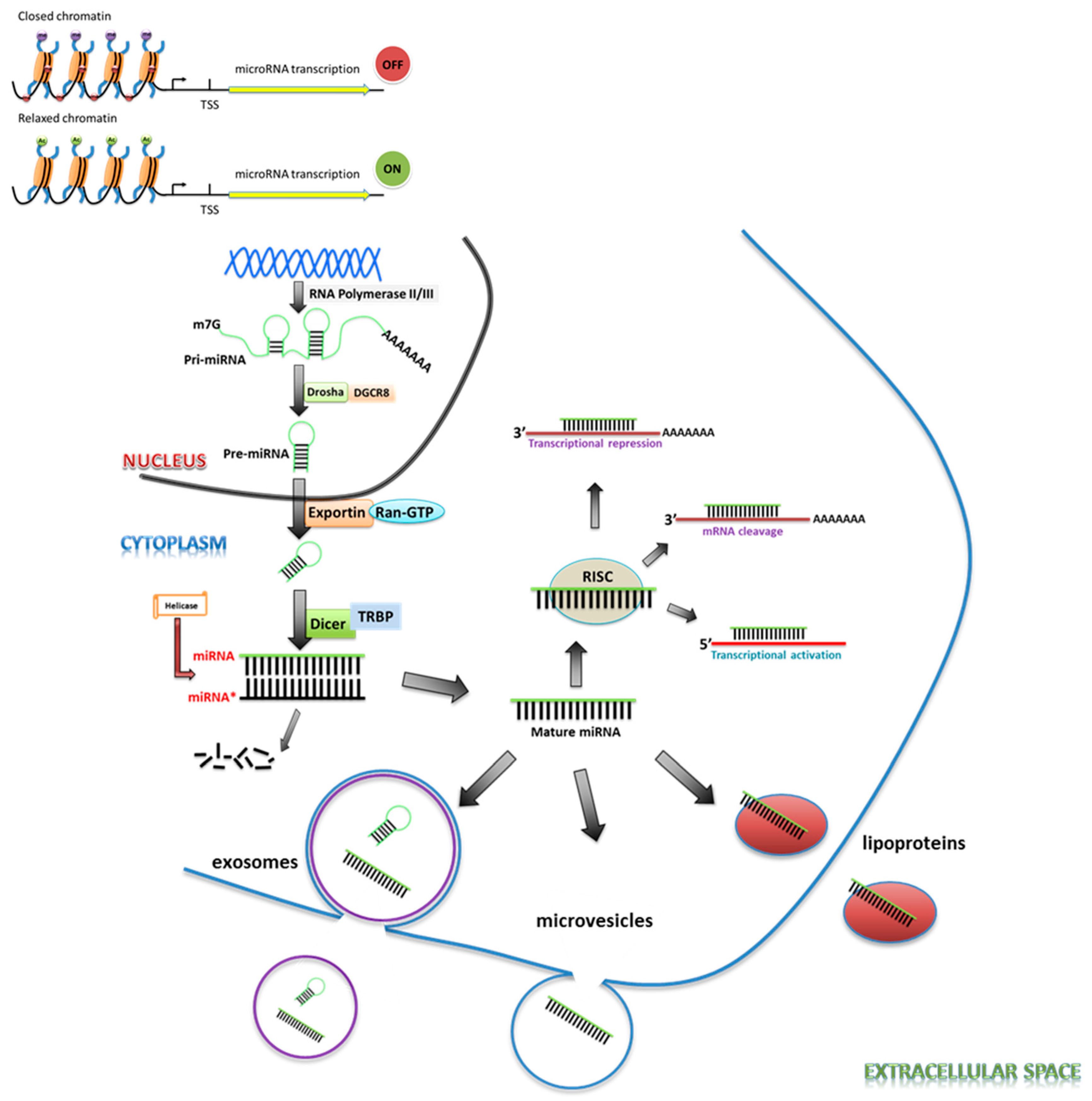
2. miRNA Biogenesis
3. miRNA Regulation
4. Gliomas
5. Defining Hallmarks of Gliomas
5.1. Angiogenesis
5.1.1. miR-296
5.1.2. miR-7
5.2. Invasion
5.2.1. miR-21
5.2.2. miR-34a
5.2.3. miR-10b
5.3. Metabolism
5.3.1. miR-153
5.3.2. miR-451
6. Prognostic and Predictive miRNA Biomarkers
7. miRNA Therapeutics
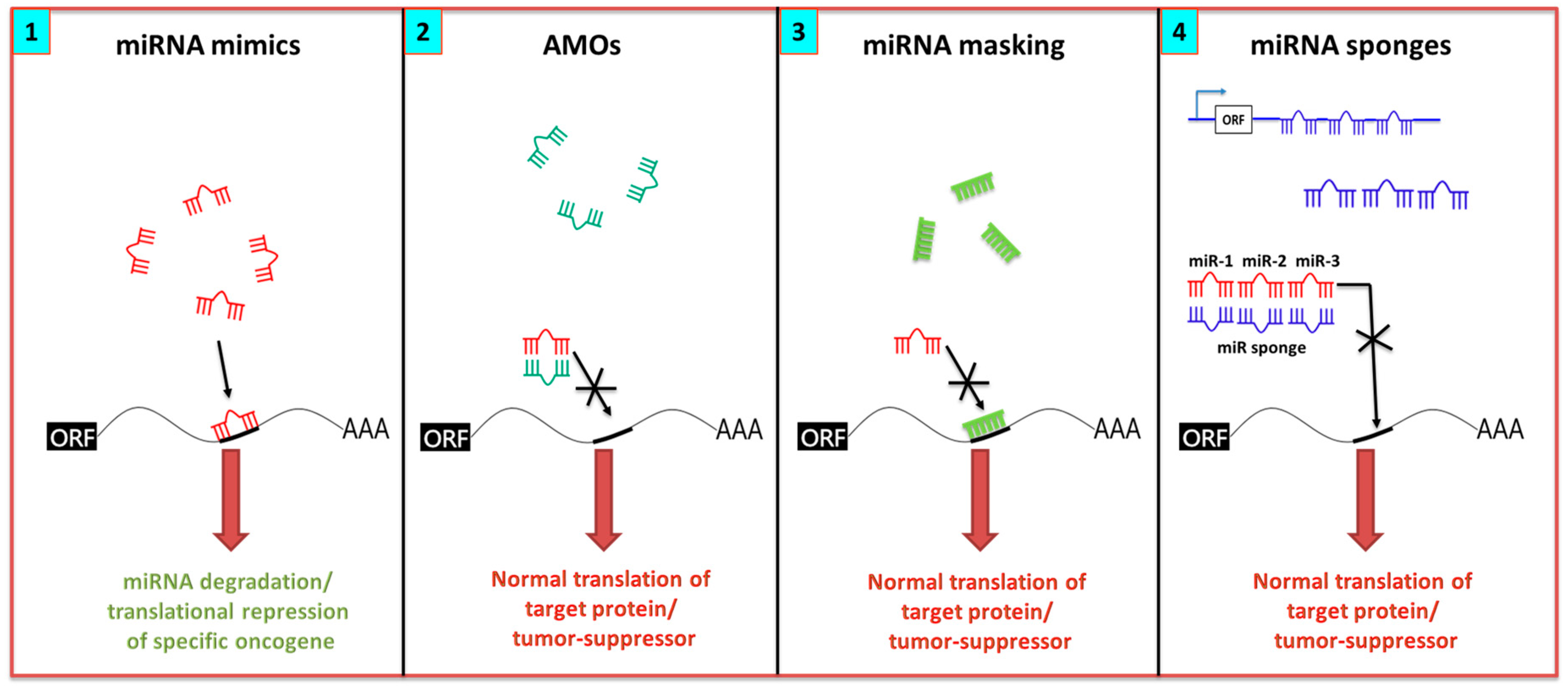
7.1. miRNA Inhibition Strategies
7.2. miRNA Replenishment Therapy
7.3. Delivery Systems
8. miRNAs in Clinical Trials
9. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Pang, C.; Guan, Y.; Zhao, K.; Chen, L.; Bao, Y.; Cui, R.; Li, G.; Wang, Y. Up-regulation of microRNA-15b correlates with unfavorable prognosis and malignant progression of human glioma. Int. J. Clin. Exp. Pathol. 2015, 8, 4943–4952. [Google Scholar] [PubMed]
- Zheng, X.; Chopp, M.; Lu, Y.; Buller, B.; Jiang, F. miR-15b and miR-152 reduce glioma cell invasion and angiogenesis via NRP-2 and MMP-3. Cancer Lett. 2013, 329, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, P.; Li, X.Y.; Zhang, L.Y.; Xiong, W.; Zhou, M.; Xiao, L.; Zeng, F.; Li, X.L.; Wu, M.H.; et al. Grade-specific expression profiles of miRNAs/mRNAs and docking study in human grade I–III astrocytomas. OMICS 2011, 15, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.; Thomson, D.; Giles, K.; Maleki, S.; Mreich, E.; Wheeler, H.; Leedman, P.; Biggs, M.; Cook, R.; Little, N.; et al. Mir-124a is frequently down-regulated in glioblastoma and is involved in migration and invasion. Eur. J. Cancer 2011, 47, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, Y.; Liu, M.; Jiang, Y. MicroRNA-181b inhibits cellular proliferation and invasion of glioma cells via targeting sal-like protein 4. Oncol. Res. 2016, 25, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.M.; Wang, X.F.; Qian, X.; Tao, T.; Wang, L.; Chen, Q.D.; Wang, X.R.; Cao, L.; Wang, Y.Y.; Zhang, J.X.; et al. miRNA-181b suppresses igf-1r and functions as a tumor suppressor gene in gliomas. RNA 2013, 19, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Lou, S.; Dai, Q.; Mao, D.; Ji, J.; Sun, X. Tumor suppressor mir-181c attenuates proliferation, invasion, and self-renewal abilities in glioblastoma. Neuroreport 2015, 26, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y. Regulation of cell proliferation and migration in glioblastoma: New therapeutic approach. Front. Oncol. 2013, 3, 53. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. Causes and consequences of microRNA dysregulation. Cancer J. 2012, 18, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Hubert, C.G.; Miller, T.E.; Taylor, M.D.; Rich, J.N. An epigenetic gateway to brain tumor cell identity. Nat. Neurosci. 2016, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P.; Palanichamy, J.K.; Singh, A.; Das, P.; Bhagat, M.; Kassab, M.A.; Sinha, S.; Chattopadhyay, P. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA 2014, 20, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of drosha, export in 5, and dicer in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [PubMed]
- Romero-Cordoba, S.L.; Salido-Guadarrama, I.; Rodriguez-Dorantes, M.; Hidalgo-Miranda, A. miRNA biogenesis: Biological impact in the development of cancer. Cancer Biol. Ther. 2014, 15, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ramasubramanian, B.; Kanji, S.; Chakraborty, A.R.; Haque, S.J.; Chakravarti, A. Circulating microRNAs in cancer: Hope or hype? Cancer Lett. 2016, 381, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′utr of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free micrornas in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G. Circulating miRNAs: Roles in cancer diagnosis, prognosis and therapy. Adv. Drug Deliv. Rev. 2015, 81, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Danquah, M.; Chaudhary, A.K.; Mahato, R.I. Small molecules targeting microRNA for cancer therapy: Promises and obstacles. J. Control Release 2015, 219, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Schanen, B.C.; Li, X. Transcriptional regulation of mammalian miRNA genes. Genomics 2011, 97, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-mediated histone h3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef]
- Kang, N.; Choi, S.Y.; Kim, Y.K.; Yoo Ie, R.; Han, D.H.; Lee, D.S.; Kim, Y.S.; Hong, S.H.; Kang, J.H.; Lee, K.Y.; et al. Silencing of miR-137 by aberrant promoter hypermethylation in surgically resected lung cancer. Lung Cancer 2015, 89, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Stresemann, C.; Kuner, R.; Mund, C.; Musch, T.; Meister, M.; Sultmann, H.; Lyko, F. The human let-7a-3 locus contains an epigenetically regulated microRNA gene with oncogenic function. Cancer Res. 2007, 67, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Sun, Q.; Wu, J.; Zheng, P.; Zhao, G. Sodium butyrate upregulates miR-203 expression to exert anti-proliferation effect on colorectal cancer cells. Cell. Physiol. Biochem. 2016, 39, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Li, S.; Wang, R.; Xiao, M.; Meng, Y.; Zeng, C.; Fang, J.H.; Yang, J.; Zhuang, S.M. Expression of microRNA-195 is transactivated by sp1 but inhibited by histone deacetylase 3 in hepatocellular carcinoma cells. Biochim. Biophys. Acta 2016, 1859, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. Cbtrus statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-oncology 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 who classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Foote, M.B.; Papadopoulos, N.; Diaz, L.A., Jr. Genetic classification of gliomas: Refining histopathology. Cancer Cell 2015, 28, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Mischel, P.S.; Reifenberger, G. Molecular classification of gliomas. Handb. Clin. Neurol. 2016, 134, 97–120. [Google Scholar] [PubMed]
- Perry, A.; Wesseling, P. Histologic classification of gliomas. Handb. Clin. Neurol. 2016, 134, 71–95. [Google Scholar] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro-oncology 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus procarbazine, ccnu, and vincristine in low-grade glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Ren, X.; Zhang, C.; Han, S.; Wu, A. Expression and prognostic value of microRNAs in lower-grade glioma depends on IDH1/2 status. J. Neurooncol. 2017, 132, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Ichikawa, T.; Kurozumi, K.; Date, I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011, 28, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Marsden, P.A. Angiogenesis in glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Moller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A systematic review of microRNA in glioblastoma multiforme: Micro-modulators in the mesenchymal mode of migration and invasion. Mol. Neurobiol. 2013, 47, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Rolle, K. miRNA multiplayers in glioma. From bench to bedside. Acta Biochim. Pol. 2015, 62, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dutta, A.; Abounader, R. The role of microRNAs in glioma initiation and progression. Front. Biosci. 2012, 17, 700–712. [Google Scholar] [CrossRef]
- Karsy, M.; Arslan, E.; Moy, F. Current progress on understanding microRNAs in glioblastoma multiforme. Genes Cancer 2012, 3, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Nikaki, A.; Piperi, C.; Papavassiliou, A.G. Role of microRNAs in gliomagenesis: Targeting miRNAs in glioblastoma multiforme therapy. Expert Opin. Investig. Drugs 2012, 21, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Silber, J.; James, C.D.; Hodgson, J.G. MicroRNAs in gliomas: Small regulators of a big problem. Neuromol. Med. 2009, 11, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; Tannous, B.A.; Saydam, O.; Skog, J.; Grau, S.; Soutschek, J.; Weissleder, R.; Breakefield, X.O.; Krichevsky, A.M. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 2008, 14, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Sedo, A.; Mentlein, R. Glioma Cell Biology; Springer: New York, NY, USA, 2014. [Google Scholar]
- Babae, N.; Bourajjaj, M.; Liu, Y.; Van Beijnum, J.R.; Cerisoli, F.; Scaria, P.V.; Verheul, M.; Van Berkel, M.P.; Pieters, E.H.; Van Haastert, R.J.; et al. Systemic miRNA-7 delivery inhibits tumor angiogenesis and growth in murine xenograft glioblastoma. Oncotarget 2014, 5, 6687–6700. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.W.; Wang, X.; Yang, Y.; Mao, Q. Role of micro-RNA (miRNA) in pathogenesis of glioblastoma. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1630–1639. [Google Scholar] [PubMed]
- Chen, L.; Kang, C. miRNA interventions serve as ‘magic bullets’ in the reversal of glioblastoma hallmarks. Oncotarget 2015, 6, 38628–38642. [Google Scholar] [PubMed]
- Fang, L.; Deng, Z.; Shatseva, T.; Yang, J.; Peng, C.; Du, W.W.; Yee, A.J.; Ang, L.C.; He, C.; Shan, S.W.; et al. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-beta8. Oncogene 2011, 30, 806–821. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Westphal, M. Glioma invasion in the central nervous system. Neurosurgery 1996, 39, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.J.; Burns, T.C.; Zhang, Y.; Abounader, R. Targeting MET for glioma therapy. Neurosurg. Focus 2014, 37, E10. [Google Scholar] [CrossRef] [PubMed]
- Areeb, Z.; Stylli, S.S.; Koldej, R.; Ritchie, D.S.; Siegal, T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. MicroRNA as potential biomarkers in glioblastoma. J. Neurooncol. 2015, 125, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, J.; Xiang, Q.; Liang, Y.; Zhao, N.; Zhang, Z.; Liu, Q.; Cui, Y. Prognostic role of microRNA-21 expression in gliomas: A meta-analysis. J. Neurooncol. 2016, 130, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Wei, W.; Zhang, Z.; He, C.; Yang, R.; Zhang, J.; Wu, Z.; Huang, Q.; Jiang, Q. Identification of microRNAs associated with glioma diagnosis and prognosis. Oncotarget 2017, 8, 26394–26403. [Google Scholar] [CrossRef] [PubMed]
- Lages, E.; Guttin, A.; El Atifi, M.; Ramus, C.; Ipas, H.; Dupre, I.; Rolland, D.; Salon, C.; Godfraind, C.; deFraipont, F.; et al. MicroRNA and target protein patterns reveal physiopathological features of glioma subtypes. PLoS ONE 2011, 6, e20600. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Yue, X.; Zhou, X.; Lan, F.M.; You, G.; Zhang, W.; Zhang, K.L.; Zhang, C.Z.; Cheng, J.Q.; Yu, S.Z.; et al. microRNA-21 expression is regulated by beta-catenin/stat3 pathway and promotes glioma cell invasion by direct targeting reck. CNS Neurosci. Ther. 2012, 18, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, G.; Wurdinger, T.; Kesari, S.; Esau, C.C.; Burchard, J.; Linsley, P.S.; Krichevsky, A.M. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell. Biol. 2008, 28, 5369–5380. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guessous, F.; Zhang, Y.; Dipierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jiang, J.; Yang, Y.; Schmittgen, T.D.; et al. microRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef] [PubMed]
- Rathod, S.S.; Rani, S.B.; Khan, M.; Muzumdar, D.; Shiras, A. Tumor suppressive miRNA-34a suppresses cell proliferation and tumor growth of glioma stem cells by targeting akt and wnt signaling pathways. FEBS Open Bio 2014, 4, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Ogawa, S.; Kawamata, N.; Leiter, A.; Ham, M.; Li, D.; Doan, N.B.; Said, J.W.; Black, K.L.; Phillip Koeffler, H. miR-34a functions as a tumor suppressor modulating EGFR in glioblastoma multiforme. Oncogene 2013, 32, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Knight, R.A. Mir-34: From bench to bedside. Oncotarget 2014, 5, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase i study of mrx34, a liposomal mir-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, G.; Yi, M.; Narayan, R.S.; Niers, J.M.; Wurdinger, T.; Imitola, J.; Ligon, K.L.; Kesari, S.; Esau, C.; Stephens, R.M.; et al. Human glioma growth is controlled by microRNA-10b. Cancer Res. 2011, 71, 3563–3572. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, T.; Nishihara, M.; Kondoh, T.; Hosoda, K.; Kohmura, E. MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, UPAR and RHOC. Int. J. Cancer 2009, 125, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yan, W.; Wang, Y.; Sun, G.; Luo, H.; Zhang, J.; Wang, X.; You, Y.; Yang, Z.; Liu, N. MicroRNA-10b induces glioma cell invasion by modulating MMP-14 and UPAR expression via HOXD10. Brain Res. 2011, 1389, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Teo, S.; Lam, D.H.; Jeyaseelan, K.; Wang, S. MicroRNA-10b pleiotropically regulates invasion, angiogenicity and apoptosis of tumor cells resembling mesenchymal subtype of glioblastoma multiforme. Cell Death Dis. 2012, 3, e398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, J.; Hao, J.; Shi, Z.; Wang, Y.; Han, L.; Yu, S.; You, Y.; Jiang, T.; Wang, J.; et al. High level of miR-221/222 confers increased cell invasion and poor prognosis in glioma. J. Transl. Med. 2012, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Qi, Y.; Ng, S.S.; Chen, X.; Li, D.; Chen, S.; Ge, R.; Jiang, S.; Li, G.; Chen, Y.; et al. MicroRNA-146b inhibits glioma cell migration and invasion by targeting mmps. Brain Res. 2009, 1269, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Huang, Q.; Chen, K.; Liu, L.; Lin, C.; Dai, T.; Yu, C.; Wu, Z.; Li, J. miR-218 inhibits the invasive ability of glioma cells by direct downregulation of IKK-beta. Biochem. Biophys. Res. Commun. 2010, 402, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; diPierro, C.G.; Li, Y.; Chiocca, E.A.; et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 2009, 29, 15161–15168. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, S. The warburg hypothesis fifty years later. Z. Krebsforsch. Klin. Onkol. Cancer Res. Clin. Oncol. 1976, 87, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Ru, P.; Williams, T.M.; Chakravarti, A.; Guo, D. Tumor metabolism of malignant gliomas. Cancers (Basel) 2013, 5, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Pelicano, H.; Huang, P. Cancer metabolism: Is glutamine sweeter than glucose? Cancer Cell 2010, 18, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CPG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. Cancer metabolism as a central driving force of glioma pathogenesis. Brain Tumor Pathol. 2016, 33, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Lokody, I. Metabolism: Reprogramming metabolic flux in glioma. Nat. Rev. Cancer 2014, 14, 706–707. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.F. Drug target miRNAs: Chances and challenges. Trends Biotechnol. 2014, 32, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, J.; Li, Y.; Fan, J.; Chen, L.; Xu, R. MicroRNA-153 regulates glutamine metabolism in glioblastoma through targeting glutaminase. Tumour Biol. 2017, 39, 1010428317691429. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Bronisz, A.; Nowicki, M.O.; Chiocca, E.A.; Lawler, S. MicroRNA-451: A conditional switch controlling glioma cell proliferation and migration. Cell Cycle 2010, 9, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; de Lay, M.; van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. microRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase m2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro-oncology 2010, 12, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.W.; Lu, Q.; Wang, L.X.; Zhao, W.Y.; Cao, Y.Q.; Li, Y.N.; Han, G.S.; Liu, J.M.; Yue, Z.J. Decreased miR-106a inhibits glioma cell glucose uptake and proliferation by targeting SLC2a3 in GBM. BMC Cancer 2013, 13, 478. [Google Scholar] [CrossRef] [PubMed]
- De Planell-Saguer, M.; Rodicio, M.C. Detection methods for microRNAs in clinic practice. Clin. Biochem. 2013, 46, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Phillips, H.S.; Brennan, C.W. Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 2011, 59, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yoshimoto, K.; Guan, Y.; Hata, N.; Mizoguchi, M.; Sagata, N.; Murata, H.; Kuga, D.; Amano, T.; Nakamizo, A.; et al. Associations between microRNA expression and mesenchymal marker gene expression in glioblastoma. Neuro-oncology 2012, 14, 1153–1162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Papagiannakopoulos, T.; Friedmann-Morvinski, D.; Neveu, P.; Dugas, J.C.; Gill, R.M.; Huillard, E.; Liu, C.; Zong, H.; Rowitch, D.H.; Barres, B.A.; et al. Pro-neural miR-128 is a glioma tumor suppressor that targets mitogenic kinases. Oncogene 2012, 31, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Marziali, G.; Buccarelli, M.; Giuliani, A.; Ilari, R.; Grande, S.; Palma, A.; D’Alessandris, Q.G.; Martini, M.; Biffoni, M.; Pallini, R.; et al. A three-microRNA signature identifies two subtypes of glioblastoma patients with different clinical outcomes. Mol. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, R.; Gao, K.; Luo, H.; Wang, X.; Shi, Y.; Dong, Q.; Luan, W.; You, Y. Identification of intrinsic subtype-specific prognostic microRNAs in primary glioblastoma. J. Exp. Clin. Cancer Res. 2014, 33, 9. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Thygesen, H.; Gregory, W.; Westhead, D.R.; French, P.J.; Van Den Bent, M.J.; Lawler, S.E.; Short, S.C. A validated microRNA profile with predictive potential in glioblastoma patients treated with bevacizumab. Mol. Oncol. 2016, 10, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Son, J.C.; Jeong, H.O.; Park, D.; No, S.G.; Lee, E.K.; Lee, J.; Chung, H.Y. miR-10a and miR-204 as a potential prognostic indicator in low-grade gliomas. Cancer Inform. 2017, 16. [Google Scholar] [CrossRef][Green Version]
- Niyazi, M.; Zehentmayr, F.; Niemoller, O.M.; Eigenbrod, S.; Kretzschmar, H.; Schulze-Osthoff, K.; Tonn, J.C.; Atkinson, M.; Mortl, S.; Belka, C. miRNA expression patterns predict survival in glioblastoma. Radiat. Oncol. 2011, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Patric, I.R.; Somasundaram, K. A ten-microRNA expression signature predicts survival in glioblastoma. PLoS ONE 2011, 6, e17438. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, H.; Liu, X.; Lu, Z.; Li, G.; Lu, M.; Tao, X. MicroRNA signatures predict prognosis of patients with glioblastoma multiforme through the cancer genome atlas. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Thygesen, H.; Tumilson, C.; Droop, A.; Boissinot, M.; Hughes, T.A.; Westhead, D.; Alder, J.E.; Shaw, L.; Short, S.C.; et al. Prediction of clinical outcome in glioblastoma using a biologically relevant nine-microRNA signature. Mol. Oncol. 2015, 9, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yu, Q.; Chen, B.; Lu, X.; Li, Q. The prognostic value of a seven-microRNA classifier as a novel biomarker for the prediction and detection of recurrence in glioma patients. Oncotarget 2016, 7, 53392–53413. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Slaby, O.; Lakomy, R.; Fadrus, P.; Hrstka, R.; Kren, L.; Lzicarova, E.; Smrcka, M.; Svoboda, M.; Dolezalova, H.; Novakova, J.; et al. MicroRNA-181 family predicts response to concomitant chemoradiotherapy with temozolomide in glioblastoma patients. Neoplasma 2010, 57, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Haemmig, S.; Baumgartner, U.; Gluck, A.; Zbinden, S.; Tschan, M.P.; Kappeler, A.; Mariani, L.; Vajtai, I.; Vassella, E. miR-125b controls apoptosis and temozolomide resistance by targeting TNFAIP3 and NKIRAS2 in glioblastomas. Cell Death Dis. 2014, 5, e1279. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Moniuszko, M.; Utikal, J.; Niklinski, J.; Allgayer, H. microRNAs as novel targets and tools in cancer therapy. Cancer Lett. 2017, 387, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Sood, A.K.; Hong, D.S. RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat. Rev. 2016, 50, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Tan, C. Combination of microRNA therapeutics with small-molecule anticancer drugs: Mechanism of action and co-delivery nanocarriers. Adv. Drug Deliv. Rev. 2015, 81, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Osman, A. microRNAs in health and disease—Basic science and clinical applications. Clin. Lab. 2012, 58, 393–402. [Google Scholar] [PubMed]
- Velu, C.S.; Grimes, H.L. Utilizing antagomiR (antisense microRNA) to knock down microRNA in murine bone marrow cells. Methods Mol. Biol. 2012, 928, 185–195. [Google Scholar] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nature reviews. Drug Dis. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Calin, G.A.; Berindan-Neagoe, I. MicroRNAs and cancer therapy—From bystanders to major players. Curr. Med. Chem. 2013, 20, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Liu, L.; Li, X.; Han, W. Decitabine, a new star in epigenetic therapy: The clinical application and biological mechanism in solid tumors. Cancer Lett. 2014, 354, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Thaci, B.; Ahmed, A.; Ulasov, I.; Lesniak, M.S. microRNA targeting as a therapeutic strategy against glioma. Curr. Mol. Med. 2013, 13, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, D.Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Petrocca, F.; Lieberman, J. Promise and challenge of RNA interference-based therapy for cancer. J. Clin. Oncol. 2011, 29, 747–754. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stylianopoulos, T.; Jain, R.K. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 18632–18637. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhao, X.; Lee, L.J.; Lee, R.J. Targeted delivery systems for oligonucleotide therapeutics. AAPS J. 2009, 11, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J. Strategies to use microRNAs as therapeutic targets. Best practice & research. Clin. Endocrinol. Metab. 2016, 30, 551–561. [Google Scholar]
- Sela, H.; Cohen, H.; Elia, P.; Zach, R.; Karpas, Z.; Zeiri, Y. Spontaneous penetration of gold nanoparticles through the blood brain barrier (BBB). J. Nanobiotechnol. 2015, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Fadda, A.M.; Sinico, C. Liposomes for brain delivery. Expert Opin. Drug. Deliv. 2013, 10, 1003–1022. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. Available online: https://clinicaltrials.gov (accessed on 29 June 2017).
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013, 31, 577. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.L.; Jiang, Q.Y.; Jin, X.; Shen, J.; Wang, K.; Li, Y.B.; Xu, F.J.; Tang, G.P.; Li, Z.H. Cationic microRNA-delivering nanovectors with bifunctional peptides for efficient treatment of panc-1 xenograft model. Biomaterials 2013, 34, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing cd44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.H.; Kim, H.; Jang, B.; Cho, H.; Ryu, J.; Kim, B.; Park, Y.; Kim, J.; Lee, J.B.; Lee, H. Technological development of structural DNA/RNA-based RNAi systems and their applications. Adv. Drug Deliv. Rev. 2016, 104, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.M.; Kruger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Chu, J.; Wu, L.C.; Mao, H.; Peng, Y.; Alvarez-Breckenridge, C.A.; Hughes, T.; Wei, M.; Zhang, J.; Yuan, S.; et al. MicroRNAs activate natural killer cells through toll-like receptor signaling. Blood 2013, 121, 4663–4671. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, A.M.; Bezman, N.A.; Lee, J.E.; Matloubian, M.; Sun, J.C.; Lanier, L.L. MicroRNA function in nk-cell biology. Immunol. Rev. 2013, 253, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.A.; Usher, L.; Lindsay, M.A. Clinical potential of oligonucleotide-based therapeutics in the respiratory system. Pharmacol. Ther. 2017, 169, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Phase 2, open-label, clinical trial of miRavirsen sodium in combination with telaprevir and ribavirin in null responders to pegylated-interferon alpha plus ribavirin subjects with chronic hepatitis c virus genotype 1 infection. Available online: https://clinicaltrials.gov/ct2/show/NCT02452814 (accessed on 10 July 2017).
- Xu, W.; San Lucas, A.; Wang, Z.; Liu, Y. Identifying microRNA targets in different gene regions. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| miRNA | Regulation in Gliomas | Function | Targets | Validation | Reference |
|---|---|---|---|---|---|
| miR-7 | Down-regulated | Inhibits angiogenesis | EGFR, IRS-1, IRS-2, FAK, OGR | Overexpression decreases cell proliferation and angiogenesis in U-87 cells in vitro and in tumor xenograft model | [43] |
| miR-296 | Up-regulated | Promotes angiogenesis | HGS | Overexpression promotes angiogenesis in vitro and in a tumor xenograft model in vivo | [41] |
| miR-15b | Up-regulated | Inhibits angiogenesis | NRP-2 | Overexpression reduces capillary tube formation in cultured endothelial cells | [46,47] |
| miR-93 | Up-regulated | Promotes angiogenesis | Integrin B8 | Overexpression increases cell migration and tube formation of co-cultured endothelial cells in vitro and in vivo | [48] |
| miRNA | Regulation in Gliomas | Function | Targets | Validation | Reference |
|---|---|---|---|---|---|
| miR-10b | Up-regulated | Promotes invasion | TP53, Pax6, Notch1, HOXD10 | Overexpression increases invasion and its inhibition decreases invasion in vitro | [67] |
| miR-21 | Up-regulated | Promotes invasion | PTEN, RECK, MARCKS | Inhibition elevates RECK and TIMP3 expression and decreases invasion in vitro and in vivo | [57] |
| miR-34a | Dow-regulated | Inhibits invasion | c-Met, Notch1/2, CDK6 | Overexpression decreases invasion in glioma cells by targeting HGF/c-MET and Notch1/2 signaling; translated into clinical trial | [58] |
| miR-221/222 | Up-regulated | Promotes invasion | p27, p57, EGFR/PTEN/AKT signaling, TIMP3, protein tyrosine phosphatase u | Overexpression increases invasion in multiple glioma cell lines | [68] |
| miR-124 | Down-regulated | Inhibits invasion | IQGAP1 | Overexpression in glioma cells inhibits cell migration and invasion | [69,70] |
| miR-181 | Down-regulated | Inhibits invasion | IQGAP1, LAMC1, ITGB1 | Overexpression decreases invasion in glioma cells | [71,72,73] |
| miR-451 | Down-regulated | Inhibits invasion | BCL2, SALL4 | Overexpression reduces invasion in glioma cells | [74,75] |
| miR-146 | Down-regulated | Inhibits invasion | PI3K/AKT signaling, CAB39 | Overexpression inhibits migration and invasion of glioma cells | [76] |
| miR-218 | Down-regulated | Inhibits invasion | MMP16 | Overexpression decreases invasion and inhibition increases invasion in glioma cells | [77] |
| miR-326 | Down-regulated | Inhibits invasion | IKKB, MMP9 | Overexpression inhibits invasion in glioma cells | [78] |
| miRNA | Regulation in Gliomas | Function | Target | Validation | Reference |
|---|---|---|---|---|---|
| miR-153 | Down-regulated | Restrained glutamine utilization and glutamate generation | Glutaminase | Overexpression decreased cell proliferation and glutamine utilization in glioma cells | [93] |
| miR-451 | Up-regulated | Inhibition helps cancer cells escape metabolic stress | CAB39 | Overexpression sensitized glioma cells to glucose deprivation | [74,94] |
| miR-326 | Down-regulated | Decreases metabolism | PKM2, Notch signaling | Overexpression induces apoptosis and reduces metabolic activity in glioma cells | [78,95] |
| miR-106a | Down-regulated | SLC2A3 | Glucose uptake | Inhibition decreases glucose uptake and proliferation in glioma cells | [96] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyer, S.; Fleming, J.; Meng, W.; Singh, R.; Haque, S.J.; Chakravarti, A. The Role of miRNAs in Angiogenesis, Invasion and Metabolism and Their Therapeutic Implications in Gliomas. Cancers 2017, 9, 85. https://doi.org/10.3390/cancers9070085
Beyer S, Fleming J, Meng W, Singh R, Haque SJ, Chakravarti A. The Role of miRNAs in Angiogenesis, Invasion and Metabolism and Their Therapeutic Implications in Gliomas. Cancers. 2017; 9(7):85. https://doi.org/10.3390/cancers9070085
Chicago/Turabian StyleBeyer, Sasha, Jessica Fleming, Wei Meng, Rajbir Singh, S. Jaharul Haque, and Arnab Chakravarti. 2017. "The Role of miRNAs in Angiogenesis, Invasion and Metabolism and Their Therapeutic Implications in Gliomas" Cancers 9, no. 7: 85. https://doi.org/10.3390/cancers9070085
APA StyleBeyer, S., Fleming, J., Meng, W., Singh, R., Haque, S. J., & Chakravarti, A. (2017). The Role of miRNAs in Angiogenesis, Invasion and Metabolism and Their Therapeutic Implications in Gliomas. Cancers, 9(7), 85. https://doi.org/10.3390/cancers9070085