
The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma

Abstract
:1. Introduction
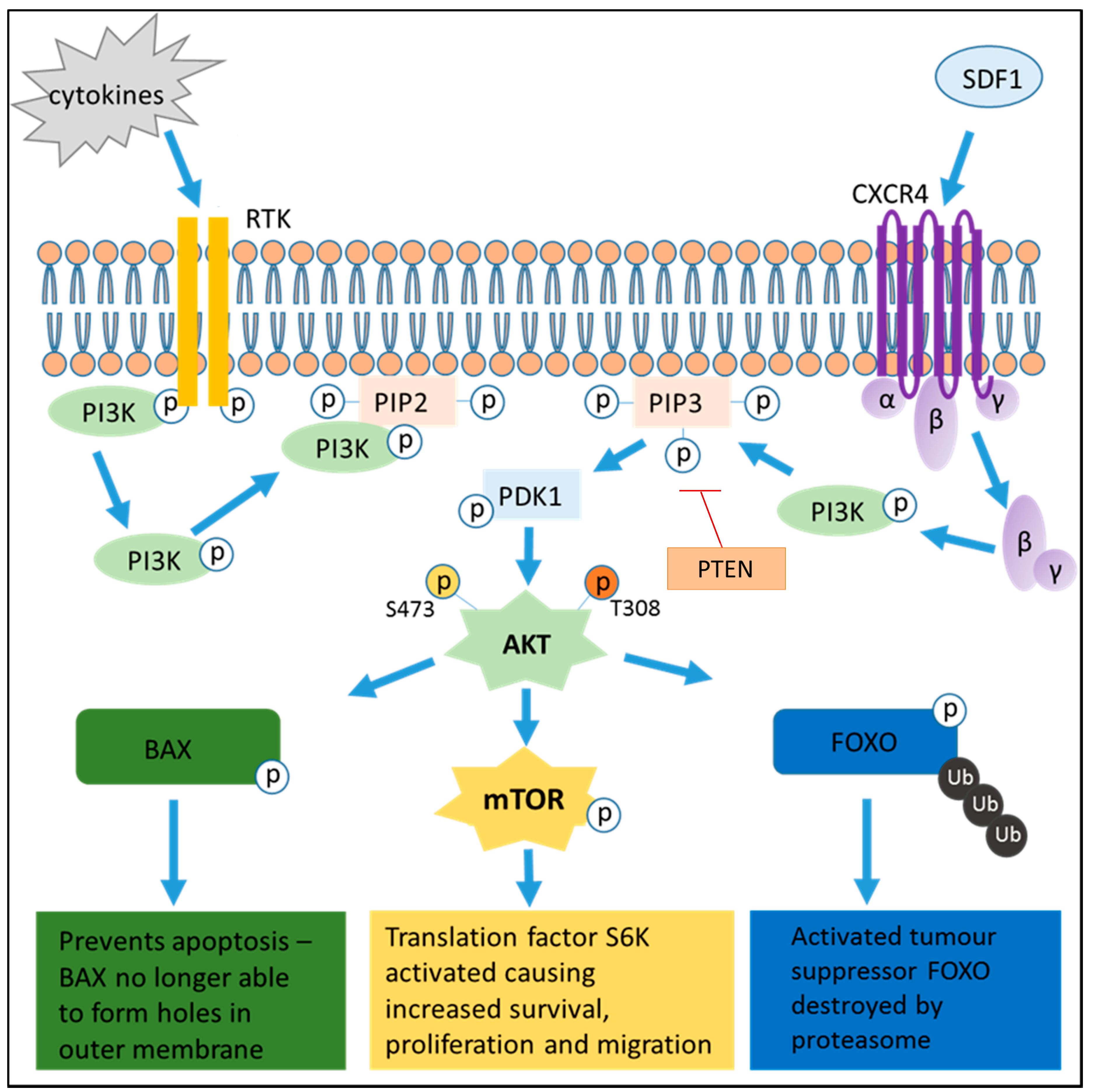
2. Phosphatidylinositol 3-Kinase (PI3K) Activation in Cancer
2.1. p110α
2.2. p110β
2.3. p110δ
2.4. p110γ
3. PI3K Pathway Activation
4. The Protective Effect of the Bone Marrow Microenvironment
5. Clinical Implications
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Maiso, P.; Liu, Y.; Morgan, B.; Azab, A.K.; Ren, P.; Martin, M.B.; Zhang, Y.; Liu, Y.; Sacco, A.; Ngo, H.; et al. Defining the role of TORC1/2 in multiple myeloma. Blood 2011, 118, 6860–6870. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef] [PubMed]
- Pillinger, G.; Loughran, N.V.; Piddock, R.E.; Shafat, M.S.; Zaitseva, L.; Abdul-Aziz, A.; Lawes, M.J.; Bowles, K.M.; Rushworth, S.A. Targeting PI3Kδ and PI3Kγ signalling disrupts human AML survival and bone marrow stromal cell mediated protection. Oncotarget 2016, 7, 39784–39795. [Google Scholar] [PubMed]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Perrone, G.; Miura, N.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Santo, L.; Vallet, S.; et al. PI3K/p110δ is a novel therapeutic target in multiple myeloma. Blood 2010, 116, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Billottet, C.; Grandage, V.L.; Gale, R.E.; Quattropani, A.; Rommel, C.; Vanhaesebroeck, B.; Khwaja, A. A selective inhibitor of the p110[delta] isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 2006, 25, 6648–6659. [Google Scholar] [CrossRef] [PubMed]
- Lannutti, B.J.; Meadows, S.A.; Herman, S.E.M.; Kashishian, A.; Steiner, B.; Johnson, A.J.; Byrd, J.C.; Tyner, J.W.; Loriaux, M.M.; Deininger, M.; et al. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011, 117, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, J.; Klein-Hitpass, L.; Carpinteiro, A.; Führer, A.; Sellmann, L.; Stilgenbauer, S.; Dührsen, U.; Dürig, J. Bone marrow fibroblasts induce expression of PI3K/NF-κB pathway genes and a pro-angiogenic phenotype in CLL cells. Leuk. Res. 2008, 32, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Chen, Z.; Li, Z.-Y.; Yeh, N.T.; Bancroft, C.C.; Van Waes, C. Hepatocyte Growth Factor/Scatter Factor-induced Activation of MEK and PI3K Signal Pathways Contributes to Expression of Proangiogenic Cytokines Interleukin-8 and Vascular Endothelial Growth Factor in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2001, 61, 5911–5918. [Google Scholar] [PubMed]
- Yang, L.; Wang, L.; Lin, H.-K.; Kan, P.-Y.; Xie, S.; Tsai, M.-Y.; Wang, P.-H.; Chen, Y.-T.; Chang, C. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem. Biophys. Res. Commun. 2003, 305, 462–469. [Google Scholar] [CrossRef]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal. 2010. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.; Sottero, T.; Yang, X.; Mueller, S.; James, C.D.; Weiss, W.A.; Polley, M.-Y.; Ozawa, T.; Berger, M.S.; Aftab, D.T.; et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-Oncology 2011, 13, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Edgar, K.A.; Guan, J.; Berry, M.; Prior, W.W.; Lee, L.; Lesnick, J.D.; Lewis, C.; Nonomiya, J.; Pang, J.; et al. GDC-0980 Is a Novel Class I PI3K/mTOR Kinase Inhibitor with Robust Activity in Cancer Models Driven by the PI3K Pathway. Mol. Cancer Ther. 2011, 10, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.M. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Kyle, R.A.; Larson, D.R.; Witzig, T.E.; Kumar, S.; Dispenzieri, A.; Morice, W.G.; Rajkumar, S.V. High levels of peripheral blood circulating plasma cells as a specific risk factor for progression of smoldering multiple myeloma. Leukemia 2013, 27, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Feller, N.; Schuurhuis, G.J.; van der Pol, M.A.; Westra, G.; Weijers, G.W.D.; van Stijn, A.; Huijgens, P.C.; Ossenkoppele, G.J. High percentage of CD34-positive cells in autologous AML peripheral blood stem cell products reflects inadequate in vivo purging and low chemotherapeutic toxicity in a subgroup of patients with poor clinical outcome. Leukemia 2003, 17, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The Bone Marrow Microenvironment as a Tumor Sanctuary and Contributor to Drug Resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.Y.; Rushworth, S.A.; Zaitseva, L.; Bowles, K.M.; Macewan, D.J. Attenuation of dexamethasone-induced cell death in multiple myeloma is mediated by miR-125b expression. Cell Cycle 2013, 12, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.M.; Rushworth, S.A.; Murray, M.Y.; Bowles, K.M.; MacEwan, D.J. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget 2013, 4, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Zlei, M.; Egert, S.; Wider, D.; Ihorst, G.; Wäsch, R.; Engelhardt, M. Characterization of in vitro growth of multiple myeloma cells. Exp. Hematol. 2007, 35, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Barrett, A.J.; Dutra, A.; Pak, E.; Miner, S.; Keyvanfar, K.; Hensel, N.F.; Rezvani, K.; Muranski, P.; Liu, P.; et al. Long term maintenance of myeloid leukemic stem cells cultured with unrelated human mesenchymal stromal cells. Stem Cell Res. 2015, 14, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Eckert, C.; Biondi, A.; Seeger, K.; Cazzaniga, G.; Hartmann, R.; Beyermann, B.; Pogodda, M.; Proba, J.; Henze, G. Prognostic value of minimal residual disease in relapsed childhood acute lymphoblastic leukaemia. Lancet 2001, 358, 1239–1241. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Martı́nez, A.; Macedo, A.; Vidriales, M.B.; López-Berges, C.; González, M.; Caballero, D.; Garcı́a-Marcos, M.A.; Ramos, F.; Fernández-Calvo, J.; et al. Immunophenotyping Investigation of Minimal Residual Disease Is a Useful Approach for Predicting Relapse in Acute Myeloid Leukemia Patients. Blood 1997, 90, 2465–2470. [Google Scholar] [PubMed]
- Cavo, M.; Terragna, C.; Martinelli, G.; Ronconi, S.; Zamagni, E.; Tosi, P.; Lemoli, R.M.; Benni, M.; Pagliani, G.; Bandini, G.; et al. Molecular monitoring of minimal residual disease in patients in long-term complete remission after allogeneic stem cell transplantation for multiple myeloma. Blood 2000, 96, 355–357. [Google Scholar] [PubMed]
- Murray, M.Y.; Zaitseva, L.; Auger, M.J.; Craig, J.I.; MacEwan, D.J.; Rushworth, S.A.; Bowles, K.M. Ibrutinib inhibits BTK-driven NF-kappaB p65 activity to overcome bortezomib-resistance in multiple myeloma. Cell Cycle 2015, 14, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.-P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Koyasu, S. The role of PI3K in immune cells. Nat. Immunol. 2003, 4, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-Y.; Wei, S.-J.; Lin, Y.-C.; Lung, J.-C.; Chang, T.-C.; Whang-Peng, J.; Liu, J.M.; Yang, D.-M.; Yang, W.K.; Shen, C.-Y. PIK3CA as an oncogene in cervical cancer. Oncogene 2000, 19, 2739–2744. [Google Scholar] [CrossRef] [PubMed]
- Bachman, K.E.; PArgani; Samuels, Y.; Silliman, N.; Ptak, J.; Szabo, S.; Konishi, H.; Karakas, B.; Blair, B.G.; Lin, C.; et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 2004, 3, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Xing, M.; Mambo, E.; Huang, X.; Liu, J.; Guo, Z.; Chatterjee, A.; Goldenberg, D.; Gollin, S.M.; Sukumar, S. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005, 7, R609–R616. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A.; Bogomolniy, F.; Yee, C.J.; Lash, A.; Barakat, R.R.; Borgen, P.I.; Boyd, J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin. Cancer Res. 2005, 11, 2875–2878. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, R.; Gonzalez-Paz, N.; Kyle, R.; Santana-Davila, R.; Price-Troska, T.; van Wier, S.; Chng, W.; Ketterling, R.P.; Gertz, M.; Henderson, K. Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008, 22, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B.; et al. Measurement of PIP3 Levels Reveals an Unexpected Role for p110β in Early Adaptive Responses to p110α-Specific Inhibitors in Luminal Breast Cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.E.; Dopheide, S.M.; Yuan, Y.; et al. PI 3-kinase p110beta: A new target for antithrombotic therapy3-kinase p110beta: A new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.H.; Okkenhaug, K.; Vanhaesebroeck, B. The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110γ. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Youn, H.; Tang, J.; Tawfik, O.; Dennis, K.; Terranova, P.F.; Du, J.; Raynal, P.; Thrasher, J.B.; Li, B. Phosphoinositide 3-OH kinase p85α and p110β are essential for androgen receptor transactivation and tumor progression in prostate cancers. Oncogene 2008, 27, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Wiederschain, D.; Maira, S.-M.; Loo, A.; Miller, C.; deBeaumont, R.; Stegmeier, F.; Yao, Y.-M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. USA 2008, 105, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Stühmer, T.; Schmiedl, N.; Wetzker, R.; Mottok, A.; Rosenwald, A.; Langer, C.; Zovko, J.; Chatterjee, M.; Einsele, H. PI3K-dependent multiple myeloma cell survival is mediated by the PIK3CA isoform. Br. J. Haematol. 2014, 166, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Doepfner, K.T.; Spertini, O.; Arcaro, A. Autocrine insulin-like growth factor-I signaling promotes growth and survival of human acute myeloid leukemia cells via the phosphoinositide 3-kinase//Akt pathway. Leukemia 2007, 21, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T Cell Antigen Receptor Signaling in p110δ PI 3-Kinase Mutant Mice. Science 2002, 297, 1031–1034. [Google Scholar] [PubMed]
- Lucas, C.L.; Kuehn, H.S.; Zhao, F.; Niemela, J.E.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110[delta] result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Sujobert, P.; Bardet, V.; Cornillet-Lefebvre, P.; Hayflick, J.S.; Prie, N.; Verdier, F.; Vanhaesebroeck, B.; Muller, O.; Pesce, F.; Ifrah, N.; et al. Essential role for the p110δ isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005, 106, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Piddock, R.E.; Loughran, N.; Marlein, C.R.; Robinson, S.D.; Edwards, D.R.; Yu, S.; Pillinger, G.E.; Zhou, Z.; Zaitseva, L.; Auger, M.J.; et al. PI3Kdelta and PI3Kgamma isoforms have distinct functions in regulating pro-tumoural signalling in the multiple myeloma microenvironment. Blood Cancer J. 2017, 7, e539. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central Role for G Protein-Coupled Phosphoinositide 3-Kinase γ in Inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Belloni, E.; Veronesi, G.; Rotta, L.; Volorio, S.; Sardella, D.; Bernard, L.; Pece, S.; di Fiore, P.P.; Fumagalli, C.; Barberis, M.; et al. Whole exome sequencing identifies driver mutations in asymptomatic computed tomography-detected lung cancers with normal karyotype. Cancer Genet. 2015, 208, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Gili, E.; Fruciano, M.; Korfei, M.; Fagone, E.; Iemmolo, M.; Furno, D.L.; Giuffrida, R.; Crimi, N.; Guenther, A.; et al. PI3K p110[gamma] overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: In vitro effects of its inhibition. Lab. Investig. 2013, 93, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J. Clin. Investig. 2007, 117, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.A.; Olsen, G.S.; Tallquist, M.D. Disruption of PDGFRα-initiated PI3K activation and migration of somite derivatives leads to spina bifida. Development 2008, 135, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Maulik, G.; Madhiwala, P.; Brooks, S.; Ma, P.; Kijima, T.; Tibaldi, E.; Schaefer, E.; Parmar, K.; Salgia, R. Activated c-Met signals through PI3K with dramatic effects on cytoskeletal functions in small cell lung cancer. J. Cell. Mol. Med. 2002, 6, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Torres-Arzayus, M.I.; de Mora, J.F.; Yuan, J.; Vazquez, F.; Bronson, R.; Rue, M.; Sellers, W.R.; Brown, M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 2004, 6, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Rottapel, R.; Turck, C.W.; Casteran, N.; Liu, X.; Birnbaum, D.; Pawson, T.; Dubreuil, P. Substrate specificities and identification of a putative binding site for PI3K in the carboxy tail of the murine Flt3 receptor tyrosine kinase. Oncogene 1994, 9, 1755–1765. [Google Scholar] [PubMed]
- Abu-Duhier, F.; Goodeve, A.; Wilson, G.; Gari, M.; Peake, I.; Rees, D.; Vandenberghe, E.; Winship, P.; Reilly, J. FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br. J. Haematol. 2000, 111, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Ocana, A.; Vera-Badillo, F.; Al-Mubarak, M.; Templeton, A.J.; Corrales-Sanchez, V.; Diez-Gonzalez, L.; Cuenca-Lopez, M.D.; Seruga, B.; Pandiella, A.; Amir, E. Activation of the PI3K/mTOR/AKT Pathway and Survival in Solid Tumors: Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e95219. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Xu, Q.; Liu, B.; Zhang, G.; Chen, L.; Pan, H. The expression of the PI3K/AKT/mTOR pathway in gastric cancer and its role in gastric cancer prognosis. Onco Targets Ther. 2015, 8, 2427–2433. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.D.; Dexter, T.M. The essential cells of the hemopoietic microenvironment. Exp. Hematol. 1984, 12, 517–521. [Google Scholar] [PubMed]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic Cells Create Bone Marrow Niches That Disrupt the Behavior of Normal Hematopoietic Progenitor Cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gaillard, J.P.; Robillard, N.; Lu, Z.Y.; Gu, Z.J.; Jourdan, M.; Boiron, J.; Bataille, R.; Klein, B. Reproducible obtaining of human myeloma cell lines as a model for tumor stem cell study in human multiple myeloma. Blood 1994, 83, 3654–3663. [Google Scholar] [PubMed]
- Nachbaur, D.M.; Herold, M.; Maneschg, A.; Huber, H. Serum levels of interleukin-6 in multiple myeloma and other hematological disorders: Correlation with disease activity and other prognostic parameters. Ann. Hematol. 1991, 62, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Schuringa, J.J.; Wierenga, A.T.; Kruijer, W.; Vellenga, E. Constitutive Stat3, Tyr705, and Ser727 phosphorylation in acute myeloid leukemia cells caused by the autocrine secretion of interleukin-6. Blood 2000, 95, 3765–3770. [Google Scholar] [PubMed]
- Lee, H.J.; Daver, N.; Kantarjian, H.M.; Verstovsek, S.; Ravandi, F. The Role of JAK Pathway Dysregulation in the Pathogenesis and Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2013, 19, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Trikha, M.; Corringham, R.; Klein, B.; Rossi, J.-F. Targeted Anti-Interleukin-6 Monoclonal Antibody Therapy for Cancer. A Review of the Rationale and Clinical Evidence. Clin. Cancer Res. 2003, 9, 4653–4665. [Google Scholar] [PubMed]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.M.; Perentesis, J.P.; Gamis, A.S.; Alonzo, T.A.; Gerbing, R.B.; Ballard, J.; Adlard, K.; Howard, D.S.; Smith, F.O.; Jenkins, G.; et al. A Phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Kortlepel, K.; Bendall, L.J.; Gottlieb, D.J. Human acute myeloid leukaemia cells express adhesion proteins and bind to bone marrow fibroblast monolayers and extracellular matrix proteins. Leukemia 1993, 7, 1174–1179. [Google Scholar] [PubMed]
- Landowski, T.H.; Olashaw, N.E.; Agrawal, D.; Dalton, W.S. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-κB (RelB/p50) in myeloma cells. Oncogene 2003, 22, 2417–2421. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.A.; Dalton, W.S. Mechanisms Associated with cell Adhesion Mediated Drug Resistance (CAM-DR) in Hematopoietic Malignancies. Cancer Metastasis Rev. 2001, 20, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Corre, J.; Mahtouk, K.; Attal, M.; Gadelorge, M.; Huynh, A.; Fleury-Cappellesso, S.; Danho, C.; Laharrague, P.; Klein, B.; Rème, T. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia 2007, 21, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, A.M.; Shafat, M.S.; Mehta, T.K.; di Palma, F.; Lawes, M.J.; Rushworth, S.A.; Bowles, K.M. MIF-Induced Stromal PKCβ/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Alsayed, Y.; Ngo, H.; Runnels, J.; Leleu, X.; Singha, U.K.; Pitsillides, C.M.; Spencer, J.A.; Kimlinger, T.; Ghobrial, J.M.; Jia, X.; et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood 2007, 109, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, E.J.C.; Pavic, B.; Löwenberg, B.; Ploemacher, R.E. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood 2004, 104, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, J.J.; Clay, D.; Dupuy, C.; Rigal, S.; Jasmin, C.; Bourin, P.; Le Bousse-Kerdiles, M.C. Chemokine SDF-1 enhances circulating CD34(+) cell proliferation in synergy with cytokines: Possible role in progenitor survival. Blood 2000, 95, 756–768. [Google Scholar] [PubMed]
- Nefedova, Y.; Landowski, T.H.; Dalton, W.S. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia 2003, 17, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009, 113, 6206–6214. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, M.; Steimberg, N.; Ponzoni, M.; Belloni, D.; Berenzi, A.; Girlanda, S.; Caligaris-Cappio, F.; Mazzoleni, G.; Ferrero, E. Ex Vivo dynamic 3-D culture of human tissues in the RCCS™ bioreactor allows the study of Multiple Myeloma biology and response to therapy. PLoS ONE 2013, 8, e71613. [Google Scholar] [CrossRef]
- Antonelli, A.; Noort, W.A.; Jaques, J.; de Boer, B.; de Jong-Korlaar, R.; Brouwers-Vos, A.Z.; Lubbers-Aalders, L.; van Velzen, J.F.; Bloem, A.C.; Yuan, H.; et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffold-based niches. Blood 2016, 128, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [PubMed]
- Bendre, M.S.; Margulies, A.G.; Walser, B.; Akel, N.S.; Bhattacharrya, S.; Skinner, R.A.; Swain, F.; Ramani, V.; Mohammad, K.S.; Wessner, L.L.; et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res. 2005, 65, 11001–11009. [Google Scholar] [CrossRef] [PubMed]
- Cacalano, G.; Lee, J.; Kikly, K.; Ryan, A.; Pitts-Meek, S.; Hultgren, B.; Wood, W.; Moore, M. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 1994, 265, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.J.; Bird, G.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.; Eagles, J.R.; Le, P.N.; et al. XactMice: Humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016, 35, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Belnoue, E.; Pihlgren, M.; McGaha, T.L.; Tougne, C.; Rochat, A.-F.; Bossen, C.; Schneider, P.; Huard, B.; Lambert, P.-H.; Siegrist, C.-A. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood 2008, 111, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Roosnek, E.; Burjanadze, M.; Dietrich, P.Y.; Matthes, T.; Passweg, J.; Huard, B. Tumors that look for their springtime in APRIL. Crit. Rev. Oncol./Hematol. 2009, 72, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of Neuronal Survival by the Serine-Threonine Protein Kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Sprynski, A.C.; Hose, D.; Caillot, L.; Reme, T.; Shaughnessy, J.D., Jr.; Barlogie, B.; Seckinger, A.; Moreaux, J.; Hundemer, M.; Jourdan, M.; et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood 2009, 113, 4614–4626. [Google Scholar] [CrossRef] [PubMed]
- Glassford, J.; Rabin, N.; Lam, E.W.; Yong, K.L. Functional regulation of D-type cyclins by insulin-like growth factor-I and serum in multiple myeloma cells. Br. J. Haematol. 2007, 139, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, N.; Tamburini, J.; Cornillet-Lefebvre, P.; Gillot, L.; Bardet, V.; Willems, L.; Park, S.; Green, A.S.; Ifrah, N.; Dreyfus, F.; et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: Therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 2010, 95, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Piddock, R.E.; Abdul-Aziz, A.M.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. Macrophage Migration Inhibitory Factor Drives Multiple Myeloma IL-6/8 Pro-Survival Signals in the Tumor Microenvironment. Blood 2015, 126, 2988–2988. [Google Scholar]
- Fan, F.; Vallet, S.; Sattler, M.; Tonon, G.; Bashari, M.H.; Bakiri, L.; Goldschmidt, H.; Wagner, E.F.; Jaeger, D.; Podar, K. The AP-1 Transcription Factor JunB Promotes Multiple Myeloma (MM) Cell Proliferation, Survival and Drug Resistance in the Bone Marrow Microenvironment. Blood 2014, 124, 3446–3446. [Google Scholar] [CrossRef]
- Hideshima, T.; Nakamura, N.; Chauhan, D.; Anderson, K.C. Biologic sequelae of interleukin-6 induced PI3-K/Akt signaling in multiple myeloma. Oncogene 2001, 20, 5991–6000. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Bjartell, A.; Culig, Z.; Persson, J.L. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int. J. Cancer 2008, 122, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.B.; Garcia-Gomez, A.; Garayoa, M.; Corchete, L.A.; Hernandez, J.M.; San Miguel, J.; Gutierrez, N.C. Effects of IL-8 Up-Regulation on Cell Survival and Osteoclastogenesis in Multiple Myeloma. Am. J. Pathol. 2016, 186, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Schinke, C.; Giricz, O.; Li, W.; Shastri, A.; Gordon, S.; Barreyro, L.; Bhagat, T.; Bhattacharyya, S.; Ramachandra, N.; Bartenstein, M.; et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 2015, 125, 3144–3152. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Lu, Z.; Zhang, Y.; Wang, M.; Li, W.; Hu, Z.; Wang, S.; Lin, Y. Interleukin-8 upregulates integrin β3 expression and promotes estrogen receptor-negative breast cancer cell invasion by activating the PI3K/Akt/NF-κB pathway. Cancer Lett. 2015, 364, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Politou, M.; Viniou, N.; Rahemtulla, A. Significance of macrophage inflammatory protein-1 alpha (MIP-1alpha) in multiple myeloma. Leuk. Lymphoma 2005, 46, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; CKato; Manno, M.; Ogaki, M.; Satou, T.; Itoh, T.; Kusunoki, T.; Tanimori, Y.; Fujiwara, K.; Matsuoka, H.; et al. Macrophage inflammatory protein-1α (MIP-1α) enhances a receptor activator of nuclear factor κB ligand (RANKL) expression in mouse bone marrow stromal cells and osteoblasts through MAPK and PI3K/Akt pathways. Mol. Cell. Biochem. 2007, 304, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Zaitseva, L.; Murray, M.Y.; Shafat, M.S.; Lawes, M.J.; MacEwan, D.J.; Bowles, K.M.; Rushworth, S.A. Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget 2014, 5, 9930–9938. [Google Scholar] [CrossRef] [PubMed]
- Voermans, C.; Van Heese, W.; De Jong, I.; Gerritsen, W.; van Der Schoot, C. Migratory behavior of leukemic cells from acute myeloid leukemia patients. Leukemia 2002, 16, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Barbero, S.; Bonavia, R.; Bajetto, A.; Porcile, C.; Pirani, P.; Ravetti, J.L.; Zona, G.L.; Spaziante, R.; Florio, T.; Schettini, G. Stromal Cell-derived Factor 1α Stimulates Human Glioblastoma Cell Growth through the Activation of Both Extracellular Signal-regulated Kinases 1/2 and Akt. Cancer Res. 2003, 63, 1969–1974. [Google Scholar] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Richardson, P.G.; Hideshima, T.; Munshi, N.; Treon, S.P.; Anderson, K.C. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: Therapeutic applications. Blood 2002, 99, 4079–4086. [Google Scholar] [CrossRef] [PubMed]
- Oster, W.; Cicco, N.A.; Klein, H.; Hirano, T.; Kishimoto, T.; Lindemann, A.; Mertelsmann, R.H.; Herrmann, F. Participation of the cytokines interleukin 6, tumor necrosis factor-alpha, and interleukin 1-beta secreted by acute myelogenous leukemia blasts in autocrine and paracrine leukemia growth control. J. Clin. Investig. 1989, 84, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.V.; Lee, M.J.; Islam, A.S.; Rohrer, J.L.; Goldberg, V.M.; Beidelschies, M.A.; Greenfield, E.M. Inhibition of the PI3K-Akt signaling pathway reduces tumor necrosis factor-alpha production in response to titanium particles in vitro. J. Bone Jt. Surg. Am. 2007, 89, 1019–1027. [Google Scholar]
- Gupta, D.; Treon, S.; Shima, Y.; Hideshima, T.; Podar, K.; Tai, Y.; Lin, B.; Lentzsch, S.; Davies, F.; Chauhan, D. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: Therapeutic applications. Leukemia 2001, 15, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar] [PubMed]
- Fiedler, W.; Graeven, U.; Ergün, S.; Verago, S.; Kilic, N.; Stockschläder, M.; Hossfeld, D.K. Vascular Endothelial Growth Factor, a Possible Paracrine Growth Factor in Human Acute Myeloid Leukemia. Blood 1997, 89, 1870–1875. [Google Scholar] [PubMed]
- Takahashi, M.; Matsui, A.; Inao, M.; Mochida, S.; Fujiwara, K. ERK/MAPK-dependent PI3K/Akt phosphorylation through VEGFR-1 after VEGF stimulation in activated hepatic stellate cells. Hepatol. Res. 2003, 26, 232–236. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Hess, G.; Herbrecht, R.; Romaguera, J.; Verhoef, G.; Crump, M.; Gisselbrecht, C.; Laurell, A.; Offner, F.; Strahs, A.; Berkenblit, A.; et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2009, 27, 3822–3829. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Mothe-Satney, I.; Yang, D.; Fadden, P.; Haystead, T.A.J.; Lawrence, J.C. Multiple Mechanisms Control Phosphorylation of PHAS-I in Five (S/T)P Sites That Govern Translational Repression. Mol. Cell. Biol. 2000, 20, 3558–3567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Schmid, T.; Frank, R.; Brüne, B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1α from pVHL-independent degradation. J. Biol. Chem. 2004, 279, 13506–13513. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.D.; McCready, J.; Jay, D.G. Extracellular heat shock protein (Hsp) 70 and Hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE 2011, 6, e18848. [Google Scholar] [CrossRef] [PubMed]
- Cornford, P.A.; Dodson, A.R.; Parsons, K.F.; Desmond, A.D.; Woolfenden, A.; Fordham, M.; Neoptolemos, J.P.; Ke, Y.; Foster, C.S. Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res. 2000, 60, 7099–7105. [Google Scholar] [PubMed]
- Kimura, E.; Enns, R.E.; Alcaraz, J.E.; Arboleda, J.; Slamon, D.J.; Howell, S.B. Correlation of the survival of ovarian cancer patients with mRNA expression of the 60-kD heat-shock protein HSP-60. J. Clin. Oncol. 1993, 11, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Jain, S.; Stuhmer, T.; Andrulis, M.; Ungethum, U.; Kuban, R.J.; Lorentz, H.; Bommert, K.; Topp, M.; Kramer, D.; et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 2007, 109, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Nepstad, I.; Sulen, A.; Gjertsen, B.T.; Hatfield, K.J.; Bruserud, O. Increased antileukemic effects in human acute myeloid leukemia by combining HSP70 and HSP90 inhibitors. Expert Opin. Investig. Drugs 2013, 22, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Du, Z.; Sun, L.; Foley, K.P.; Proia, D.A.; Blackman, R.K.; Zhou, D.; Inoue, T.; Tatsuta, N.; Sang, J.; et al. Ganetespib, a Unique Triazolone-Containing Hsp90 Inhibitor, Exhibits Potent Antitumor Activity and a Superior Safety Profile for Cancer Therapy. Mol. Cancer Ther. 2012, 11, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Nishiuchi, R.; Kitamura, T.; Kersey, J.H. Human leukemias with mutated FLT3 kinase are synergistically sensitive to FLT3 and Hsp90 inhibitors: The key role of the STAT5 signal transduction pathway. Leukemia 2005, 19, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Gandhi, S.; Joy, A.A.; Edwards, S.; Gorr, M.; Hopkins, S.; Kondejewski, J.; Ayoub, J.P.; Califaretti, N.; Rayson, D.; et al. Novel agents and associated toxicities of inhibitors of the pi3k/Akt/mtor pathway for the treatment of breast cancer. Curr. Oncol. 2015, 22, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Modi, P.; Newcomb, T.; Queva, C.; Gandhi, V. Idelalisib: First-in-Class PI3K Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; O’Brien, S. Initial treatment of CLL: Integrating biology and functional status. Blood 2015, 126, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Li, L.; Sari, D.; Petkova, V.; Boussiotis, V.A. PD-1 Increases PTEN Phosphatase Activity While Decreasing PTEN Protein Stability by Inhibiting Casein Kinase 2. Mol. Cell. Biol. 2013, 33, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Banerjee, L.; Cheung, C.; Mansour, M.; Jenkinson, S.; Gale, R.; Khwaja, A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.A.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, K.; Peluso, M.; Fu, M.; Rosin, N.Y.; Burger, J.A.; Wierda, W.G.; Keating, M.J.; Faia, K.; O’Brien, S.; Kutok, J.L.; et al. The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia 2015, 29, 1811–1822. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Soluble Factor | Involvement in MM Pathogenesis | Involvement in AML Pathogenesis | Involvement in PI3K/Akt Pathway |
|---|---|---|---|
| APRIL | Survival factor for BM plasmablasts [86] | [87] | |
| BAFF | Normal plasma cell development [88] | [87,89] | |
| IGF-1 | Induces growth in all MM cell lines, promotes cell cycle progression [90,91] | [92] | |
| IL-6 | Promotes drug resistance, high levels associated with poor prognosis [93,94] | Upregulates STAT3 increasing AML proliferation and survival [63] | [95,96] |
| IL-8 | Increases osteoclastogenesis and promotes angiogenesis [97] | Increases survival, invasion and proliferation of AML cells [98] | [99] |
| MIP-1a | Increased survival and osteoclast production [100] | [101] | |
| SDF-1 | Mediates MM homing to the BM; tumour growth; drug resistance [74] | Mediates AML migration, BTK shown to be involved [102,103] | [104] |
| TNFα | Induces expression of pro-survival factors [105] | Stimulates AML blast growth via colony stimulating factor [106] | [107] |
| VEGF | Angiogenesis, promotes MM survival and attenuates apoptosis [108] | Upregulated, increases rate of angiogenesis [109,110] | [111] |
| Phase | Status | Drug Name | Target | Mono/Co Therapy | ClinicalTrials.Gov. Number |
|---|---|---|---|---|---|
| I | Completed | Idelalisib | p110δ | Mono | NCT01555281 |
| I | Recruiting | CUDC-907 | p110α + HDAC1/2/3/10 | Mono | NCT01742988 |
| I/II | Completed | Afuresertib | Akt | Co | NCT01476137 |
| I/II | Recruiting | Nelfinavir | pan PI3K | Co | NCT01555281 |
| I/II | Recruiting | ACP-319 | p110δ | Co | NCT02328014 |
| Ib/II | Completed | BYL719 | p110α | Co | NCT02144038 |
| II | Recruiting | Gedatolisib | p110α/γ + mTOR | Mono | NCT02438761 |
| III | Ongoing, not recruiting | Duvelisib | p110δ/γ | Co | NCT02004522 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piddock, R.E.; Bowles, K.M.; Rushworth, S.A. The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma. Cancers 2017, 9, 29. https://doi.org/10.3390/cancers9040029
Piddock RE, Bowles KM, Rushworth SA. The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma. Cancers. 2017; 9(4):29. https://doi.org/10.3390/cancers9040029
Chicago/Turabian StylePiddock, Rachel E., Kristian M. Bowles, and Stuart A. Rushworth. 2017. "The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma" Cancers 9, no. 4: 29. https://doi.org/10.3390/cancers9040029
APA StylePiddock, R. E., Bowles, K. M., & Rushworth, S. A. (2017). The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma. Cancers, 9(4), 29. https://doi.org/10.3390/cancers9040029