
Pancreatic Cancer Chemoresistance to Gemcitabine



Abstract
1. Introduction
2. Chemoresistance in Pancreatic Cancer
2.1. Desmoplastic Stroma in Chemoresistance
2.2. Stromal Barrier or Drug Metabolism?
3. Gemcitabine Pharmacology
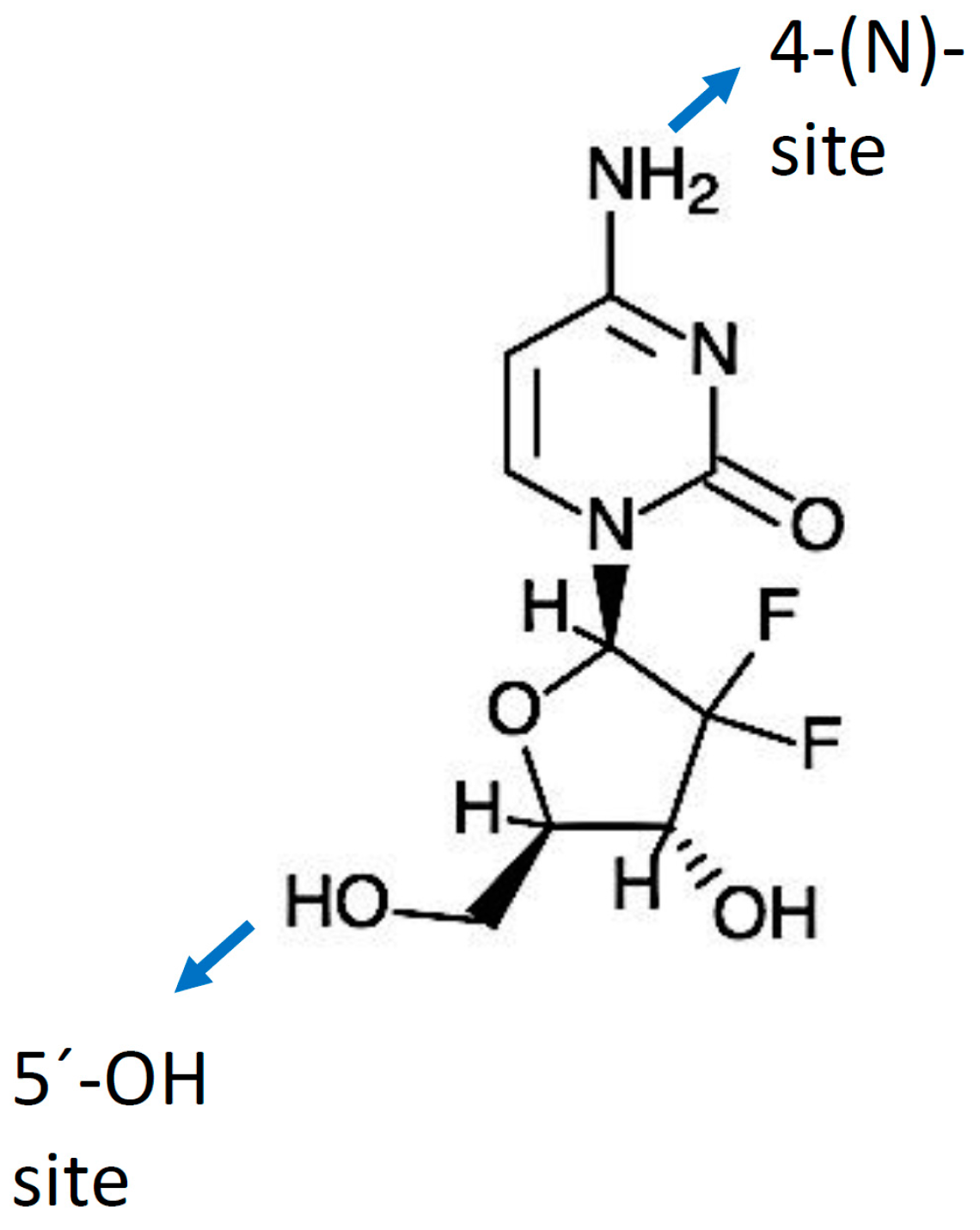
3.1. Chemical Structure and Properties
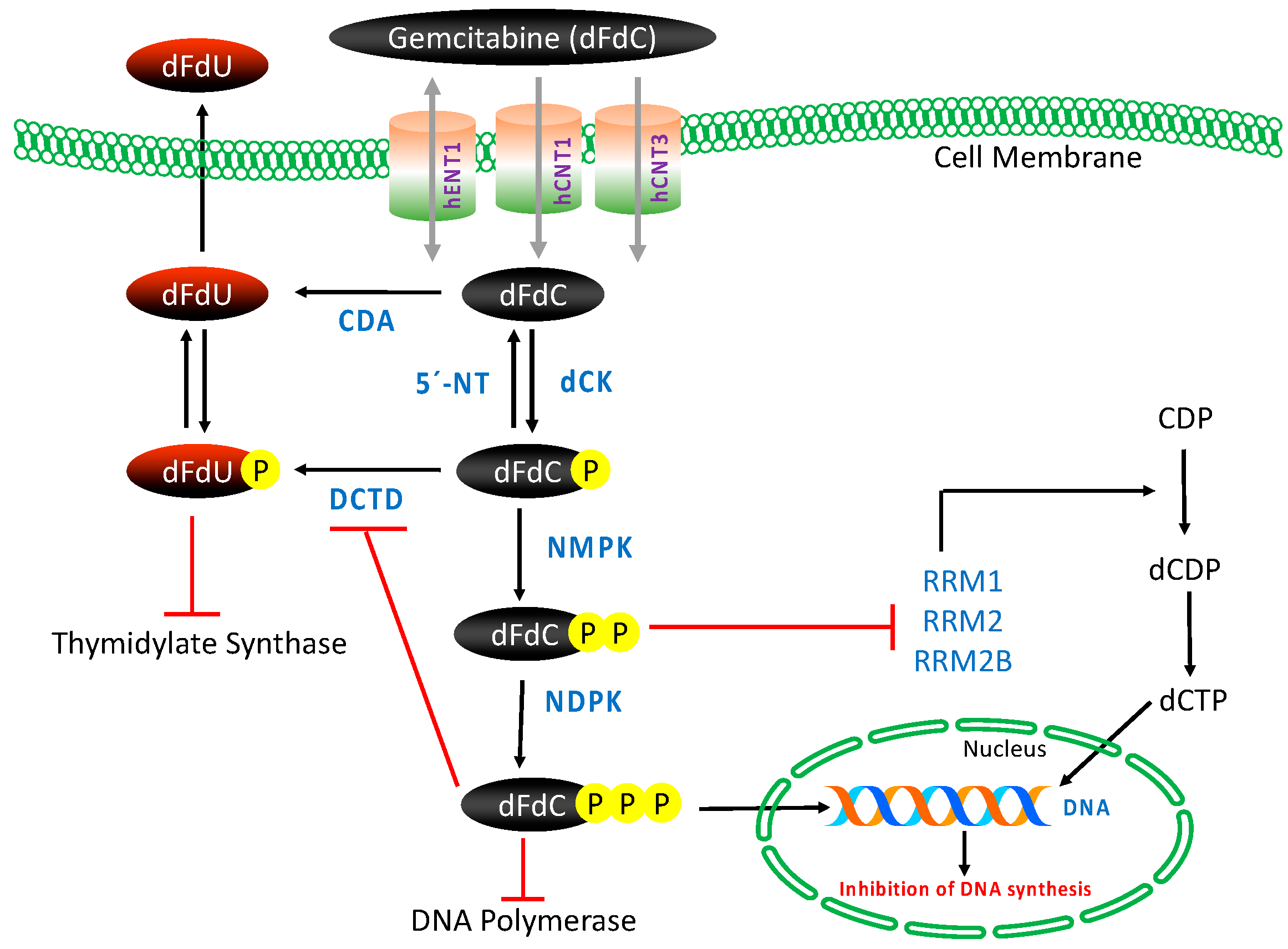
3.2. Transport, Metabolism and Mechanism of Action
4. Gemcitabine Metabolism-Associated Entities in Chemoresistance
4.1. Nucleoside Transporters
4.2. Deoxycytidine Kinase
4.3. Cytidine Deaminase
4.4. 5′-Nucletidase
4.5. Ribonucleotide Reductase
4.6. Thymidylate Synthase
5. Potential Ways to Improve Gemcitabine Delivery and Efficacy
5.1. Prodrug Approach
5.1.1. Modifications at the 5′-OH Position
5.1.2. Other Modifications
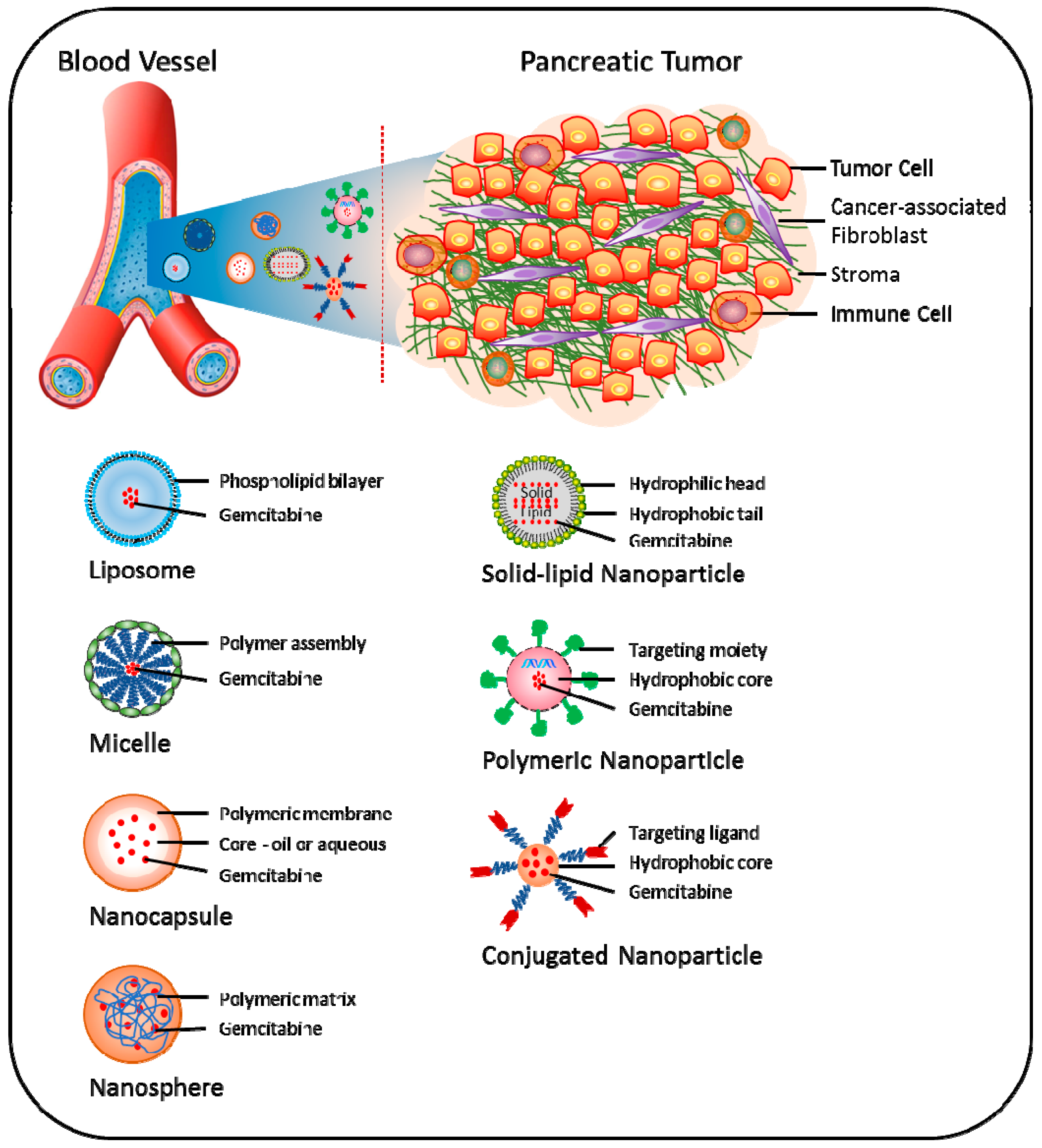
5.2. Nano-Carrier Approach
6. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 5′-NT | 5′-nucleosidase |
| CAFs | cancer-associated fibroblasts |
| CDA | cytidine deaminase |
| CNT | concentrative nucleoside transporter |
| dCK | deoxycytidine kinase |
| DCTD | deoxycytidylate deaminase |
| dCTP | deoxycytidine triphosphate |
| dFdC | 2′,2′-difluorodeoxycytidine |
| dFdU | 2′,2′- difluorodeoxyuridine |
| DFS | disease-free survival |
| ECM | extracellular matrix |
| ENT | equilibrative nucleoside transporter |
| NT | nucleoside transporters |
| NP | nanoparticle |
| OS | overall survival |
| PDAC | pancreatic ductal adenocarcinoma |
| PEG | polyethylene glycol |
| PSCs | pancreatic stellate cells |
| RR | ribonucleotide reductase |
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society Key Statistics for Pancreatic Cancer. Available online: https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html (accessed on 1 October 2017).
- Seer Cancer Stat Facts: Pancreas Cancer. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 1 October 2017).
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Zijlstra, M.; Bernards, N.; de Hingh, I.H.; van de Wouw, A.J.; Goey, S.H.; Jacobs, E.M.; Lemmens, V.E.; Creemers, G.J. Does long-term survival exist in pancreatic adenocarcinoma? Acta. Oncol. 2016, 55, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, L.E.; Connor, A.A.; Gallinger, S. Molecular events in the natural history of pancreatic cancer. Trends Cancer 2017, 3, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Ellenrieder, V.; König, A.; Seufferlein, T. Current standard and future perspectives in first- and second-line treatment of metastatic pancreatic adenocarcinoma. Digestion 2016, 94, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Labori, K.J.; Katz, M.H.; Tzeng, C.W.; Bjornbeth, B.A.; Cvancarova, M.; Edwin, B.; Kure, E.H.; Eide, T.J.; Dueland, S.; Buanes, T.; et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma—A population-based cohort study. Acta Oncol. 2016, 55, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, V.; Borella, S.; Calderazzo, F.; Ferraro, P.; Chieco Bianchi, L.; Reichard, P. Inhibition of ribonucleotide reductase by 2′-substituted deoxycytidine analogs: Possible application in aids treatment. Proc. Natl. Acad. Sci. USA 1994, 91, 8403–8407. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Soo, R.A.; Yong, W.P.; Innocenti, F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab. Rev. 2009, 41, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [PubMed]
- Kadaba, R.; Birke, H.; Wang, J.; Hooper, S.; Andl, C.D.; Di Maggio, F.; Soylu, E.; Ghallab, M.; Bor, D.; Froeling, F.E.; et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J. Pathol. 2013, 230, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Li, L.; Li, Z.; Xie, K. Targeted destruction of the orchestration of the pancreatic stroma and tumor cells in pancreatic cancer cases: Molecular basis for therapeutic implications. Cytokine Growth Factor Rev. 2012, 23, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.; Sun, W. Novel approaches in the management of pancreatic ductal adenocarcinoma: Potential promises for the future. J. Hematol. Oncol. 2015, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Dimou, A.; Syrigos, K.N.; Saif, M.W. Overcoming the stromal barrier: Technologies to optimize drug delivery in pancreatic cancer. Ther. Adv. Med. Oncol. 2012, 4, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; DelGiorno, K.E.; Greenberg, P.D.; Hingorani, S.R. Stromal reengineering to treat pancreas cancer. Carcinogenesis 2014, 35, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Samuel, S.; Lopez-Casas, P.; Grizzle, W.; Hidalgo, M.; Kovar, J.; Oelschlager, D.; Zinn, K.; Warram, J.; Buchsbaum, D. Sparc-independent delivery of nab-paclitaxel without depleting tumor stroma in patient-derived pancreatic cancer xenografts. Mol. Cancer Ther. 2016, 15, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Plaza, C.; Musteanu, M.; Illei, P.; Brachmann, C.B.; Heise, C.; Pierce, D.; Lopez-Casas, P.P.; Menendez, C.; Tabernero, J.; et al. Sparc expression did not predict efficacy of nab-paclitaxel plus gemcitabine or gemcitabine alone for metastatic pancreatic cancer in an exploratory analysis of the phase III MPACT trial. Clin. Cancer Res. 2015, 21, 4811–4818. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, R.; Bachet, J.B.; Mackey, J.R.; Dalban, C.; Demetter, P.; Graham, K.; Couvelard, A.; Svrcek, M.; Bardier-Dupas, A.; Hammel, P.; et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology 2012, 143, 664–674. [Google Scholar]
- Greenhalf, W.; Ghaneh, P.; Neoptolemos, J.P.; Palmer, D.H.; Cox, T.F.; Lamb, R.F.; Garner, E.; Campbell, F.; Mackey, J.R.; Costello, E.; et al. Pancreatic cancer hENT1 expression and survival from gemcitabine in patients from the ESPAC-3 trial. J. Natl. Cancer Inst. 2014, 106, djt347. [Google Scholar] [CrossRef] [PubMed]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Gnanamony, M.; Gondi, C.S. Chemoresistance in pancreatic cancer: Emerging concepts. Oncol. Lett. 2017, 13, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Binenbaum, Y.; Na′ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Rueff, J.; Rodrigues, A.S. Cancer drug resistance: A brief overview from a genetic viewpoint. Methods Mol. Biol. 2016, 1395, 1–18. [Google Scholar] [PubMed]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Tjomsland, V.; Pomianowska, E.; Aasrum, M.; Sandnes, D.; Verbeke, C.S.; Gladhaug, I.P. Profile of mmp and timp expression in human pancreatic stellate cells: Regulation by 1L-1 α and TGF α and implications for migration of pancreatic cancer cells. Neoplasia 2016, 18, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M. Understanding the stroma of pancreatic cancer: Co-evolution of the microenvironment with epithelial carcinogenesis. J. Pathol. 2013, 231, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology 2016, 151, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1α signaling mediates the effects of PSCS on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097. [Google Scholar] [CrossRef] [PubMed]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin d receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Schmitt, T.M.; Hulbert, A.; Brockenbrough, J.S.; Nguyen, H.; Cuevas, C.; Dotson, A.M.; Tan, X.; Hotes, J.L.; Greenberg, P.D.; et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 2015, 28, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mab FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar] [PubMed]
- Gandhi, V.; Plunkett, W. Modulatory activity of 2′,2′-difluorodeoxycytidine on the phosphorylation and cytotoxicity of arabinosyl nucleosides. Cancer Res. 1990, 50, 3675–3680. [Google Scholar] [PubMed]
- Hertel, L.W.; Boder, G.B.; Kroin, J.S.; Rinzel, S.M.; Poore, G.A.; Todd, G.C.; Grindey, G.B. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine). Cancer Res 1990, 50, 4417–4422. [Google Scholar] [PubMed]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. 5), v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, A.K.; Marsh, S.; Murry, D.J.; Hurley, T.D.; McLeod, H.L. Identification and analysis of single-nucleotide polymorphisms in the gemcitabine pharmacologic pathway. Pharmacogenomics J 2004, 4, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar] [PubMed]
- De Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Ohuchida, K.; Mizumoto, K.; Itaba, S.; Ito, T.; Nakata, K.; Yu, J.; Kayashima, T.; Souzaki, R.; Tajiri, T.; et al. Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine-based adjuvant chemotherapy. Neoplasia 2010, 12, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Shipley, L.A.; Brown, T.J.; Cornpropst, J.D.; Hamilton, M.; Daniels, W.D.; Culp, H.W. Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab. Dispos. 1992, 20, 849–855. [Google Scholar] [PubMed]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: A mechanism of self-potentiation. Cancer Res. 1992, 52, 533–539. [Google Scholar] [PubMed]
- Moysan, E.; Bastiat, G.; Benoit, J.P. Gemcitabine versus modified gemcitabine: A review of several promising chemical modifications. Mol. Pharm. 2013, 10, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol. Pharmacol. 1990, 38, 567–572. [Google Scholar] [PubMed]
- Spratlin, J.; Sangha, R.; Glubrecht, D.; Dabbagh, L.; Young, J.D.; Dumontet, C.; Cass, C.; Lai, R.; Mackey, J.R. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin. Cancer Res. 2004, 10, 6956–6961. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Hung, S.W.; Patel, B.; Lovin, D.; Govindarajan, R. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Res. 2011, 71, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Saiki, Y.; Yoshino, Y.; Fujimura, H.; Manabe, T.; Kudo, Y.; Shimada, M.; Mano, N.; Nakano, T.; Lee, Y.; Shimizu, S.; et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, W.; Fu, M.; Yang, A.; Huang, H.; Xie, J. Establishment of human pancreatic cancer gemcitabine-resistant cell line with ribonucleotide reductase overexpression. Oncol. Rep. 2015, 33, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.G.; Farrell, M.P.; Schmitz, J.C. Thymidylate synthase: A critical target for cancer chemotherapy. Clin. Colorectal Cancer 2002, 1, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Réjiba, S.; Bigand, C.; Parmentier, C.; Hajri, A. Gemcitabine-based chemogene therapy for pancreatic cancer using AD-dCK::UMK GDEPT and TSRR siRNA strategies. Neoplasia 2009, 11, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, R.; Mackey, J.R.; Lai, R.; Demetter, P.; Peeters, M.; Polus, M.; Cass, C.E.; Young, J.; Salmon, I.; Devière, J.; et al. Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clin. Cancer Res. 2009, 15, 2913–2919. [Google Scholar]
- Nakano, Y.; Tanno, S.; Koizumi, K.; Nishikawa, T.; Nakamura, K.; Minoguchi, M.; Izawa, T.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br. J. Cancer 2007, 96, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Giovannetti, E.; Del Tacca, M.; Mey, V.; Funel, N.; Nannizzi, S.; Ricci, S.; Orlandini, C.; Boggi, U.; Campani, D.; Del Chiaro, M.; et al. Transcription analysis of human equilibrative nucleoside transporter-1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Res. 2006, 66, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Ciccolini, J.; Mercier, C.; Dahan, L.; André, N. Integrating pharmacogenetics into gemcitabine dosing--time for a change? Nat. Rev. Clin. Oncol. 2011, 8, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.J.; Elsaleh, H.; Garcia, M.; Lai, R.; Ammar, A.; Regine, W.F.; Abrams, R.; Benson, A.B.; Macdonald, J.; Cass, C.E.; et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 2009, 136, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Achiwa, H.; Oguri, T.; Sato, S.; Maeda, H.; Niimi, T.; Ueda, R. Determinants of sensitivity and resistance to gemcitabine: The roles of human equilibrative nucleoside transporter 1 and deoxycytidine kinase in non-small cell lung cancer. Cancer Sci. 2004, 95, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Aho, U.; Nilsson, B.I.; Peters, G.J.; Pastor-Anglada, M.; Rasch, W.; Sandvold, M.L. Gemcitabine chemoresistance in pancreatic cancer: Molecular mechanisms and potential solutions. Scand. J. Gastroenterol. 2009, 44, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Yao, S.Y.; Smith, K.M.; Karpinski, E.; Baldwin, S.A.; Cass, C.E.; Young, J.D. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J. Natl. Cancer Inst. 1999, 91, 1876–1881. [Google Scholar] [CrossRef] [PubMed]
- García-Manteiga, J.; Molina-Arcas, M.; Casado, F.J.; Mazo, A.; Pastor-Anglada, M. Nucleoside transporter profiles in human pancreatic cancer cells: Role of hCNT1 in 2′,2′-difluorodeoxycytidine- induced cytotoxicity. Clin. Cancer. Res. 2003, 9, 5000–5008. [Google Scholar] [PubMed]
- Chaturvedi, P.; Singh, A.P.; Moniaux, N.; Senapati, S.; Chakraborty, S.; Meza, J.L.; Batra, S.K. MUC4 mucin potentiates pancreatic tumor cell proliferation, survival, and invasive properties and interferes with its interaction to extracellular matrix proteins. Mol. Cancer Res. 2007, 5, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Bafna, S.; Kaur, S.; Momi, N.; Batra, S.K. Pancreatic cancer cells resistance to gemcitabine: The role of MUC4 mucin. Br. J. Cancer 2009, 101, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Duchêne, B.; Hebbar, M.; Leteurtre, E.; van Seuningen, I.; Jonckheere, N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the concentrative nucleoside transporter family. Oncogene 2013, 32, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, N.; Skrypek, N.; Merlin, J.; Dessein, A.F.; Dumont, P.; Leteurtre, E.; Harris, A.; Desseyn, J.L.; Susini, C.; Frénois, F.; et al. The mucin MUC4 and its membrane partner ERBB2 regulate biological properties of human capan-2 pancreatic cancer cells via different signalling pathways. PLoS ONE 2012, 7, e32232. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Vasseur, R.; Vincent, A.; Duchêne, B.; Van Seuningen, I.; Jonckheere, N. The oncogenic receptor ERBB2 modulates gemcitabine and irinotecan/SN-38 chemoresistance of human pancreatic cancer cells via hCNT1 transporter and multidrug-resistance associated protein MRP-2. Oncotarget 2015, 6, 10853–10867. [Google Scholar] [CrossRef] [PubMed]
- Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernanke, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. TGF-β-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis 2016, 37, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Koay, E.J.; Baio, F.E.; Ondari, A.; Truty, M.J.; Cristini, V.; Thomas, R.M.; Chen, R.; Chatterjee, D.; Kang, Y.; Zhang, J.; et al. Intra-tumoral heterogeneity of gemcitabine delivery and mass transport in human pancreatic cancer. Phys. Biol. 2014, 11, 065002. [Google Scholar] [CrossRef] [PubMed]
- Koay, E.J.; Truty, M.J.; Cristini, V.; Thomas, R.M.; Chen, R.; Chatterjee, D.; Kang, Y.; Bhosale, P.R.; Tamm, E.P.; Crane, C.H.; et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J. Clin. Investig. 2014, 124, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Ohhashi, S.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Egami, T.; Yu, J.; Toma, H.; Sadatomi, S.; Nagai, E.; Tanaka, M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008, 28, 2205–2212. [Google Scholar] [PubMed]
- Kroep, J.R.; Loves, W.J.; van der Wilt, C.L.; Alvarez, E.; Talianidis, I.; Boven, E.; Braakhuis, B.J.; van Groeningen, C.J.; Pinedo, H.M.; Peters, G.J. Pretreatment deoxycytidine kinase levels predict in vivo gemcitabine sensitivity. Mol. Cancer Ther. 2002, 1, 371–376. [Google Scholar] [PubMed]
- Funamizu, N.; Okamoto, A.; Kamata, Y.; Misawa, T.; Uwagawa, T.; Gocho, T.; Yanaga, K.; Manome, Y. Is the resistance of gemcitabine for pancreatic cancer settled only by overexpression of deoxycytidine kinase? Oncol. Rep. 2010, 23, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, V.; Ricci, F.; Rubio-Viqueira, B.; Rubio-Viquiera, B.; Kulesza, P.; Yeo, C.J.; Hidalgo, M.; Klein, A.; Laheru, D.; Iacobuzio-Donahue, C.A. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: Relationship to molecular mechanisms of gemcitabine resistance and survival. Clin. Cancer Res. 2006, 12, 2492–2497. [Google Scholar] [CrossRef] [PubMed]
- Costantino, C.L.; Witkiewicz, A.K.; Kuwano, Y.; Cozzitorto, J.A.; Kennedy, E.P.; Dasgupta, A.; Keen, J.C.; Yeo, C.J.; Gorospe, M.; Brody, J.R. The role of hur in gemcitabine efficacy in pancreatic cancer: HuR UP-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009, 69, 4567–4572. [Google Scholar] [CrossRef] [PubMed]
- Richards, N.G.; Rittenhouse, D.W.; Freydin, B.; Cozzitorto, J.A.; Grenda, D.; Rui, H.; Gonye, G.; Kennedy, E.P.; Yeo, C.J.; Brody, J.R.; et al. HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients. Ann. Surg. 2010, 252, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, M.; Blanco, F.F.; Huang, Y.H.; Telonis, A.G.; Screnci, B.A.; Cosma, G.L.; Alexeev, V.; Gonye, G.E.; Yeo, C.J.; Sawicki, J.A.; et al. Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells. Oncotarget 2015, 6, 27312–27331. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.K.; Costantino, C.L.; Bildzukewicz, N.A.; Richards, N.G.; Rittenhouse, D.W.; Einstein, L.; Cozzitorto, J.A.; Keen, J.C.; Dasgupta, A.; Gorospe, M.; et al. Pp32 (anp32a) expression inhibits pancreatic cancer cell growth and induces gemcitabine resistance by disrupting hur binding to mRNAs. PLoS ONE 2010, 5, e15455. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, J.L.; Grunewald, R.; Weeks, E.A.; Gravel, D.; Adams, T.; Nowak, B.; Mineishi, S.; Tarassoff, P.; Satterlee, W.; Raber, M.N. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Eda, H.; Ura, M.; F-Ouchi, K.; Tanaka, Y.; Miwa, M.; Ishitsuka, H. The antiproliferative activity of DMDC is modulated by inhibition of cytidine deaminase. Cancer Res. 1998, 58, 1165–1169. [Google Scholar] [PubMed]
- Dumontet, C.; Bauchu, E.C.; Fabianowska, K.; Lepoivre, M.; Wyczechowska, D.; Bodin, F.; Rolland, M.O. Common resistance mechanisms to nucleoside analogues in variants of the human erythroleukemic line K562. Adv. Exp. Med. Biol. 1999, 457, 571–577. [Google Scholar] [PubMed]
- Hunsucker, S.A.; Spychala, J.; Mitchell, B.S. Human cytosolic 5′-Nucleotidase I characterization and role in nucleoside analog resistance. J. Biol. Chem. 2001, 276, 10498–10504. [Google Scholar] [CrossRef] [PubMed]
- Sève, P.; Mackey, J.R.; Isaac, S.; Trédan, O.; Souquet, P.J.; Pérol, M.; Cass, C.; Dumontet, C. Cn-II expression predicts survival in patients receiving gemcitabine for advanced non-small cell lung cancer. Lung Cancer 2005, 49, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, S.; Nakamori, S.; Tsujie, M.; Takahashi, Y.; Okami, J.; Yoshioka, S.; Yamasaki, M.; Marubashi, S.; Takemasa, I.; Miyamoto, A.; et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer 2007, 120, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Shinsato, Y.; Yamamoto, M.; Takahashi, H.; Zhang, S.; Nishizawa, Y.; Tabata, S.; Ikeda, R.; Kawahara, K.; Tsujikawa, K.; et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J Pharmacol. Sci. 2015, 127, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Itoi, T.; Sofuni, A.; Fukushima, N.; Itokawa, F.; Tsuchiya, T.; Kurihara, T.; Moriyasu, F.; Tsuchida, A.; Kasuya, K. Ribonucleotide reductase subunit M2 mRNA expression in pretreatment biopsies obtained from unresectable pancreatic carcinomas. J. Gastroenterol. 2007, 42, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Komori, S.; Osada, S.; Yoshida, K. Novel strategy with gemcitabine for advanced pancreatic cancer. ISRN Oncol. 2011, 2011, 936893. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Sasaki, T.; Sasahira, N.; Tsujino, T.; Toda, N.; Kogure, H.; Matsubara, S.; Ito, Y.; Togawa, O.; et al. A multicentre randomised phase II trial of gemcitabine alone vs gemcitabine and S-1 combination therapy in advanced pancreatic cancer: GEMSAP study. Br. J. Cancer 2012, 106, 1934–1939. [Google Scholar] [CrossRef] [PubMed]
- Komori, S.; Osada, S.; Mori, R.; Matsui, S.; Sanada, Y.; Tomita, H.; Tokuyama, Y.; Takahashi, T.; Yamaguchi, K.; Yoshida, K. Contribution of thymidylate synthase to gemcitabine therapy for advanced pancreatic cancer. Pancreas 2010, 39, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Daman, Z.; Ostad, S.; Amini, M.; Gilani, K. Preparation, optimization and in vitro characterization of stearoyl-gemcitabine polymeric micelles: A comparison with its self-assembled nanoparticles. Int. J. Pharm. 2014, 468, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Sloat, B.R.; Sandoval, M.A.; Li, D.; Chung, W.G.; Lansakara-P, D.S.; Proteau, P.J.; Kiguchi, K.; DiGiovanni, J.; Cui, Z. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int. J. Pharm. 2011, 409, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wonganan, P.; Lansakara-P, D.S.; O′Mary, H.L.; Li, Y.; Cui, Z. The effect of the acid-sensitivity of 4-(N)-stearoyl gemcitabine-loaded micelles on drug resistance caused by RRM1 overexpression. Biomaterials 2013, 34, 2327–2339. [Google Scholar] [CrossRef] [PubMed]
- Vandana, M.; Sahoo, S.K. Long circulation and cytotoxicity of pegylated gemcitabine and its potential for the treatment of pancreatic cancer. Biomaterials 2010, 31, 9340–9356. [Google Scholar] [CrossRef] [PubMed]
- Chitkara, D.; Mittal, A.; Behrman, S.W.; Kumar, N.; Mahato, R.I. Self-assembling, amphiphilic polymer-gemcitabine conjugate shows enhanced antitumor efficacy against human pancreatic adenocarcinoma. Bioconjug. Chem. 2013, 24, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Du, F.; Xu, Y.; Meng, H.; Huang, J.; Zhang, X.; Lu, W.; Liu, S.; Yu, J. Enhanced cellular uptake and intracellular drug controlled release of vesylated gemcitabine prodrug nanocapsules. Colloids Surf. B Biointerfaces 2015, 128, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Sakarchi, W.A.; Gupta, P.N.; Curtis, A.D.M.; Hoskins, C. Synthesis and characterization of TPGS– gemcitabine prodrug micelles for pancreatic cancer therapy. RSC Adv. 2016, 6, 60126–60137. [Google Scholar] [CrossRef]
- Xu, Y.; Meng, H.; Du, F.; Lu, W.; Liu, S.; Huang, J.; Yu, J. Preparation of intravenous injection nanoformulation of vesylated gemcitabine by co-assembly with tpgs and its anti-tumor activity in pancreatic tumor-bearing mice. Int. J. Pharm. 2015, 495, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Réjiba, S.; Reddy, L.H.; Bigand, C.; Parmentier, C.; Couvreur, P.; Hajri, A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine 2011, 7, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.M.; Khan, A.R.; Ahmad, M.U.; Chen, P.; Sheikh, S.; Ahmad, I. Synthesis and biological evaluation of gemcitabine-lipid conjugate (NEO6002). Bioorg. Med. Chem. Lett. 2005, 15, 2571–2574. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chien, P.Y.; Khan, A.R.; Sheikh, S.; Ali, S.M.; Ahmad, M.U.; Ahmad, I. In-vitro and in-vivo anti-cancer activity of a novel gemcitabine-cardiolipin conjugate. Anticancer Drugs 2006, 17, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.M.; Adema, A.D.; Balzarini, J.; Bruheim, S.; Fichtner, I.; Noordhuis, P.; Fodstad, O.; Myhren, F.; Sandvold, M.L.; Hendriks, H.R.; et al. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Investig. New Drugs 2011, 29, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Stuurman, F.E.; Lolkema, M.P.; Huitema, A.D.; Soetekouw, P.M.; Rosing, H.; Rolfe, L.; Kaur, P.; Beijnen, J.H.; van Tinteren, H.; Voest, E.E.; et al. A phase I comparative pharmacokinetic and cardiac safety study of two intravenous formulations of CO-101 in patients with advanced solid tumors. J. Clin. Pharmacol. 2013, 53, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Sigmond, J.; Peters, G.J.; Borch, R.F. Synthesis and biological activity of a gemcitabine phosphoramidate prodrug. J. Med. Chem. 2007, 50, 3743–3746. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Habib, N.A.; Wasan, H.S.; Gabra, H.; Jiao, L.R.; Slusarczyk, M.; Chabot, J.A.; Saif, M.W. A phosphoramidate protide (NUC-1031) and acquired and intrinsic resistance to gemcitabine. J. Clin. Oncol. 2011, 29, e13540. [Google Scholar] [CrossRef]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of protide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Incecayir, T.; Song, X.; Hilfinger, J.M.; Amidon, G.L. The development of orally administrable gemcitabine prodrugs with D-enantiomer amino acids: Enhanced membrane permeability and enzymatic stability. Eur. J. Pharm. Biopharm. 2014, 86, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Borras Bermejo, B.; Amidon, G.L. The dipeptide monoester prodrugs of floxuridine and gemcitabine-feasibility of orally administrable nucleoside analogs. Pharmaceuticals 2014, 7, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Castelli, F.; Sarpietro, M.G.; Ceruti, M.; Rocco, F.; Cattel, L. Characterization of lipophilic gemcitabine prodrug-liposomal membrane interaction by differential scanning calorimetry. Mol. Pharm. 2006, 3, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Brusa, P.; Immordino, M.L.; Rocco, F.; Cattel, L. Antitumor activity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. Anticancer Res. 2007, 27, 195–199. [Google Scholar] [PubMed]
- Hung, S.W.; Mody, H.R.; Govindarajan, R. Overcoming nucleoside analog chemoresistance of pancreatic cancer: A therapeutic challenge. Cancer Lett. 2012, 320, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Poplin, E.; Wasan, H.; Rolfe, L.; Raponi, M.; Ikdahl, T.; Bondarenko, I.; Davidenko, I.; Bondar, V.; Garin, A.; Boeck, S.; et al. Randomized, multicenter, phase II study of CO-101 versus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma: Including a prospective evaluation of the role of hENT1 in gemcitabine or CO-101 sensitivity. J. Clin. Oncol. 2013, 31, 4453–4461. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, B.; Awada, A.; Evans, T.R.; Dueland, S.; Hendlisz, A.; Rasch, W.; Hernes, K.; Hagen, S.; Aamdal, S. A first-in-human phase I and pharmacokinetic study of CP-4126 (CO-101), a nucleoside analogue, in patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2015, 76, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjug. Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.; He, C.; Liu, D.; Lu, K.; Lin, W. Self-assembled nanoscale coordination polymers carrying oxaliplatin and gemcitabine for synergistic combination therapy of pancreatic cancer. J. Control. Release 2015, 201, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Brusa, P.; Rocco, F.; Arpicco, S.; Ceruti, M.; Cattel, L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J. Control. Release 2004, 100, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.G.; Sandoval, M.A.; Sloat, B.R.; Lansakara-P, D.S.; Cui, Z. Stearoyl gemcitabine nanoparticles overcome resistance related to the over-expression of ribonucleotide reductase subunit M1. J. Control. Release. 2012, 157, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Stella, B.; Arpicco, S.; Rocco, F.; Marsaud, V.; Renoir, J.M.; Cattel, L.; Couvreur, P. Encapsulation of gemcitabine lipophilic derivatives into polycyanoacrylate nanospheres and nanocapsules. Int. J. Pharm. 2007, 344, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Allain, V.; Bourgaux, C.; Couvreur, P. Self-assembled nucleolipids: From supramolecular structure to soft nucleic acid and drug delivery devices. Nucleic Acids Res. 2012, 40, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Maksimenko, A.; Caron, J.; Mougin, J.; Desmaële, D.; Couvreur, P. Gemcitabine-based therapy for pancreatic cancer using the squalenoyl nucleoside monophosphate nanoassemblies. Int. J. Pharm. 2015, 482, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Chen, W.; Wang, H.; Jin, C.; Yu, X.J.; Lu, W.Y.; Cui, L.; Fu, D.L.; Ni, Q.X.; Hou, H.M. Preparation of albumin nanospheres loaded with gemcitabine and their cytotoxicity against BPXP-3 cells in vitro. Acta Pharmacol. Sin. 2009, 30, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Di, Y.; Xie, C.; Song, Y.; He, H.; Li, H.; Pu, X.; Lu, W.; Fu, D.; Jin, C. An in vitro and in vivo study of gemcitabine-loaded albumin nanoparticles in a pancreatic cancer cell line. Int. J. Nanomed. 2015, 10, 6825–6834. [Google Scholar] [CrossRef] [PubMed]
- Arya, G.; Vandana, M.; Acharya, S.; Sahoo, S.K. Enhanced antiproliferative activity of herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy. Nanomedicine 2011, 7, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Patra, C.R.; Bhattacharya, R.; Wang, E.; Katarya, A.; Lau, J.S.; Dutta, S.; Muders, M.; Wang, S.; Buhrow, S.A.; Safgren, S.L.; et al. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer Res. 2008, 68, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Joubert, F.; Martin, L.; Perrier, S.; Pasparakis, G. Development of a gemcitabine-polymer conjugate with prolonged cytotoxicity against a pancreatic cancer cell line. ACS Macro. Lett. 2017, 6, 535–540. [Google Scholar] [CrossRef]
- Utama, R.H.; Jiang, Y.; Zetterlund, P.B.; Stenzel, M.H. Biocompatible glycopolymer nanocapsules via inverse miniemulsion periphery RAFT polymerization for the delivery of gemcitabine. Biomacromolecules 2015, 16, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Mizuma, M.; Feldmann, G.; Ottenhof, N.A.; Hong, S.M.; Pramanik, D.; Chenna, V.; Karikari, C.; Sharma, R.; Goggins, M.G.; et al. Systemic administration of polymeric nanoparticle-encapsulated curcumin (NanoCurc) blocks tumor growth and metastases in preclinical models of pancreatic cancer. Mol. Cancer Ther. 2010, 9, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Aryal, S.; Hu, C.M.; Zhang, L. Combinatorial drug conjugation enables nanoparticle dual-drug delivery. Small 2010, 6, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Zhang, C.H.; Schwarz, A.M.; Hinz, S.; Wang, C.G.; Williams, N.S.; Schwarz, M.A.; Schwarz, R.E. Comparative benefits of nab-paclitaxel over gemcitabine or polysorbate-based docetaxel in experimental pancreatic cancer. Carcinogenesis 2013, 34, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Al-Batran, S.E.; Geissler, M.; Seufferlein, T.; Oettle, H. Nab-paclitaxel for metastatic pancreatic cancer: Clinical outcomes and potential mechanisms of action. Oncol. Res. Treat. 2014, 37, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Nomura, A.; Saluja, A.; Banerjee, S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology 2017, 17, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The role of stroma in pancreatic cancer: Diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Coppola, S.; Carnevale, I.; Danen, E.H.J.; Peters, G.J.; Schmidt, T.; Assaraf, Y.G.; Giovannetti, E. A mechanopharmacology approach to overcome chemoresistance in pancreatic cancer. Drug Resist. Updat. 2017, 31, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Liss, A.S.; Thayer, S.P. Therapeutic targeting of pancreatic stroma. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Target Entity | Role in Gemcitabine Metabolism | Resistance Pattern | Impact on Progression of Chemoresistance to Gemcitabine | References |
|---|---|---|---|---|
| Nucleoside transporters | Drug transport | Downregulation | Level of hEN1, hCNT1 and hCNT3 are correlative of resistance to gemcitabine | [63,64] |
| Deoxycytidine kinase | Intracellular drug activation | Downregulation | Reduced levels of dCK are linked with acquired resistance to gemcitabine | [65] |
| Cytidine deaminase | Drug inactivation | Upregulation | CDA induced deamination causes degradation and excretion of gemcitabine | [26] |
| Ribonucleotide reductase | Competition in DNA synthesis | Upregulation | RR mediates DNA synthesis via generation of dCTPs | [66] |
| Thymidylate synthase | Competition in DNA synthesis | Upregulation | Regulation of early stages of DNA biosynthesis, activation of salvage pathway | [67,68] |
| Position | Target Moiety | Prodrug | Experimental Model | Outcomes | References |
|---|---|---|---|---|---|
| 4-(N) | Acyl derivative (stearoyl) | GemC18 | AsPC-1 and PANC-1 cells, murine BxPC-3 tumor xenografts | Inhibition of RRM1 and increased dFdCTP levels, enhanced anti-tumor activity | [105,106,107] |
| Polyethylene glycol (PEG) | PEG–NHS | MIA PaCa-2 and PANC-1 cells | Prolonged circulation in murine plasma, improved cytotoxicity and apoptosis | [108] | |
| PEG-PCC | MIA PaCa-2 and L3.6 cells, murine MIA PaCa-2 tumor xenografts | High anti-tumor activity and increased apoptosis | [109] | ||
| Vitamin E succinate (VES) | VES-dFdC | BxPC-3 cells | High anti-tumor activity, enhanced cellular uptake | [110] | |
| D-ɑ-tocopheryl PEG succinate | TPGS/VES-dFdC | BxPC-3 cells and murine BxPC-3 tumor xenografts | High anti-tumor activity, resistant to CDA induced deamination and superior cytotoxicity | [111,112] | |
| 1,1′,2-tris-nor-squalenoic acid (squalene) | SQ-dFdC/SQ-dFdCMP | BxPC-3, Capan-1, PANC-1 cells; murine BxPC-3, MIA PaCa-2 and PANC-1 tumor xenografts | High anti-proliferative and cytotoxic effects, reduced tumor growth and prolonged survival | [113] | |
| 5′-OH | Cardiolipin | NEO6002 | BxPC-3 cells and murine BxPC-3 tumor xenografts | High cytotoxicity independent of NT activity and high tumor growth inhibition | [114,115] |
| Elaidic acid | CP-4126 (CO-101) | Murine MIA PaCa-2, PANC-1 tumor xenografts | Transport independent of hENT1, equally effective to gemcitabine | [116,117] | |
| Phosphoramidate | Mono-phosphate | Cell lines with dCK-deficient variants: AG600 and CEM-dCK | ~4-fold more effective than gemcitabine | [118] | |
| Phosphoramidate ProTide | NUC-1031 (ProTide 6f) | BxPC-3, MIA PaCa-2, PANC-1 cells; murine MIA PaCa-2, tumor xenografts | Resistant to CDA mediated deamination and directly generates dFdCMP intracellularly; reduced tumor volume | [119,120] | |
| Other | D-amino modifications | - | AsPC-1 cells | High plasma concentration superior enzymatic stability | [121] |
| Dipeptide monoester prodrugs | - | PANC-1, and AsPC-1 cells | Enhanced uptake and anti-proliferation activity | [122] |
| Drug Combination | ClinicalTrials.gov Identifier | Disease Condition (Pancreatic Cancer) | Study Phase |
|---|---|---|---|
| Gemcitabine + Abraxane | NCT02043730 | Stage II | II |
| Gemcitabine + Erlotinib | NCT02154737 | Locally advanced | I |
| Gemcitabine + SRA737 | NCT02797977 | Locally advanced | I |
| Gemcitabine + Cisplatin, +/−Veliparib | NCT01585805 | Metastatic | II |
| Gemcitabine + Capecitabine | NCT02919787 | Locally advanced | II |
| Gemcitabine + S-1 | NCT02131493 | Locally advanced | II |
| Gemcitabine + Metformin | NCT02005419 | Stage IA, IB, IIA, IIB | II |
| Gemcitabine + All-trans retinoic acid (ATRA) | NCT03307148 | Locally advanced or metastatic | IB |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. https://doi.org/10.3390/cancers9110157
Amrutkar M, Gladhaug IP. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers. 2017; 9(11):157. https://doi.org/10.3390/cancers9110157
Chicago/Turabian StyleAmrutkar, Manoj, and Ivar P. Gladhaug. 2017. "Pancreatic Cancer Chemoresistance to Gemcitabine" Cancers 9, no. 11: 157. https://doi.org/10.3390/cancers9110157
APA StyleAmrutkar, M., & Gladhaug, I. P. (2017). Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers, 9(11), 157. https://doi.org/10.3390/cancers9110157