
An Exploration into the Origins and Pathogenesis of Anaplastic Large Cell Lymphoma, Anaplastic Lymphoma Kinase (ALK)-Positive


{kind=link}
Abstract
:1. Introduction
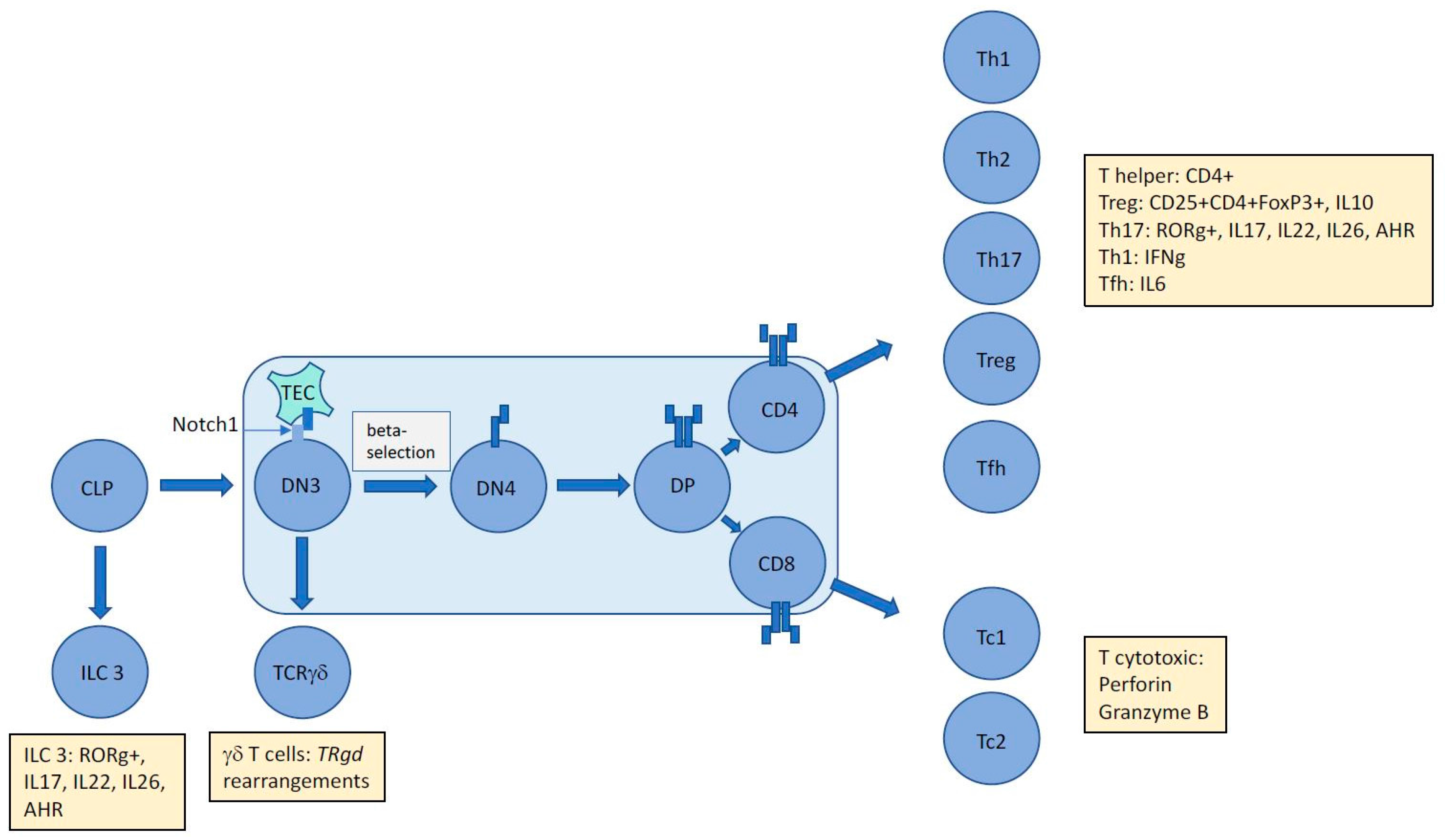
2. T-Cell Development and the Origins of ALCL
2.1. T-Cell Receptor Gene Rearrangement Status Presents a History and Time Stamp of Thymocyte Development
2.2. TCR Rearrangements in ALCL are Suggestive of Stalled Thymocyte Development of Apparently Mature T Cells Indicative of a Primitive T-Cell Origin for this Malignancy
3. NPM-ALK Induced Signaling Events May Counteract Thymic Beta-Selection
4. Accounting for the Activated Cellular Phenotype of ALCL
4.1. Is NPM-ALK, the Tumour Microenvironment, and/or the Cell of Origin Responsible for the Immunophenotype of Cells in ALCL, ALK+?
4.2. What Shapes the Phenotype of ALCL, ALK−?
4.3. Does Infection Play a Role in ALCL Lymphomagenesis and Cellular Immunophenotype?
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugieres, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 173, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Fornari, A.; Piva, R.; Chiarle, R.; Novero, D.; Inghirami, G. Anaplastic large cell lymphoma: One or more entities among T-cell lymphoma? Hematol. Oncol. 2009, 27, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Shokrollahi, K.; Rozen, W.M.; Conyers, R.; Wright, P.; Kenner, L.; Turner, S.D.; Whitaker, I.S. Anaplastic large cell lymphoma (ALCL) and breast implants: Breaking down the evidence. Mutat. Res. Rev. Mutat. Res. 2014, 762, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; p. 439. [Google Scholar]
- Malcolm, T.I.; Hodson, D.J.; Macintyre, E.A.; Turner, S.D. Challenging perspectives on the cellular origins of lymphoma. Open. Biol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, T.I.; Villarese, P.; Fairbairn, C.J.; Lamant, L.; Trinquand, A.; Hook, C.E.; Burke, G.A.; Brugieres, L.; Hughes, K.; Payet, D.; et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun. 2016, 7, 10087. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.M.; Greaves, M. ETV6-RUNX1 + acute lymphoblastic leukaemia in identical twins. Adv. Exp. Med. Biol. 2017, 962, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Le Noir, S.; Ben Abdelali, R.; Lelorch, M.; Bergeron, J.; Sungalee, S.; Payet-Bornet, D.; Villarese, P.; Petit, A.; Callens, C.; Lhermitte, L.; et al. Extensive molecular mapping of TCRalpha/delta- and TCRbeta-involved chromosomal translocations reveals distinct mechanisms of oncogene activation in T-ALL. Blood 2012, 120, 3298–3309. [Google Scholar] [CrossRef] [PubMed]
- Callens, C.; Baleydier, F.; Lengline, E.; Ben Abdelali, R.; Petit, A.; Villarese, P.; Cieslak, A.; Minard-Colin, V.; Rullier, A.; Moreau, A.; et al. Clinical impact of NOTCH1 and/or FBXW7 mutations, FLASH deletion, and TCR status in pediatric T-cell lymphoblastic lymphoma. J. Clin. Oncol. 2012, 30, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Dadi, S.; Le Noir, S.; Asnafi, V.; Beldjord, K.; Macintyre, E.A. Normal and pathological V(D)J recombination: Contribution to the understanding of human lymphoid malignancies. Adv. Exp. Med. Biol. 2009, 650, 180–194. [Google Scholar] [PubMed]
- Piccaluga, P.P.; Fuligni, F.; De Leo, A.; Bertuzzi, C.; Rossi, M.; Bacci, F.; Sabattini, E.; Agostinelli, C.; Gazzola, A.; Laginestra, M.A.; et al. Molecular profiling improves classification and prognostication of nodal peripheral T-cell lymphomas: Results of a phase III diagnostic accuracy study. J. Clin. Oncol. 2013, 31, 3019–3025. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Weisenburger, D.D.; Greiner, T.C.; Vose, J.M.; McKeithan, T.; Kucuk, C.; Geng, H.; Deffenbacher, K.; Smith, L.; Dybkaer, K.; et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood 2010, 115, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK− anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Zuniga-Pflucker, J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014, 192, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Yeung, D.; Hadfield, K.; Cook, S.J.; Alexander, D.R. The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell. Signal. 2007, 19, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Lopez, C.; Desjobert, C.; Berrebi, A.; Damm-Welk, C.; Delsol, G.; Brousset, P.; Lamant, L. Circulating t(2;5)-positive cells can be detected in cord blood of healthy newborns. Leukemia 2012, 26, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Alexander, D.R. Fusion tyrosine kinase mediated signalling pathways in the transformation of haematopoietic cells. Leukemia 2006, 20, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar] [PubMed]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.X.; Kerby, A.; McDuff, F.K.; Ye, H.; Turner, S.D. NPM-ALK inhibits the p53 tumor suppressor pathway in an MDM2 and JNK-dependent manner. Blood 2009, 113, 5217–5227. [Google Scholar] [CrossRef] [PubMed]
- Staber, P.B.; Vesely, P.; Haq, N.; Ott, R.G.; Funato, K.; Bambach, I.; Fuchs, C.; Schauer, S.; Linkesch, W.; Hrzenjak, A.; et al. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood 2007, 110, 3374–3383. [Google Scholar] [CrossRef] [PubMed]
- Leventaki, V.; Drakos, E.; Medeiros, L.J.; Lim, M.S.; Elenitoba-Johnson, K.S.; Claret, F.X.; Rassidakis, G.Z. NPM-ALK oncogenic kinase promotes cell-cycle progression through activation of JNK/cJun signaling in anaplastic large-cell lymphoma. Blood 2007, 110, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dorken, B.; et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.Y.; Ouyang, T.; Miething, C.; Morris, S.W.; Peschel, C.; Duyster, J. Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood 2000, 96, 4319–4327. [Google Scholar] [PubMed]
- Marzec, M.; Kasprzycka, M.; Liu, X.; Raghunath, P.N.; Wlodarski, P.; Wasik, M.A. Oncogenic tyrosine kinase NPM/ALK induces activation of the MEK/ERK signaling pathway independently of c-Raf. Oncogene 2007, 26, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D.; Tooze, R.; Maclennan, K.; Alexander, D.R. Vav-promoter regulated oncogenic fusion protein NPM-ALK in transgenic mice causes B-cell lymphomas with hyperactive Jun kinase. Oncogene 2003, 22, 7750–7761. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Nakamura, S.; Ichinohasama, R.; Abe, M.; Akagi, T.; Takeshita, M.; Mori, N.; Fujimoto, J.; Miyauchi, J.; Mikata, A.; et al. Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: A distinct clinicopathologic entity. Blood 1995, 86, 1954–1960. [Google Scholar] [PubMed]
- Kamstrup, M.R.; Biskup, E.; Gjerdrum, L.M.; Ralfkiaer, E.; Niazi, O.; Gniadecki, R. The importance of Notch signaling in peripheral T-cell lymphomas. Leuk. Lymphoma 2014, 55, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Osborne, B.A.; Minter, L.M. Notch signalling during peripheral T-cell activation and differentiation. Nature reviews. Immunology 2007, 7, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Bonzheim, I.; Geissinger, E.; Roth, S.; Zettl, A.; Marx, A.; Rosenwald, A.; Muller-Hermelink, H.K.; Rudiger, T. Anaplastic large cell lymphomas lack the expression of T-cell receptor molecules or molecules of proximal T-cell receptor signaling. Blood 2004, 104, 3358–3360. [Google Scholar] [CrossRef] [PubMed]
- Eckerle, S.; Brune, V.; Doring, C.; Tiacci, E.; Bohle, V.; Sundstrom, C.; Kodet, R.; Paulli, M.; Falini, B.; Klapper, W.; et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: Insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia 2009, 23, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, B.; Krantz, M.J.; Parker, J.; Longenecker, B.M. Expression of MUC1 mucin on activated human T cells: Implications for a role of MUC1 in normal immune regulation. Cancer Res. 1998, 58, 4079–4081. [Google Scholar] [PubMed]
- Ten Berge, R.L.; Snijdewint, F.G.; von Mensdorff-Pouilly, S.; Poort-Keesom, R.J.; Oudejans, J.J.; Meijer, J.W.; Willemze, R.; Hilgers, J.; Meijer, C.J. MUC1 (EMA) is preferentially expressed by ALK positive anaplastic large cell lymphoma, in the normally glycosylated or only partly hypoglycosylated form. J. Clin. Pathol. 2001, 54, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Savan, R.; McFarland, A.P.; Reynolds, D.A.; Feigenbaum, L.; Ramakrishnan, K.; Karwan, M.; Shirota, H.; Klinman, D.M.; Dunleavy, K.; Pittaluga, S.; et al. A novel role for IL-22R1 as a driver of inflammation. Blood 2011, 117, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Suzuki, H.I.; Nishimori, H.; Noguchi, M.; Yao, T.; Komatsu, N.; Mano, H.; Sugimoto, K.; Miyazono, K. miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood 2011, 118, 6881–6892. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D.; Lee, J.; Bacani, J.T.; Lai, R.; Ingham, R.J. NPM-ALK and the JunB transcription factor regulate the expression of cytotoxic molecules in ALK-positive, anaplastic large cell lymphoma. Int. J. Clin. Exp. Pathol. 2011, 4, 124–133. [Google Scholar] [PubMed]
- Merkel, O.; Hamacher, F.; Griessl, R.; Grabner, L.; Schiefer, A.I.; Prutsch, N.; Baer, C.; Egger, G.; Schlederer, M.; William Krenn, P.; et al. Oncogenic role of miR-155 in anaplastic large cell lymphoma lacking the t(2;5) translocation. J. Pathol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.; Hamacher, F.; Laimer, D.; Sifft, E.; Trajanoski, Z.; Scheideler, M.; Egger, G.; Hassler, M.R.; Thallinger, C.; Schmatz, A.; et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK− anaplastic large-cell lymphoma. Proc. Natl. Acad. Sci. USA 2010, 107, 16228–16233. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Petrus, M.N.; Xiao, W.; Nicolae, A.; Raffeld, M.; Pittaluga, S.; Bamford, R.N.; Nakagawa, M.; Ouyang, S.T.; et al. Cytokine receptor signaling is required for the survival of ALK- anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 3975–3980. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.N.; Aladily, T.N.; Prince, H.M.; Kanagal-Shamanna, R.; de Jong, D.; Fayad, L.E.; Amin, M.B.; Haideri, N.; Bhagat, G.; Brooks, G.S.; et al. Breast implant-associated anaplastic large-cell lymphoma: Long-term follow-up of 60 patients. J. Clin. Oncol. 2014, 32, 114–120. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.; Vasmel, W.L.; de Boer, J.P.; Verhave, G.; Barbe, E.; Casparie, M.K.; van Leeuwen, F.E. Anaplastic large-cell lymphoma in women with breast implants. JAMA 2008, 300, 2030–2035. [Google Scholar] [CrossRef] [PubMed]
- Kadin, M.E.; Deva, A.; Xu, H.; Morgan, J.; Khare, P.; MacLeod, R.A.; Van Natta, B.W.; Adams, W.P., Jr.; Brody, G.S.; Epstein, A.L. Biomarkers provide clues to early events in the pathogenesis of breast implant-associated anaplastic large cell lymphoma. Aesthet. Surg. J. 2016, 36, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Thompson, E.R.; Jones, K.; Arnau, G.M.; Lade, S.; Markham, J.F.; Li, J.; Deva, A.; Johnstone, R.W.; Khot, A.; et al. Whole exome sequencing reveals activating JAK1 and STAT3 mutations in breast implant-associated anaplastic large cell lymphoma anaplastic large cell lymphoma. Haematologica 2016, 101, e387–e390. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Johani, K.; Almatroudi, A.; Vickery, K.; Van Natta, B.; Kadin, M.E.; Brody, G.; Clemens, M.; Cheah, C.Y.; Lade, S.; et al. Bacterial Biofilm Infection Detected in Breast Implant-Associated Anaplastic Large-Cell Lymphoma. Plast. Reconstr. Surg. Glob Open 2016, 137, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Lamant, L.; Pileri, S.; Sabattini, E.; Brugieres, L.; Jaffe, E.S.; Delsol, G. Cutaneous presentation of ALK-positive anaplastic large cell lymphoma following insect bites: Evidence for an association in five cases. Haematologica 2010, 95, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Ascani, S.; Fraternali Orcioni, G.; Piccioli, M.; Pileri, A., Jr.; Falini, B.; Pileri, S. Anaplastic lymphoma kinase expression as a marker of malignancy. Application to a case of anaplastic large cell lymphoma with huge granulomatous reaction. Haematologica 2000, 85, 978–981. [Google Scholar] [PubMed]
- Atsaves, V.; Lekakis, L.; Drakos, E.; Leventaki, V.; Ghaderi, M.; Baltatzis, G.E.; Chioureas, D.; Jones, D.; Feretzaki, M.; Liakou, C.; et al. The oncogenic JUNB/CD30 axis contributes to cell cycle deregulation in ALK+ anaplastic large cell lymphoma. Br. J. Haematol. 2014, 167, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Martinengo, C.; Voena, C.; Tondat, F.; Riera, L.; di Celle, P.F.; Inghirami, G.; Chiarle, R. NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 2009, 69, 8611–8619. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nature Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turner, S.D. An Exploration into the Origins and Pathogenesis of Anaplastic Large Cell Lymphoma, Anaplastic Lymphoma Kinase (ALK)-Positive. Cancers 2017, 9, 141. https://doi.org/10.3390/cancers9100141
Turner SD. An Exploration into the Origins and Pathogenesis of Anaplastic Large Cell Lymphoma, Anaplastic Lymphoma Kinase (ALK)-Positive. Cancers. 2017; 9(10):141. https://doi.org/10.3390/cancers9100141
Chicago/Turabian StyleTurner, Suzanne D. 2017. "An Exploration into the Origins and Pathogenesis of Anaplastic Large Cell Lymphoma, Anaplastic Lymphoma Kinase (ALK)-Positive" Cancers 9, no. 10: 141. https://doi.org/10.3390/cancers9100141
APA StyleTurner, S. D. (2017). An Exploration into the Origins and Pathogenesis of Anaplastic Large Cell Lymphoma, Anaplastic Lymphoma Kinase (ALK)-Positive. Cancers, 9(10), 141. https://doi.org/10.3390/cancers9100141