
Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame


 and
and
Abstract
:1. Introduction: Alcohol Consumption and Its Impact on Human Health
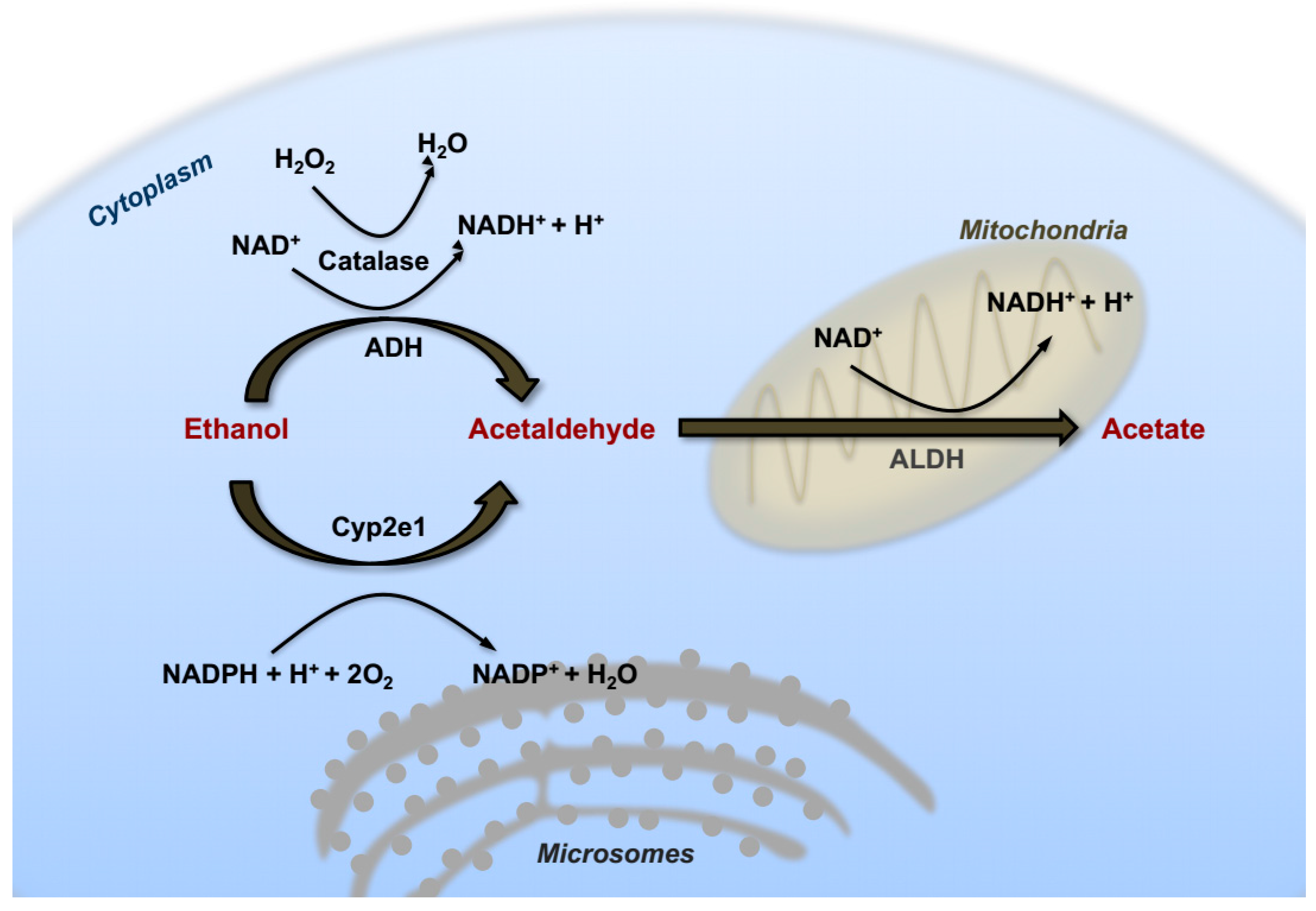
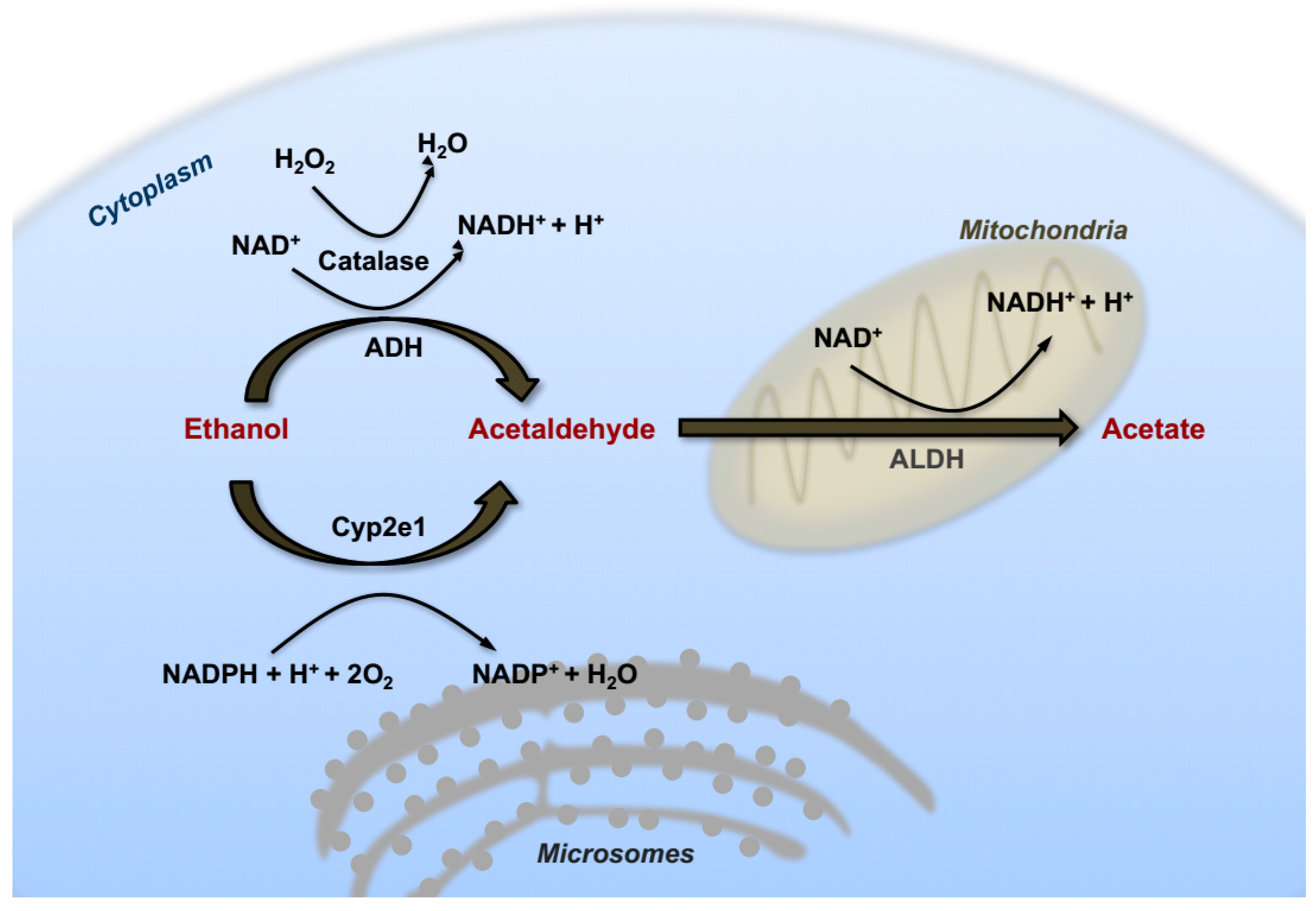
2. Alcohol Metabolism in the Liver
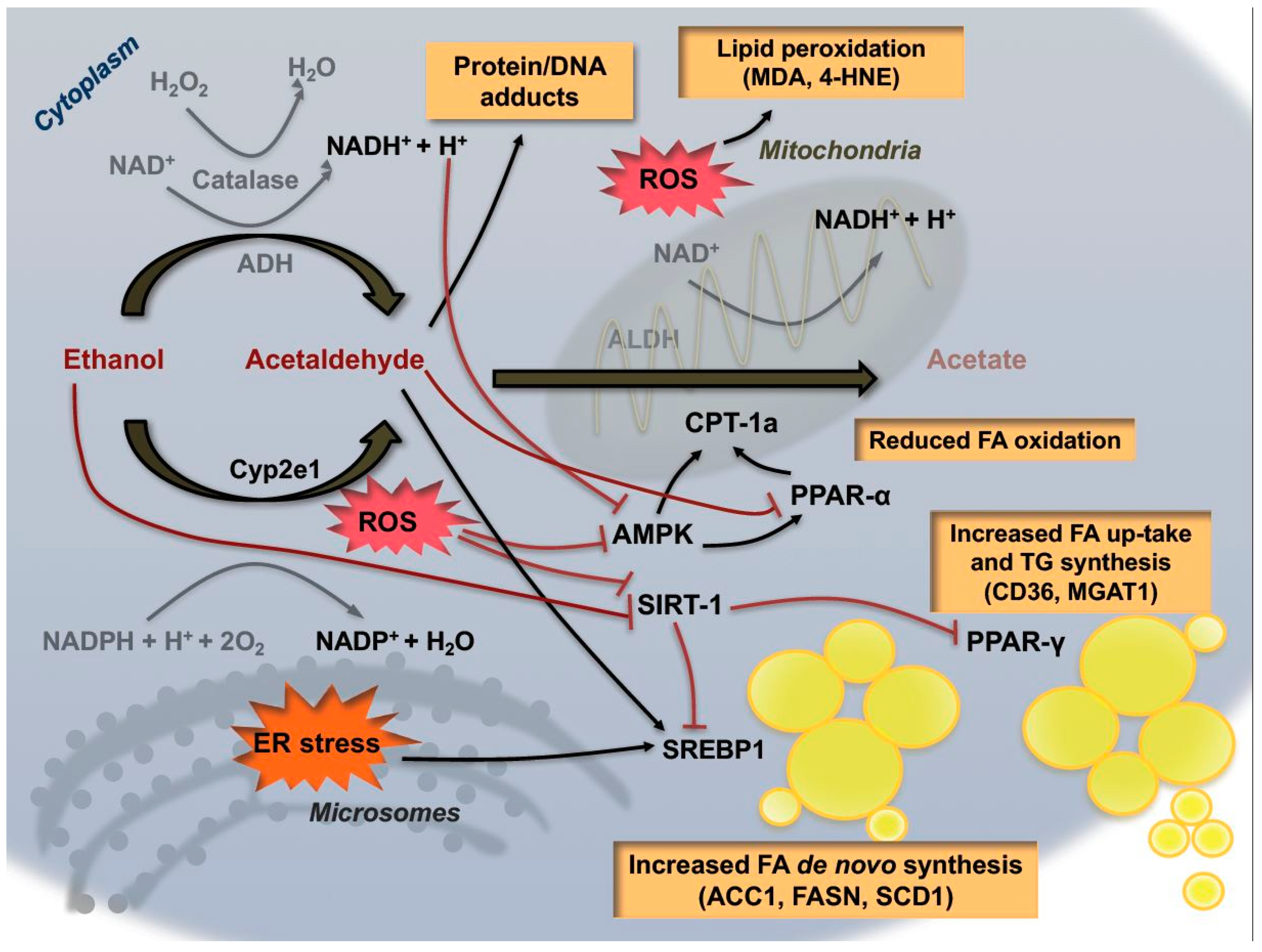
3. Pre-Carcinogenic Alterations of Hepatic Metabolism: From Steatosis to Oxidative Stress
4. Genetics and Epigenetics of Alcohol-Related Liver Cancer
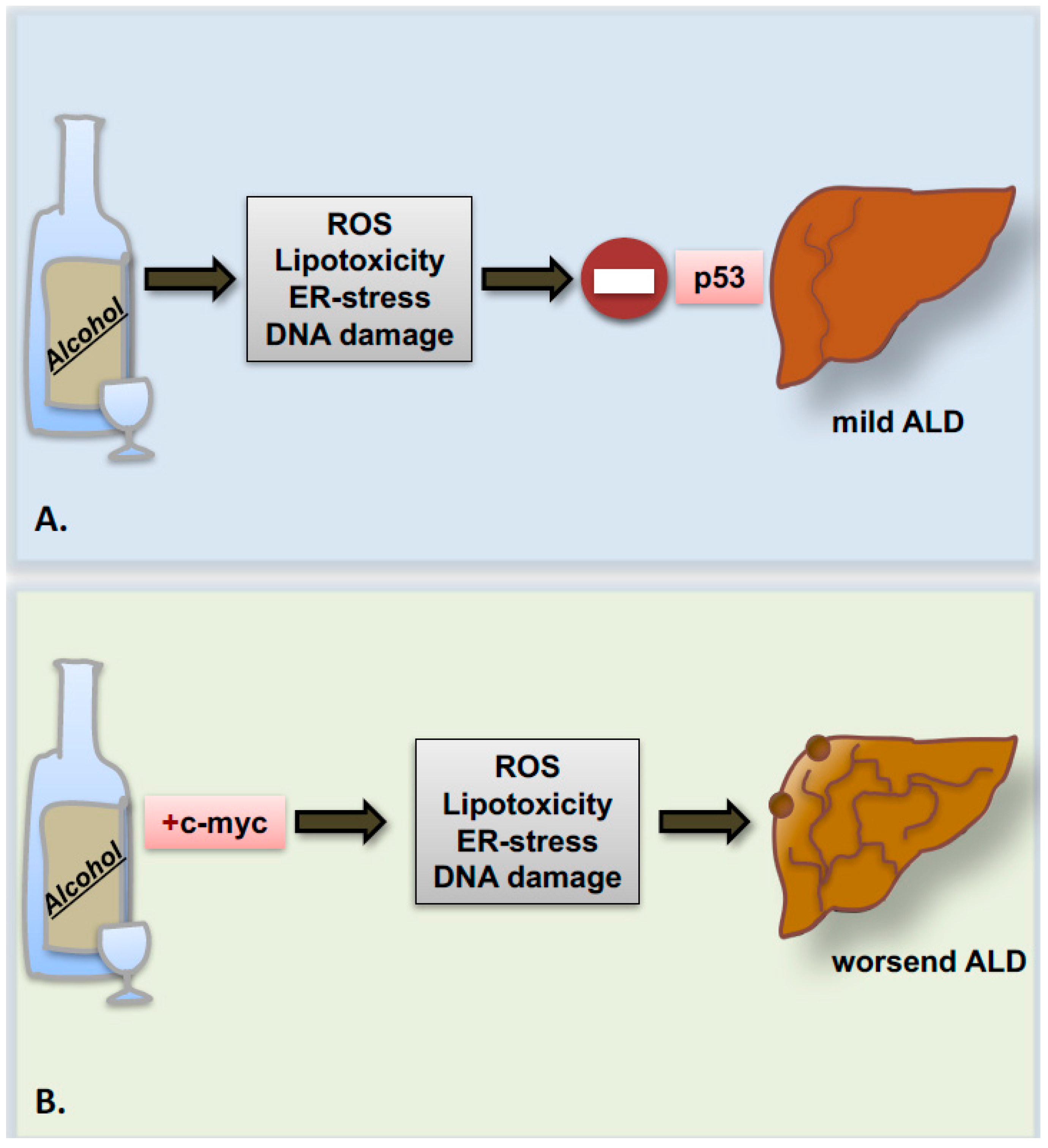
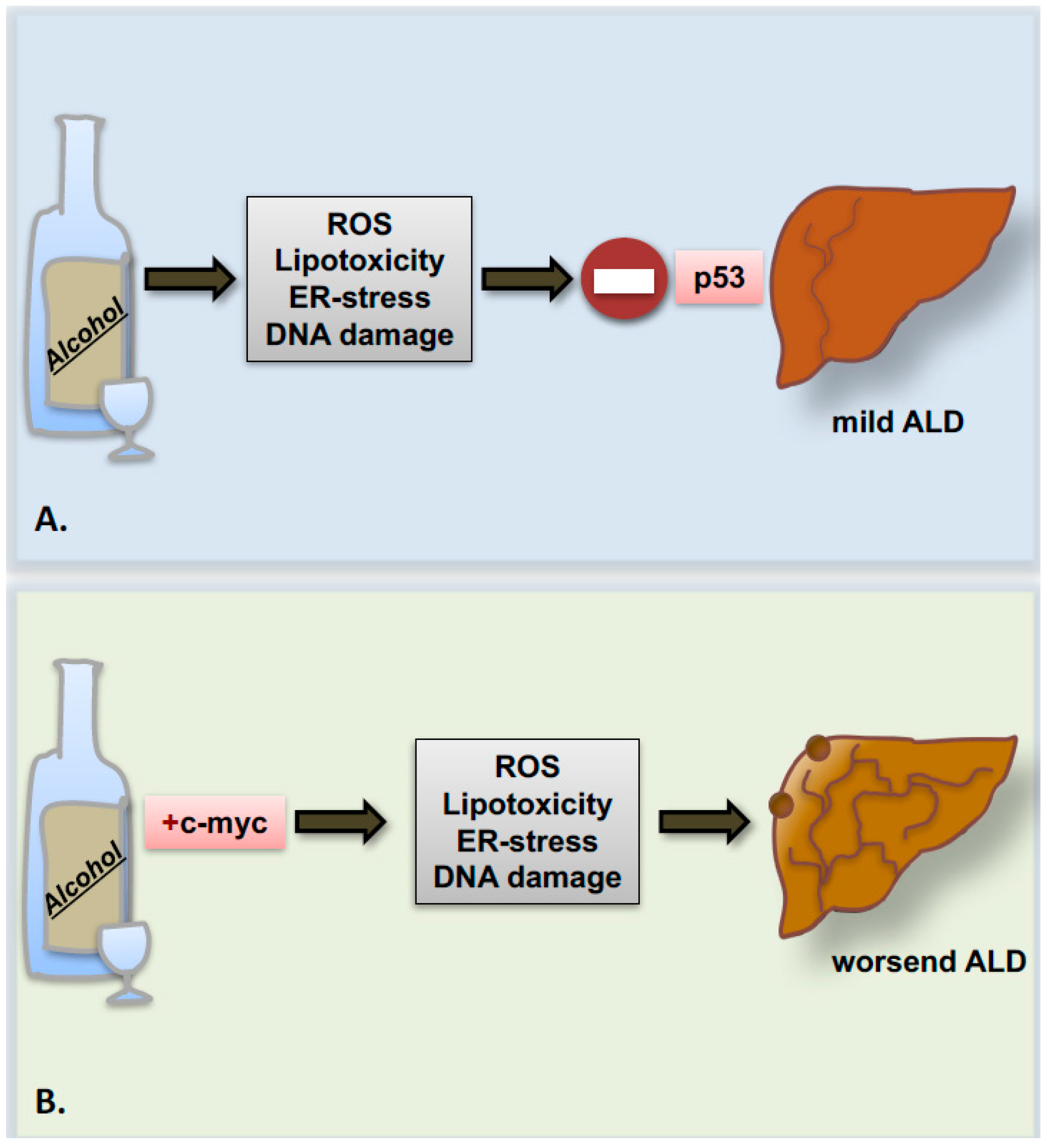
5. Alcohol and the Cell Cycle Machinery: Hepatocyte Cell Death and Proliferation
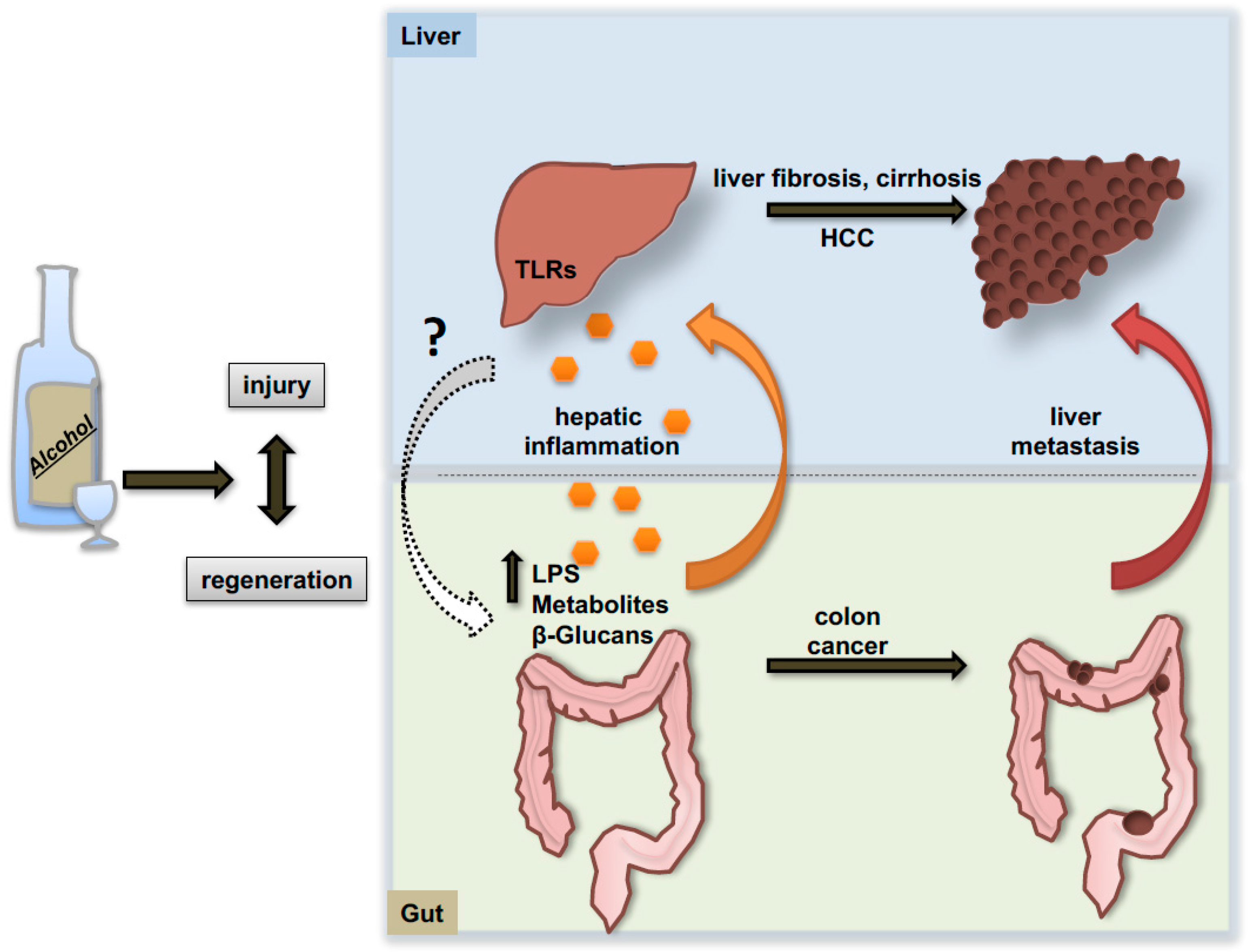
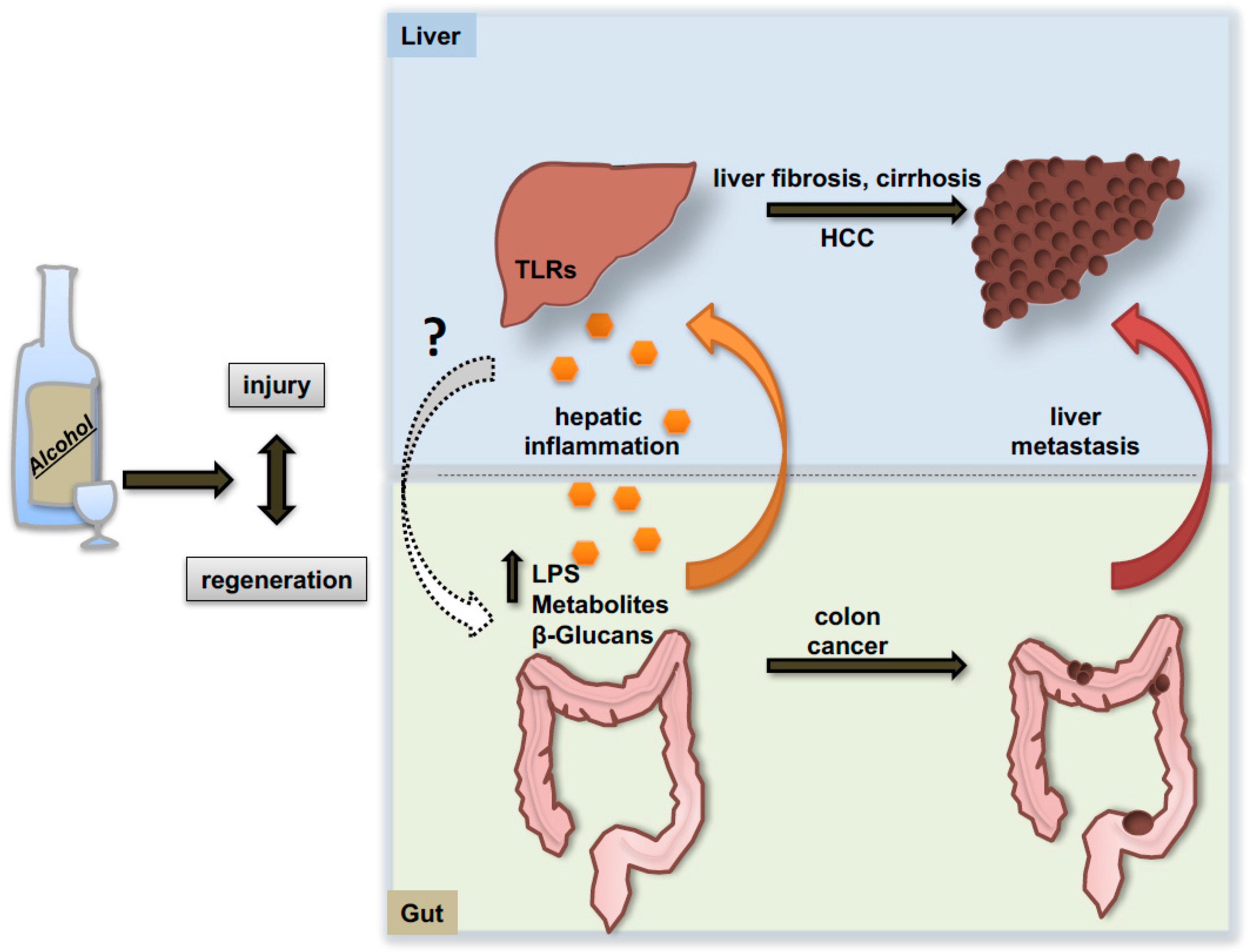
6. The Gut-Liver Axis: Alcohol Effects on Primary Tumor Development and on the Pro-Metastatic Niche
7. Extra-Hepatic Effects of Alcohol
8. Pre-Clinical Models of Alcohol Induced HCC
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stickel, F.; Datz, C.; Hampe, J.; Bataller, R. Pathophysiology and management of alcoholic liver disease: Update 2016. Gut Liver 2017, 11, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Scoccianti, C.; Cecchini, M.; Anderson, A.S.; Berrino, F.; Boutron-Ruault, M.-C.; Espina, C.; Key, T.J.; Leitzmann, M.; Norat, T.; Powers, H.; et al. European code against cancer 4th edition: Alcohol drinking and cancer. Cancer Epidemiol. 2016, 45, 181–188. [Google Scholar]
- Sheron, N. Alcohol and liver disease in Europe—Simple measures have the potential to prevent tens of thousands of premature deaths. J. Hepatol. 2016, 64, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Burra, P.; Senzolo, M.; Adam, R.; Delvart, V.; Karam, V.; Germani, G.; Neuberger, J. Liver Transplantation for Alcoholic Liver Disease in Europe: A Study from the ELTR (European Liver Transplant Registry). Am. J. Transplant. 2010, 10, 138–148. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- Blonski, W.; Kotlyar, D.S.; Forde, K.A. Non-viral causes of hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 3603–3615. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Jarl, J.; Gerdtham, U.-G. Time pattern of reduction in risk of oesophageal cancer following alcohol cessation—A meta-analysis. Addiction 2012, 107, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Lelbach, W.K. Cirrhosis in the alcoholic and its relation to the volume of alcohol abuse. Ann. N. Y. Acad. Sci. 1975, 252, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Becker, U.; Deis, A.; Sørensen, T.; Grønbaek, M.; Borch-Johnsen, K.; Müller, C.; Schnohr, P.; Jensen, G. Prediction of risk of liver disease by alcohol intake, sex, and age: A prospective population study. Hepatology 1996, 23, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Saccoccio, G.; Costa, G.; Tiribelli, C.; Manenti, F.; Sodde, M.; Croce, L.; Sasso, F.; Pozzato, G.; The Dionysos Study Group; et al. Drinking habits as cofactors of risk for alcohol induced liver damage. Gut 1997, 41, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Mahli, A.; Hellerbrand, C. Alcohol and Obesity: A Dangerous Association for Fatty Liver Disease. Dig. Dis. 2016, 34, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Crombie, I.K.; Cunningham, K.; Irvine, L.; Williams, B.; Sniehotta, F.; Norrie, J.; Melson, A.; Jones, C.; Briggs, A.; Rice, P.; et al. Modifying Alcohol Consumption to Reduce Obesity (MACRO): Development and feasibility trial of a complex community-based intervention for men. Health Technol. Assess. 2017, 21, 1–150. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Yang, H.; Su, J.; Brenner, D.; Barrett-Connor, E.; Iloeje, U.; Chen, C. Synergism between obesity and alcohol in increasing the risk of hepatocellular carcinoma: A prospective cohort study. Am. J. Epidemiol. 2013, 177, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.L.; Morrison, D.S.; Batty, G.D.; Mitchell, R.J.; Smith, G.D. Effect of body mass index and alcohol consumption on liver disease: Analysis of data from two prospective cohort studies. BMJ 2010, 340, c1240. [Google Scholar] [CrossRef] [PubMed]
- Vimaleswaran, K.S.; Cavadino, A.; Verweij, N.; Nolte, I.; Leach, I.; Auvinen, J.; Veijola, J.; Elliott, P.; Penninx, B.; Snieder, H.; et al. Interactions between uncoupling protein 2 gene polymorphisms, obesity and alcohol intake on liver function: A large meta-analysed population-based study. Eur. J. Endocrinol. 2015, 173, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Huo, T.-I.; Wu, J.-C.; Lee, S.-D. Are alcohol, tobacco and obesity genuine risk factors for hepatocellular carcinoma? J. Hepatol. 2005, 42, 941. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Metabolism of alcohol. Clin. Liver Dis. 2005, 9, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Faller, J.; Fox, I.H. Ethanol-Induced Hyperuricemia. N. Engl. J. Med. 1982, 307, 1598–1602. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Ethanol oxidation by hepatic microsomes: Adaptive increase after ethanol feeding. Science 1968, 162, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Lasker, J.M.; Rosman, A.S.; Lieber, C.S. Induction of cytochrome P-4502E1 in the human liver by ethanol is caused by a corresponding increase in encoding messenger RNA. Hepatology 1993, 17, 236–245. [Google Scholar] [PubMed]
- Tsutsumi, M.; Lasker, J.M.; Shimizu, M.; Rosman, A.S.; Lieber, C.S. The intralobular distribution of ethanol-inducible P450IIE1 in rat and human liver. Hepatology 1989, 10, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar]
- Morgan, K.; French, S.W.; Morgan, T.R. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology 2002, 36, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.U.; Kono, H.; Isayama, F.; Kosyk, O.; Wheeler, M.; Akiyama, T.; Bleye, L.; Krausz, K.; Gonzalez, F.; Koop, D.; et al. Cytochrome P450 CYP2E1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver. Hepatology 2005, 41, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-García, D.; Hernández-Muñoz, R. Catalase increases ethanol oxidation through the purine catabolism in rat liver. Biochem. Pharmacol. 2017, 137, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Agarwal, D.P.; Goedde, H.W. Aldehyde Dehydrogenase Deficiency As Cause of Facial Flushing Reaction to Alcohol in Japanese. Lancet 1981, 318, 982. [Google Scholar] [CrossRef]
- Kwon, H.-J.; Won, Y.; Park, O.; Chang, B.; Duryee, M.; Thiele, G.; Matsumoto, A.; Singh, S.; Abdelmegeed, M.; Song, B.; et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology 2014, 60, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Wands, J.R.; de la Monte, S.M. Acetaldehyde Adducts in Alcoholic Liver Disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, O. Aldehyde-protein adducts in the liver as a result of ethanol-induced oxidative stress. Front. Biosci. 1999, 4, D506–D513. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.L.; Vaca, C.E. Development of a 32P-postlabelling method for the analysis of adducts arising through the reaction of acetaldehyde with 2′-deoxyguanosine-3′-monophosphate and DNA. Carcinogenesis 1995, 16, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J.; Theruvathu, J.A. DNA adducts from acetaldehyde: Implications for alcohol-related carcinogenesis. Alcohol 2005, 35, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Espina, N.; Lima, V.; Lieber, C.S.; Garro, A.J. In vitro and in vivo inhibitory effect of ethanol and acetaldehyde on O6-methylguanine transferase. Carcinogenesis 1988, 9, 761–766. [Google Scholar] [CrossRef]
- Lluis, J.M.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology 2003, 124, 708–724. [Google Scholar] [CrossRef] [PubMed]
- Grunnet, N.; Kondrup, J.; Dich, J. Effect of ethanol on lipid metabolism in cultured hepatocytes. Biochem. J. 1985, 228, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Grunnet, N.; Kondrup, J. The effect of ethanol on the beta-oxidation of fatty acids. Alcohol. Clin. Exp. Res. 1986, 10, 64S–68S. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, G.; Grasselli, E.; Compalati, A.; Ragazzoni, M.; Cortese, K.; Gallo, G.; Voci, A.; Vergani, L. Ethanol and fatty acids impair lipid homeostasis in an in vitro model of hepatic steatosis. Food Chem. Toxicol. 2016, 90, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome proliferator-activated receptor α (PPARα) agonist treatment reverses PPARα dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J. Biol. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Ren, W.; Li, W.; Zhao, S.; Mi, H.; Wang, R.; Zhang, Y.; Wu, W.; Nan, Y.; Yu, J. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol induced steatohepatitis in mice. Lipids Health Dis. 2011, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, Q.; Zhong, W.; Sun, X.; Zhou, Z. Hepatic Peroxisome Proliferator-Activated Receptor Gamma Signaling Contributes to Alcohol-Induced Hepatic Steatosis and Inflammation in Mice. Alcohol. Clin. Exp. Res. 2016, 40, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Song, S.; Kim, A.; Choi, Y.; Seok, J.; Kim, H.; Lee, Y.; Lee, K.; Kim, J. Suppression of PPARγ-mediated monoacylglycerol O-acyltransferase 1 expression ameliorates alcoholic hepatic steatosis. Sci. Rep. 2016, 6, 29352. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wong, K.; Giles, A.; Jiang, J.; Lee, J.; Adams, A.; Kharitonenkov, A.; Yang, Q.; Gao, B.; Guarente, L.; et al. Hepatic SIRT1 Attenuates Hepatic Steatosis and Controls Energy Balance in Mice by Inducing Fibroblast Growth Factor 21. Gastroenterology 2014, 146, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Wang, F.; Li, X.; Rogers, C.; Liang, X.; Finck, B.; Mitra, M.; Zhang, R.; Mitchell, D.; You, M. Regulation of hepatic lipin-1 by ethanol: Role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology 2012, 55, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Otsuki, Y. Ethanol-induced hepatic autophagy: Friend or foe? World J. Hepatol. 2015, 7, 1154. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Du, K.; You, M.; Ding, W.-X. Critical Role of FoxO3a in Alcohol-Induced Autophagy and Hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, X.; Zhou, R.; Yang, L.; Cederbaum, A.I. Alcohol steatosis and cytotoxicity: The role of cytochrome P4502E1 and autophagy. Free Radic. Biol. Med. 2012, 53, 1346–1357. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.-M.; Ding, Y.; Ding, W.-X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, Y.; Xiao, J.; Liu, L.; Chen, S.; Mohammadi, M.; McClain, C.; Li, X.; Feng, W. FGF21 mediates alcohol-induced adipose tissue lipolysis by activation of systemic release of catecholamine in mice. J. Lipid Res. 2015, 56, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Clugston, R.D.; Yuen, J.J.; Hu, Y.; Abumrad, N.A.; Berk, P.D.; Goldberg, I.J.; Blaner, W.S.; Huang, L.S. CD36-deficient mice are resistant to alcohol- and high-carbohydrate-induced hepatic steatosis. J. Lipid Res. 2014, 55, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.M.; Peralta, G.; Yin, X.; Ahima, R.S. Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol-fed mice. PLoS ONE 2014, 9, e97118. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-J.; Cai, Y.; Wang, H.; Altamirano, J.; Chang, B.; Bertola, A.; Odena, G.; Lu, J.; Tanaka, N.; Matsusue, K.; et al. Fat-Specific Protein 27/CIDEC Promotes Development of Alcoholic Steatohepatitis in Mice and Humans. Gastroenterology 2015, 149, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Badger, T.M.; Ronis, M.J.J.; Seitz, H.K.; Albano, E.; Ingelman-Sunberg, M.; Lieber, C.S. Alcohol Metabolism: Role in Toxicity and Carcinogenesis. Alcoholism 2003, 27, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Kohgo, Y.; Takaaki, O.; Katsuya, I.; Yasuaki, S.; Yayoi, H.; Hiroyuki, S.; Junji, K. Iron accumulation in alcoholic liver diseases. Alcohol. Clin. Exp. Res. 2005, 29, 189–193. [Google Scholar] [CrossRef]
- Arteel, G.E.; Iimuro, Y.; Yin, M.; Raleigh, J.A.; Thurman, R.G. Chronic enteral ethanol treatment causes hypoxia in rat liver tissuein vivo. Hepatology 1997, 25, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.A.; Carretero, M.V.; Rodriguez, E.N.; Mato, J.M. Regulation by hypoxia of methionine adenosyltransferase activity and gene expression in rat hepatocytes. Gastroenterology 1998, 114, 364–371. [Google Scholar] [CrossRef]
- King, A.L.; Mantena, S.K.; Andringa, K.K.; Millender-Swain, T.; Dunham-Snary, K.J.; Oliva, C.R.; Griguer, C.E.; Bailey, S.M. The methyl donor S-adenosylmethionine prevents liver hypoxia and dysregulation of mitochondrial bioenergetic function in a rat model of alcohol-induced fatty liver disease. Redox Biol. 2016, 9, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Ohhira, M.; Ohtake, T.; Matsumoto, A.; Saito, H.; Ikuta, K.; Fujimoto, Y.; Ono, M.; Toyokuni, S.; Kohgo, Y. Immunohistochemical detection of 4-hydroxy-2-nonenal-modified-protein adducts in human alcoholic liver diseases. Alcohol. Clin. Exp. Res. 1998, 22, 145–149. [Google Scholar] [CrossRef]
- Mottaran, E.; Sudheer, K.M.; Kelly, K.A.; Millender-Swain, T.; Dunham-Snary, K.J.; Oliva, C.R.; Griguer, C.E.; Bailey, S.M. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic. Biol. Med. 2002, 32, 38–45. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar]
- Ye, Q.; Lian, F.; Chavez, P.R.G.; Chung, J.; Ling, W.; Qin, H.; Seitz, H.K.; Wang, H.-D. Cytochrome P450 2E1 inhibition prevents hepatic carcinogenesis induced by diethylnitrosamine in alcohol-fed rats. Hepatobiliary Surg. Nutr. 2012, 1, 5–18. [Google Scholar] [PubMed]
- Sun, Q.; Zhang, W.; Zhong, W.; Sun, X.; Zhou, Z. Pharmacological inhibition of NOX4 ameliorates alcohol-induced liver injury in mice through improving oxidative stress and mitochondrial function. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 2912–2921. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Lin, J.; Wu, D. Sulforaphane induces Nrf2 and protects against CYP2E1-dependent binge alcohol-induced liver steatosis. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Lamlé, J.; Marhenke, S.; Borlak, J.; von Wasielewski, R.; Eriksson, C.J.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology 2008, 134, 1159–1168. [Google Scholar]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandam, S.; Duran, A. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-D.; Liu, C.; Chung, J.; Stickel, F.; Seitz, H.K.; Russell, R.M. Chronic alcohol intake reduces retinoic acid concentration and enhances AP-1 (c-Jun and c-Fos) expression in rat liver. Hepatology 1998, 28, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Clugston, R.D.; Blaner, W.S. The adverse effects of alcohol on vitamin a metabolism. Nutrients 2012, 4, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Napoli, J.L. Effects of ethanol on physiological retinoic acid levels. IUBMB Life 2011, 63, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Moriwaki, H.; Muto, Y.; Yamada, Y.; Fukutomi, Y.; Shimazaki, M.; Okuno, M.; Ninomiya, M. Reduced retinoid content in hepatocellular carcinoma with special reference to alcohol consumption. Hepatology 1991, 14, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Ishijima, N.; Kanki, K.; Shimizu, H.; Shiota, G. Activation of AMP-activated protein kinase by retinoic acid sensitizes hepatocellular carcinoma cells to apoptosis induced by sorafenib. Cancer Sci. 2015, 106, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.P.; Toranelli, M.; Sasse, D. Distribution of alcohol dehydrogenase isoenzymes in the human liver acinus. Histochem. Cell Biol. 1999, 111, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Borràs, E.; Coutelle, C.; Rosell, A.; Fernandez-Muixi, F.; Broch, M.; Crosas, B.; Hjelmqvist, L.; Lorenzo, A.; Gutierrez, C.; Santos, M.; et al. Genetic polymorphism of alcohol dehydrogenase in europeans: TheADH2*2 allele decreases the risk for alcoholism and is associated withADH3*1. Hepatology 2000, 31, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Xiao, L.; Zhang, Y.; Xian, J.; Jiang, J.; Zong, W.; Huang, Z.; Yang, Y. Genetic polymorphisms of ALDH2 and ADH2 are not associated with risk of hepatocellular carcinoma among East Asians. Tumor Biol. 2012, 33, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Chen, J.; Chen, L.; Histen, G.; Lin, Z.; Gross, S.; Hixon, J.; Chen, Y.; Kung, C.; Chen, Y.; et al. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, 9088–9093. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Hara, M.; Higaki, Y.; Ichiba, M.; Horita, M.; Mizuta, T.; Eguchi, Y.; Yasutake, T.; Ozaki, I.; Yamamoto, K.; et al. Influence of alcohol consumption and gene polymorphisms of ADH2 and ALDH2 on hepatocellular carcinoma in a Japanese population. Int. J. Cancer 2006, 118, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhao, Z.; Sun, M.; Luo, J.; Xiao, Y. ALDH2 gene polymorphism in different types of cancers and its clinical significance. Life Sci. 2016, 147, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, H.; Pan, C.; Shen, J.; Liang, Y. CYP2E1 PstI/RsaI polymorphism and interaction with alcohol consumption in hepatocellular carcinoma susceptibility: Evidence from 1661 cases and 2317 controls. Tumor Biol. 2012, 33, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Fabris, C.; Toniutto, P.; Falletti, E.; Fontanini, E.; Cussigh, A.; Bitetto, D.; Fornasiere, E.; Fumolo, E.; Avellini, C.; Minisini, R.; et al. MTHFR C677T Polymorphism and Risk of HCC in Patients With Liver Cirrhosis: Role of Male Gender and Alcohol Consumption. Alcohol. Clin. Exp. Res. 2009, 33, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Kienesberger, P.C.; Oberer, M.; Lass, A.; Zechner, R. Mammalian patatin domain containing proteins: A family with diverse lipolytic activities involved in multiple biological functions. J. Lipid Res. 2008, 50, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Falleti, E.; Cussigh, A.; Cmet, S.; Fabris, C.; Toniutto, P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig. Liver Dis. 2016, 48, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Guyot, E.; Sutton, A.; Rufat, P.; Languillier, C.; Mansouri, A.; Moreau, R.; Ganne-Carrié, N.; Beaugrand, M.; Charnaux, N.; Trinchet, J.C.; et al. PNPLA3 rs738409, hepatocellular carcinoma occurrence and risk model prediction in patients with cirrhosis. J. Hepatol. 2013, 58, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Nischalke, H.D.; Lutz, P.; Krämer, B.; Söhne, J.; Müller, T.; Rosendahl, J.; Fischer, J.; Berg, T.; Hittatiya, K.; Fischer, H.-P.; et al. A common polymorphism in the NCAN gene is associated with hepatocellular carcinoma in alcoholic liver disease. J. Hepatol. 2014, 61, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, Y.A.; Cubero, F.J.; Hu, W.; Hao, F.; Haas, U.; Ramadori, P.; Gassler, N.; Hoss, M.; Strnad, P.; Zimmermann, H.M.; et al. Enhanced expression of c-myc in hepatocytes promotes initiation and progression of alcoholic liver disease. J. Hepatol. 2016, 64, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Hlady, R.A.; Tiedemann, R.L.; Puszyk, W.; Zendejas, I.; Roberts, L.R.; Choi, J.-H.; Liu, C.; Robertson1et, K.D. Epigenetic signatures of alcohol abuse and hepatitis infection during human hepatocarcinogenesis. Oncotarget 2014, 5, 9425–9443. [Google Scholar] [CrossRef] [PubMed]
- Tsuchishima, M.; George, J.; Shiroeda, H.; Arisawa, T.; Takegami, T.; Tsutsumi, M. Chronic Ingestion of Ethanol Induces Hepatocellular Carcinoma in Mice Without Additional Hepatic Insult. Dig. Dis. Sci. 2013, 58, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Shepard, B.D.; Tuma, D.J.; Tuma, P.L. Lysine acetylation induced by chronic ethanol consumption impairs dynamin-mediated clathrin-coated vesicle release. Hepatology 2012, 55, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kwon, O.K.; Ki, S.H.; Jeong, T.C.; Lee, S. Characterization of novel mechanisms for steatosis from global protein hyperacetylation in ethanol-induced mouse hepatocytes. Biochem. Biophys. Res. Commun. 2015, 463, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Shearn, C.T.; Smathers, R.L.; Backos, D.S.; Reigan, P.; Orlicky, D.J.; Petersen, D.R. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic. Biol. Med. 2013, 65, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Yeon, J.; Califano, S.; Xu, J.; Wands, J.R.; de la Monte, S.M. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology 2003, 38, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Peyrou, M.; Bourgoin, L.; Foti, M. PTEN in liver diseases and cancer. World J. Gastroenterol. 2010, 16, 4627–4633. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Lazaro, R.G.; Wang, J.; Kim, J.; Povero, D.; Williams, B.; Ho, S.B.; Stärkel, P.; Schnabl, B.; Ohno-Machado, L.; et al. Extracellular vesicles released by hepatocytes from gastric infusion model of alcoholic liver disease contain a MicroRNA barcode that can be detected in blood. Hepatology 2017, 65, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Satishchandran, A.; Szabo, G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and miR-122-mediated HIF-1α activation. Sci. Rep. 2016, 6, 21340. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, Q. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Francis, H.; McDaniel, K.; Han, Y.; Liu, X.; Kennedy, L.; Yang, F.; McCarra, J.; Zhou, T.; Glaser, S.; Venter, J.; et al. Regulation of the Extrinsic Apoptotic Pathway by MicroRNA-21 in Alcoholic Liver Injury. J. Biol. Chem. 2014, 289, 27526–27539. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, K.; Herrera, L.; Zhou, T.; Francis, H.; Han, Y.; Levine, P.; Lin, E.; Glaser, S.; Alpini, G.; Meng, F. The functional role of microRNAs in alcoholic liver injury. J. Cell. Mol. Med. 2014, 18, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fan, Z.; Tang, Y.; Ke, Z. The Resveratrol Attenuates Ethanol-Induced Hepatocyte Apoptosis Via Inhibiting ER-Related Caspase-12 Activation and PDE Activity In Vitro. Alcohol. Clin. Exp. Res. 2014, 38, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-R.; Zhu, G.-Q.; Shi, K.-Q.; Braddock, M.; Zheng, M.-H. Autophagy in ethanol-exposed liver disease. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1031–1037. [Google Scholar]
- Longato, L.; Ripp, K.; Setshedi, M.; Dostalek, M.; Akhlaghi, F.; Branda, M.; Wands, J.R.; de la Monte, S.M. Insulin Resistance, Ceramide Accumulation, and Endoplasmic Reticulum Stress in Human Chronic Alcohol-Related Liver Disease. Oxid. Med. Cell. Longev. 2012, 2012, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Czaja, M.J. Regulation of the effects of CYP2E1-induced oxidative stress by JNK signaling. Redox Biol. 2014, 3, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Higuchi, H.; Miura, S.; Azuma, T.; Inokuchi, S.; Saito, H.; Kato, S.; Ishii, H. Bax interacts with the voltage-dependent anion channel and mediates ethanol-induced apoptosis in rat hepatocytes. AJP Gastrointest. Liver Physiol. 2004, 287, G695–G705. [Google Scholar]
- Hao, F.; Cubero, F.J.; Ramadori, P.; Lijun, L.; Haas, U.; Lambertz, D.; Sonntag, R.; Bangen, J.M.; Gassler, N.; Hoss, M.; et al. Inhibition of Caspase-8 does not protect from alcohol-induced liver apoptosis but alleviates alcoholic hepatic steatosis in mice. Cell Death Dis. 2017, in press. [Google Scholar]
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and Necrosis in the Liver. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 977–1010. [Google Scholar]
- Wang, S.; Ni, H.M.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Nawabi, A.; Komatsu, M.; Huang, H.; Ding, W.X. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016, 7, 17681–17698. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, S.; McMullen, M.R.; Pisano, S.G.; Liu, X.; Nagy, L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013, 57, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Schneider, K.; Finnigan, R.; Maloney, E.; Wells, M.; Clemens, D. The Involvement of Acetaldehyde in Ethanol-Induced Cell Cycle Impairment. Biomolecules 2016, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Koteish, A.; Yang, S.; Lin, H.; Huang, J.; Diehl, A.M. Ethanol induces redox-sensitive cell-cycle inhibitors and inhibits liver regeneration after partial hepatectomy. Alcohol. Clin. Exp. Res. 2002, 26, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A.; Pietrosi, G.; Hoare, M.; Jupp, J.; Marshall, A.; Verrill, C.; Davies, S.; Bateman, A.; Sheron, N.; Allison, M.; et al. Hepatocyte Expression of the Senescence Marker p21 Is Linked to Fibrosis and an Adverse Liver-Related Outcome in Alcohol-Related Liver Disease. PLoS ONE 2013, 8, e72904. [Google Scholar] [CrossRef] [PubMed]
- Isayama, F.; Froh, M.; Yin, M.; Conzelmann, L.O.; Milton, R.J.; McKim, S.E.; Wheeler, M.D. TNF alpha-induced ras activation due to ethanol promotes hepatocyte proliferation independently of liver injury in the mouse. Hepatology 2004, 39, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Koteish, A.; Lin, H.; Huang, J.; Roskams, T.; Dawson, V.; Diehl, A.M. Oval cells compensate for damage and replicative senescence of mature hepatocytes in mice with fatty liver disease. Hepatology 2004, 39, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ferrere, G.; Wrzosek, L.; Cailleux, F.; Turpin, W.; Purchois, V.; Spatz, M.; Ciocan, D.; Rainteau, D.; Humbert, L.; Hugot, C. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 2017, 66, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Stärkel, P.; van Pijkeren, J.P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of Saturated Long-Chain Fatty Acids Maintains Intestinal Eubiosis and Reduces Ethanol-induced Liver Injury in Mice. Gastroenterology 2015, 148, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.B.U.; Chen, C.L.; Liu, J.C.; Feldman, D.E.; Sher, L.S.; French, S.; DiNorcia, J.; French, S.W.; Naini, B.V.; Junrungsee, S.; et al. TLR4 Signaling via NANOG Cooperates With STAT3 to Activate Twist1 and Promote Formation of Tumor-Initiating Stem-Like Cells in Livers of Mice. Gastroenterology 2016, 150, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Kumar, D.B.U.; Punj, V.; Xu, J.; Sher, L.S.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.-W.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.E. The Role of Innate Immunity in Alcoholic Liver Disease. Alcohol Res. 2015, 37, 237–250. [Google Scholar] [PubMed]
- Im, H.-J.; Kim, H.J.; Lee, J.S.; Kim, H.S.; Cho, J.H.; Jo, I.J.; Park, S.J.; Son, C.G. A Preclinical Model of Chronic Alcohol Consumption Reveals Increased Metastatic Seeding of Colon Cancer Cells in the Liver. Cancer Res. 2016, 76, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Wang, X.; Sun, C.; Zheng, X.; Wei, H.; Tian, Z.; Sun, R. Chronic Alcohol Consumption Promotes Diethylnitrosamine-Induced Hepatocarcinogenesis via Immune Disturbances. Sci. Rep. 2017, 7, 2567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhong, W.; Sun, X.; Sun, Q.; Tan, X.; Li, Q.; Sun, X.; Zhou, Z. Visceral White Adipose Tissue is Susceptible to Alcohol-Induced Lipodystrophy in Rats: Role of Acetaldehyde. Alcohol. Clin. Exp. Res. 2015, 39, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-I.; Lee, M.-K. Coordinated regulation of scopoletin at adipose tissue–liver axis improved alcohol-induced lipid dysmetabolism and inflammation in rats. Toxicol. Lett. 2015, 237, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.L.; Lang, C.H. Dysregulation of skeletal muscle protein metabolism by alcohol. Am. J. Physiol. 2015, 308, E699–E712. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; LeCapitaine, N.; Berner, P.; Vande Stouwe, C.; Mussell, J.C.; Allerton, T.; Primeaux, S.D.; Dufour, J.; Nelson, S.; Bagby, G.J.; et al. Chronic binge alcohol consumption alters myogenic gene expression and reduces in vitro myogenic differentiation potential of myoblasts from rhesus macaques. AJP Regul. Integr. Comp. Physiol. 2014, 306, R837–R844. [Google Scholar] [CrossRef] [PubMed]
- Thapaliya, S.; Runkana, A.; McMullen, M.R.; Nagy, L.E.; McDonald, C.; Naga Prasad, S.V.; Dasarathy, S. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy 2014, 10, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.A.; Le, T.; Tong, M.; Silbermann, E.; Gundogan, F.; de la Monte, S.M. Impaired Insulin/IGF Signaling in Experimental Alcohol-Related Myopathy. Nutrients 2012, 4, 1058–1075. [Google Scholar] [CrossRef] [PubMed]
- Merli, M.; Giusto, M.; Molfino, A.; Bonetto, A.; Rossi, M.; Ginanni Corradini, S.; Baccino, F.M.; Rossi Fanelli, F.; Costelli, P.; Muscaritoli, M. MuRF-1 and p-GSK3α expression in muscle atrophy of cirrhosis. Liver Int. 2013, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.S.; Guy, C.D.; Machado, M.V.; Wank, A.; Kadiyala, V.; Michelotti, G.; Choi, S.; Swiderska-Syn, M.; Karaca, G.; Pereira, T.A.; et al. Alcohol Activates the Hedgehog Pathway and Induces Related Procarcinogenic Processes in the Alcohol-Preferring Rat Model of Hepatocarcinogenesis. Alcohol. Clin. Exp. Res. 2014, 38, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Doyle, C.J.; McKillop, I.H.; Wentz, S.C.; Brandon-Warner, E.; Matos, J.M.; Sandrasegaran, K.; Saxena, R.; Hennig, M.E.; Wu, H.; et al. Alcohol Induces Liver Neoplasia in a Novel Alcohol-Preferring Rat Model. Alcohol. Clin. Exp. Res. 2011, 35, 2216–2225. [Google Scholar] [CrossRef] [PubMed]
- Gäbele, E.; Dostert, K.; Dorn, C.; Patsenker, E.; Stickel, F.; Hellerbrand, C. A New Model of Interactive Effects of Alcohol and High-Fat Diet on Hepatic Fibrosis. Alcohol. Clin. Exp. Res. 2011, 35, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Baker, S.S.; Moylan, C.A.; Abdelmalek, M.F.; Guy, C.D.; Zamboni, F.; Wu, D.; Lin, W.; Liu, W.; Baker, R.D.; et al. Systematic transcriptome analysis reveals elevated expression of alcohol-metabolizing genes in NAFLD livers. J. Pathol. 2016, 238, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Xu, M.J.; Zhou, Z.; Cai, Y.; Li, M.; Wang, W.; Feng, D.; Bertola, A.; Wang, H.; Kunos, G.; et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology 2015, 62, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology 2003, 125, 1818–1833. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chen, T.; Prough, R.A.; Cave, M.C.; McClain, C.J. Chronic alcohol consumption causes liver injury in high-fructose-fed male mice through enhanced hepatic inflammatory response. Alcohol. Clin. Exp. Res. 2016, 40, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Swan, R.Z.; Walling, T.L.; Iannitti, D.A.; McKillop, I.H.; Sindram, D. Obesity, but not ethanol, promotes tumor incidence and progression in a mouse model of hepatocellular carcinoma in vivo. Surg. Endosc. 2013, 27, 2782–2791. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Swan, R.Z.; Iannitti, D.A.; McKillop, I.H.; Sindram, D. Diet-induced obesity and ethanol impair progression of hepatocellular carcinoma in a mouse mesenteric vein injection model. Surg. Endosc. 2013, 27, 246–255. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Identified SNPs | Protein Functionality | ALD/HCC Association |
|---|---|---|---|
| Alcohol dehydrogenase (ADH) | ADH1B*2 (rs1229984) ADH1B*3 (rs2066702) ADH1C*1 ADH2*1 ADH2*2 (rs1229984) ADH3*2 | Increased enzymatic activity | Associated with gastric cancers, but unknown association with ALD/HCC [74,75] |
| Aldehyde dehydrogenase (ALDH) | ALDH2*2 (rs671) | Reduced enzymatic activity | Correlation with HCC development in combination with ADH2*2 in a Japanese cohort [79] |
| Cytochrome P450 Family 2 Subfamily E Member 1 (CYP2E1) | PstI/RsaI (rs2031920/rs3813867) | Increased enzymatic activity | Association with HCC development in combination with alcohol consumption [80] |
| Methylenetetrahydrofolate reductase (MTHFR) | C677T (rs1801133) | Reduced enzymatic activity | Correlation with HCC in a population of alcohol-related cirrhotic patients [81] |
| Patatin-like phospholipase 3 domain containing 3 (PNPLA3) | I148M (rs738409) | Loss of enzymatic function | Important association with ALD progression and HCC development in alcohol-related cirrhotic patients [83], [84] |
| Transmembrane 6 superfamily member 2 (TM6SF2) | E167K (rs58542926) | Loss of expression and Function | Associated with HCC development in ALD setting in combination with I148M [83] |
| Neurocan (NCAN) | NCAN (rs2228603) | Altered functionality, unclear mechanisms | Association with HCC development in patients with ALD etiology [85] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramadori, P.; Cubero, F.J.; Liedtke, C.; Trautwein, C.; Nevzorova, Y.A. Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame. Cancers 2017, 9, 130. https://doi.org/10.3390/cancers9100130
Ramadori P, Cubero FJ, Liedtke C, Trautwein C, Nevzorova YA. Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame. Cancers. 2017; 9(10):130. https://doi.org/10.3390/cancers9100130
Chicago/Turabian StyleRamadori, Pierluigi, Francisco Javier Cubero, Christian Liedtke, Christian Trautwein, and Yulia A. Nevzorova. 2017. "Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame" Cancers 9, no. 10: 130. https://doi.org/10.3390/cancers9100130
APA StyleRamadori, P., Cubero, F. J., Liedtke, C., Trautwein, C., & Nevzorova, Y. A. (2017). Alcohol and Hepatocellular Carcinoma: Adding Fuel to the Flame. Cancers, 9(10), 130. https://doi.org/10.3390/cancers9100130