
NORE1A Regulates MDM2 Via β-TrCP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
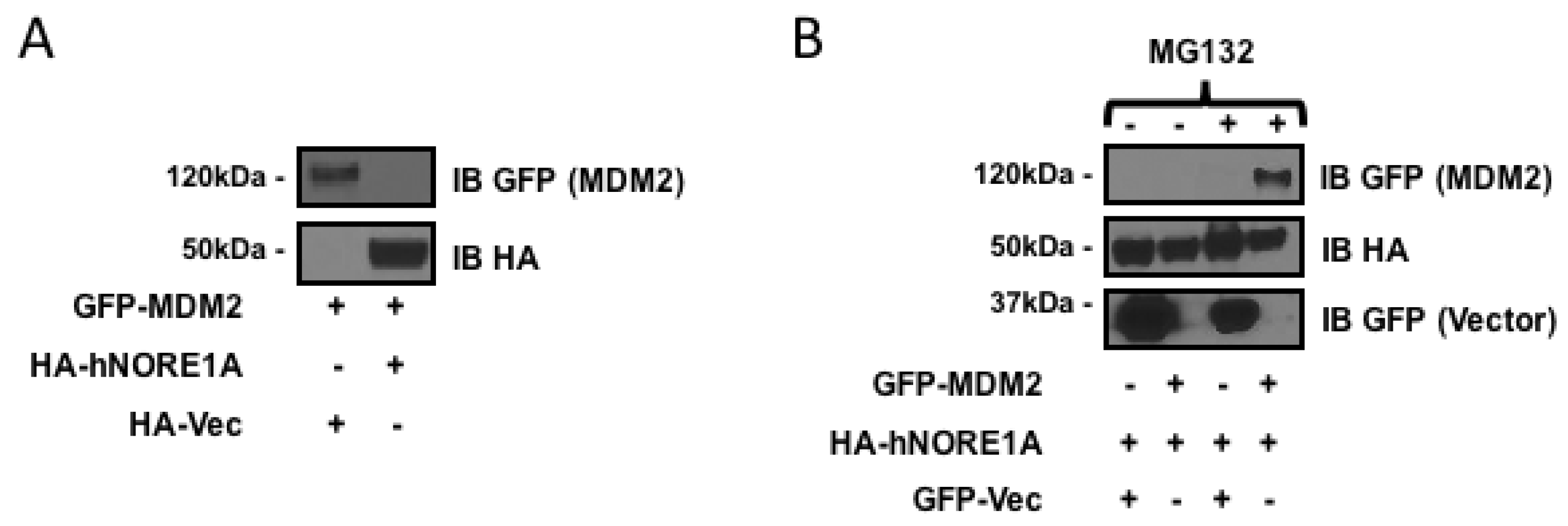
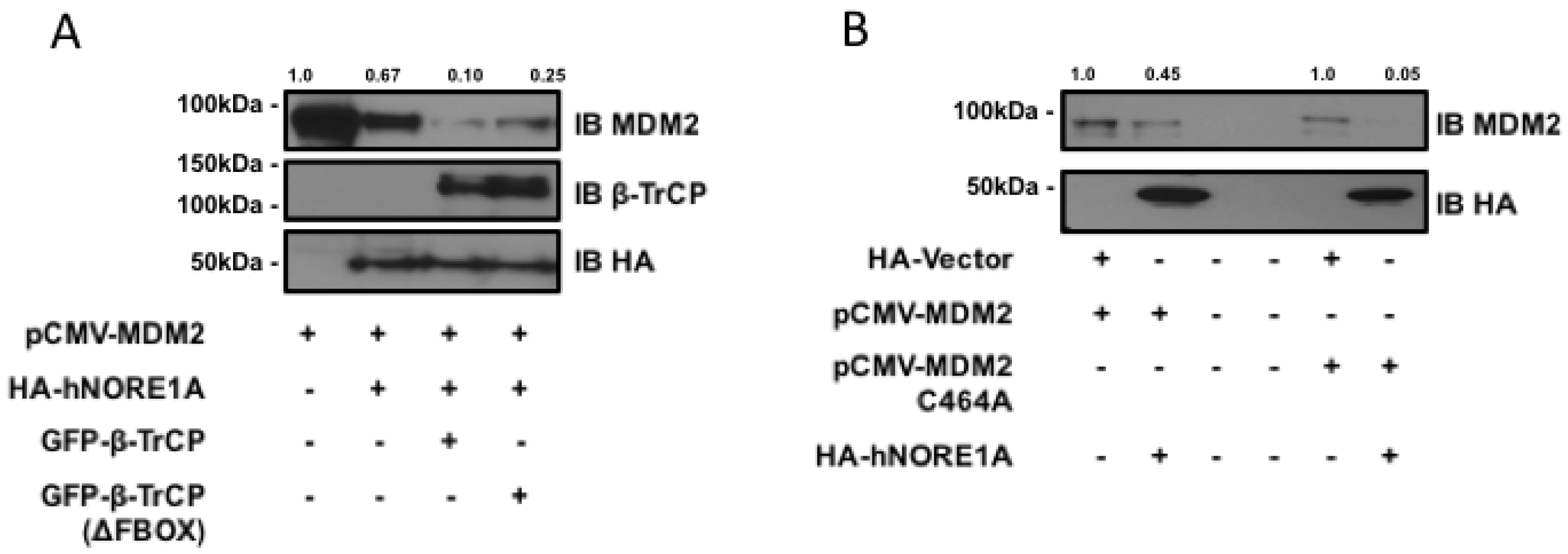
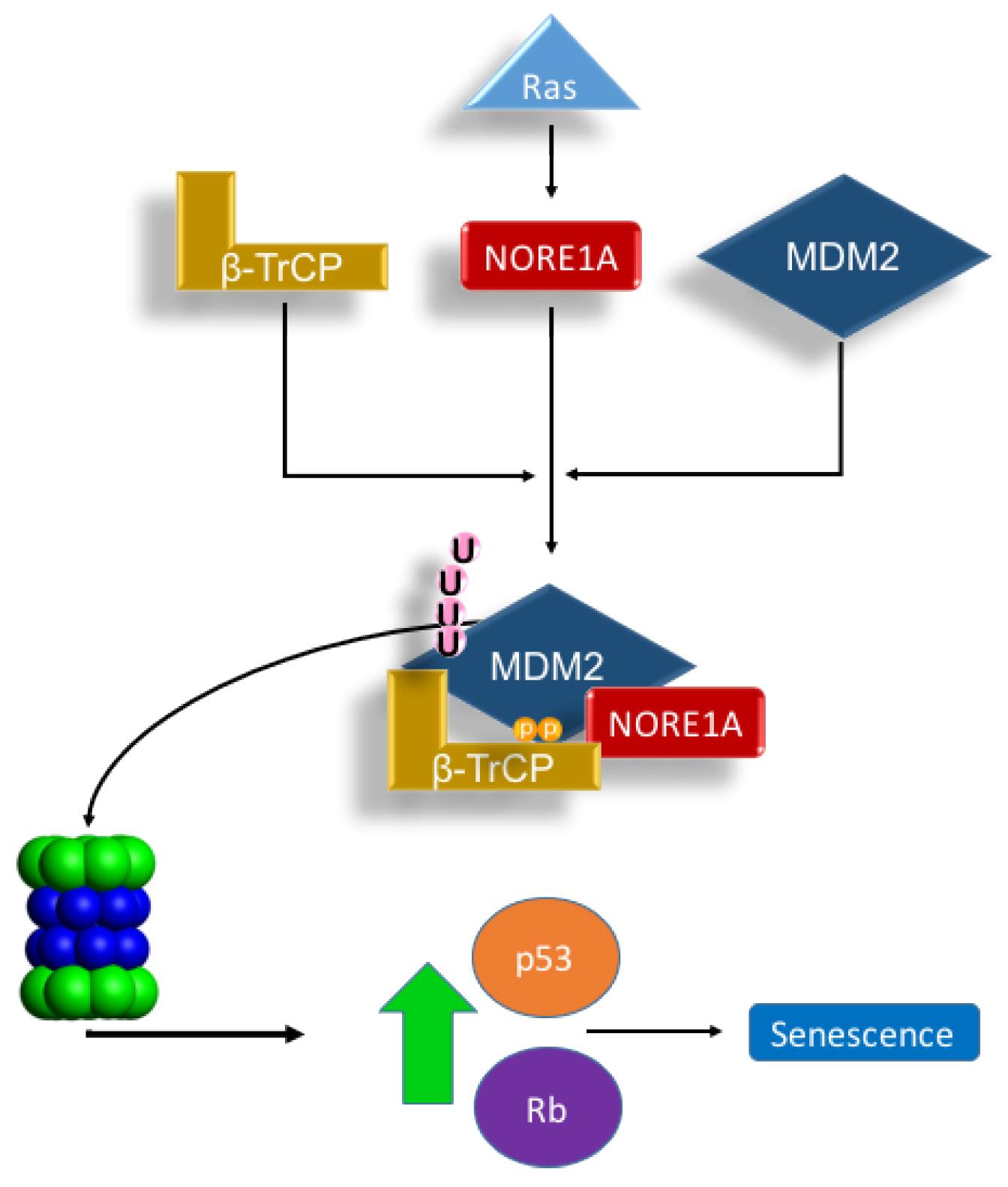
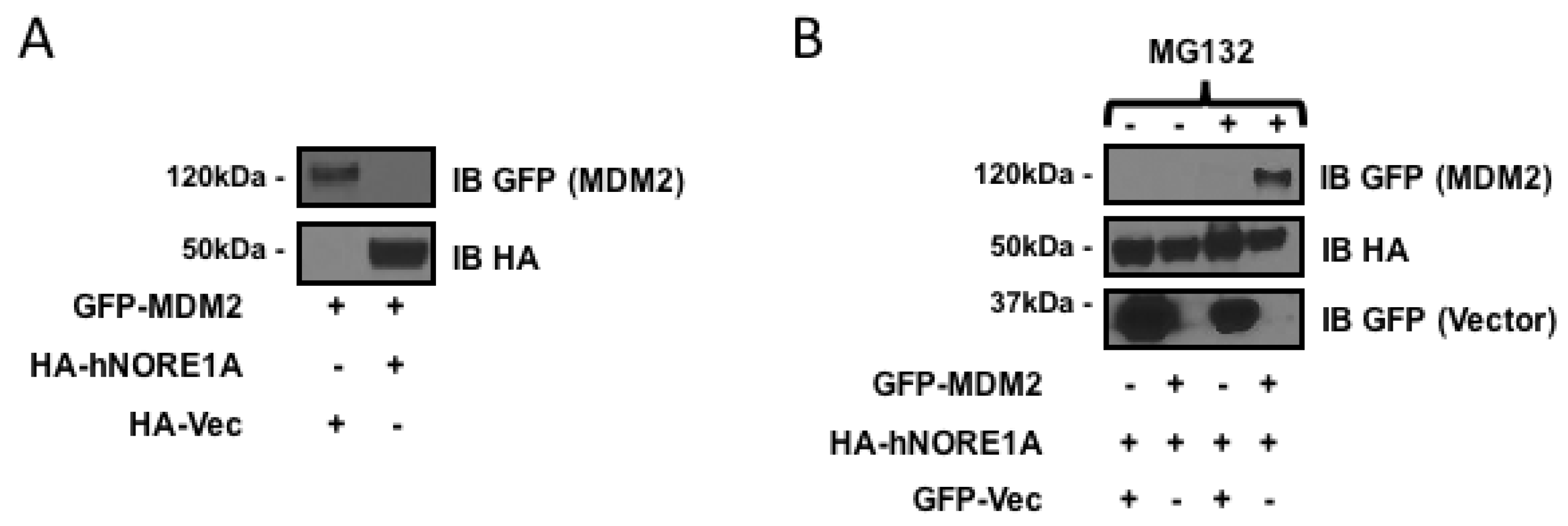
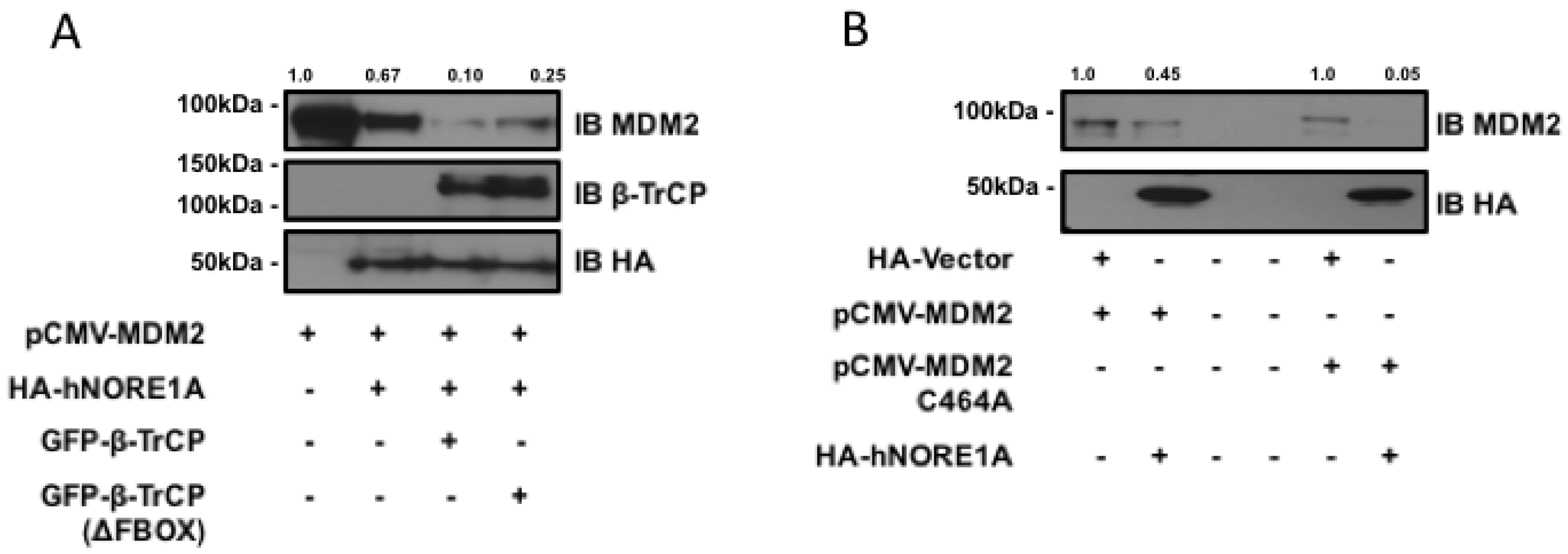
2.1. NORE1A Suppresses MDM2 Protein Expression via β-TrCP
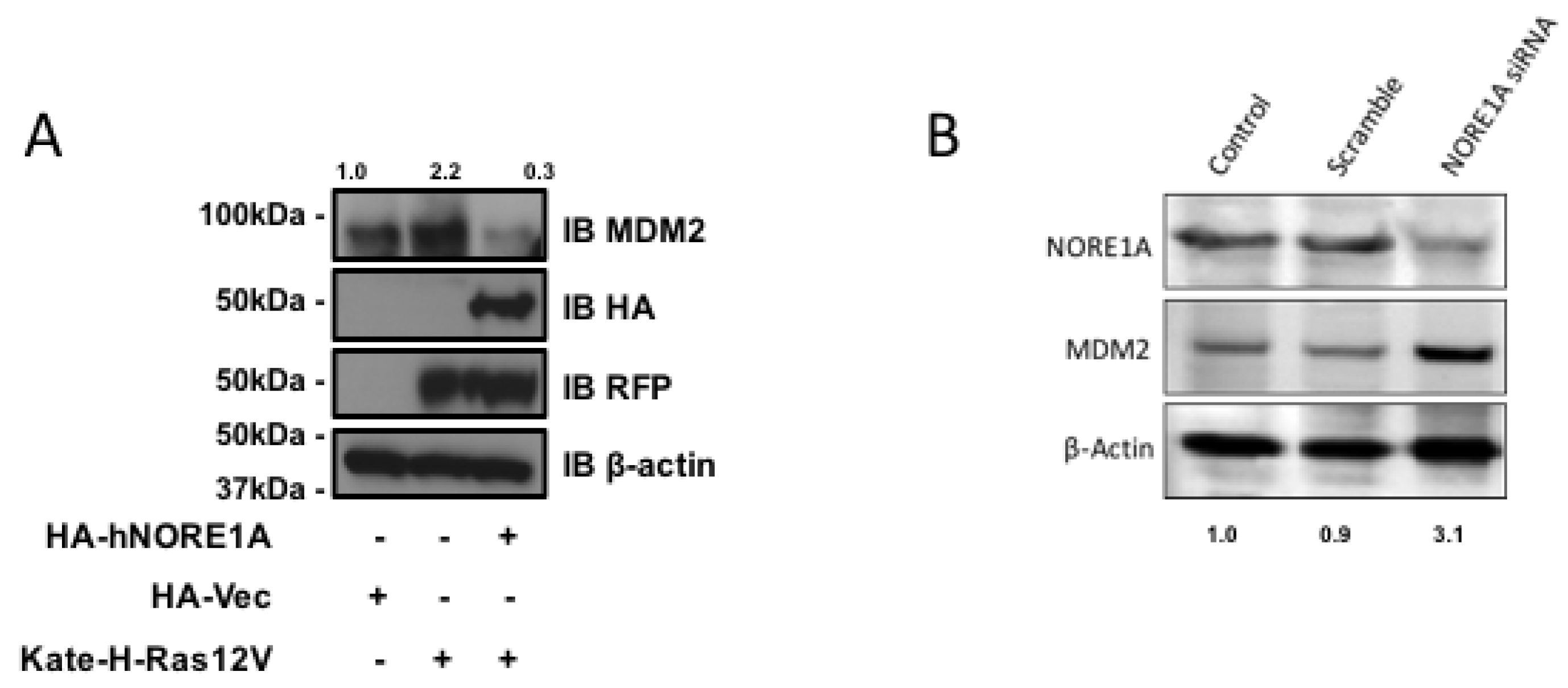
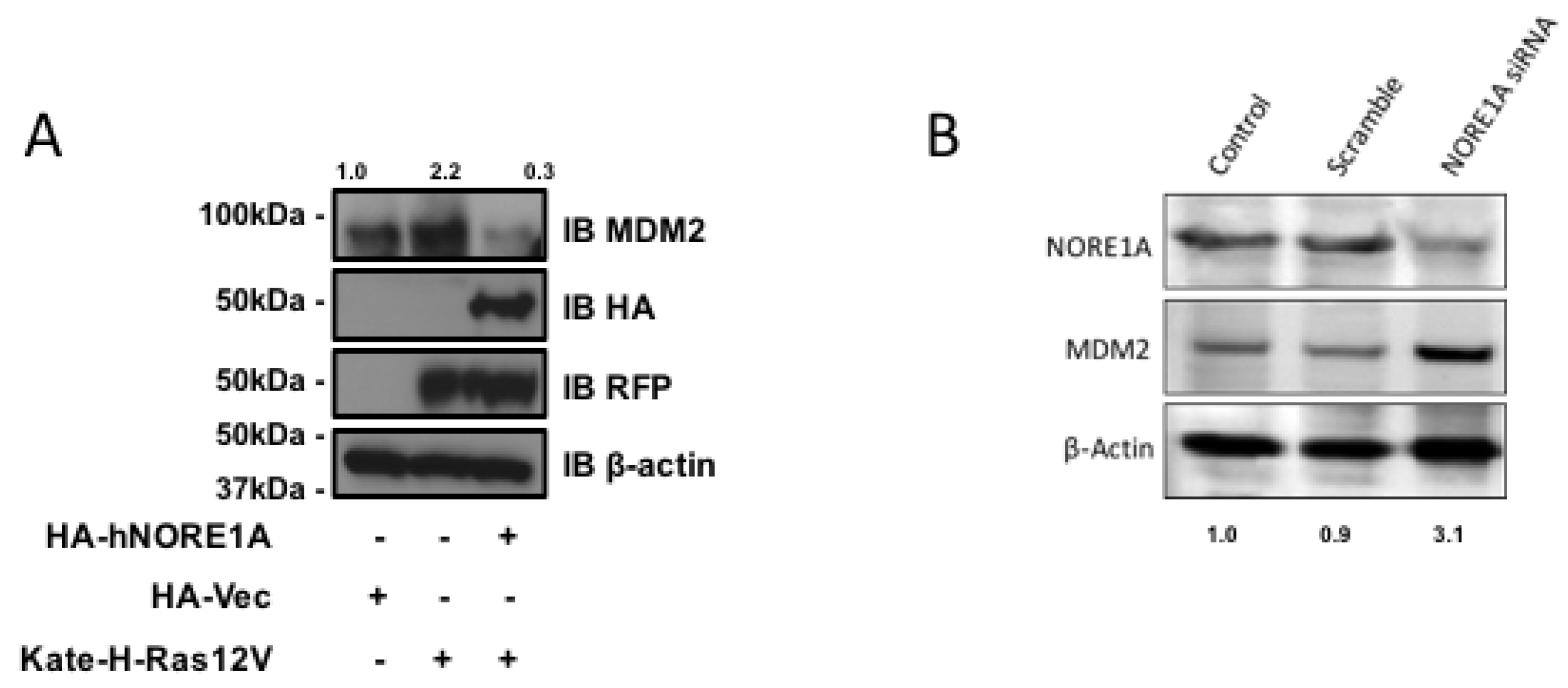
2.2. NORE1A Levels Determine if RAS Has a Negative or Positive Effect on MDM2
2.3. Inactivation of NORE1A Enhances MDM2 Expression
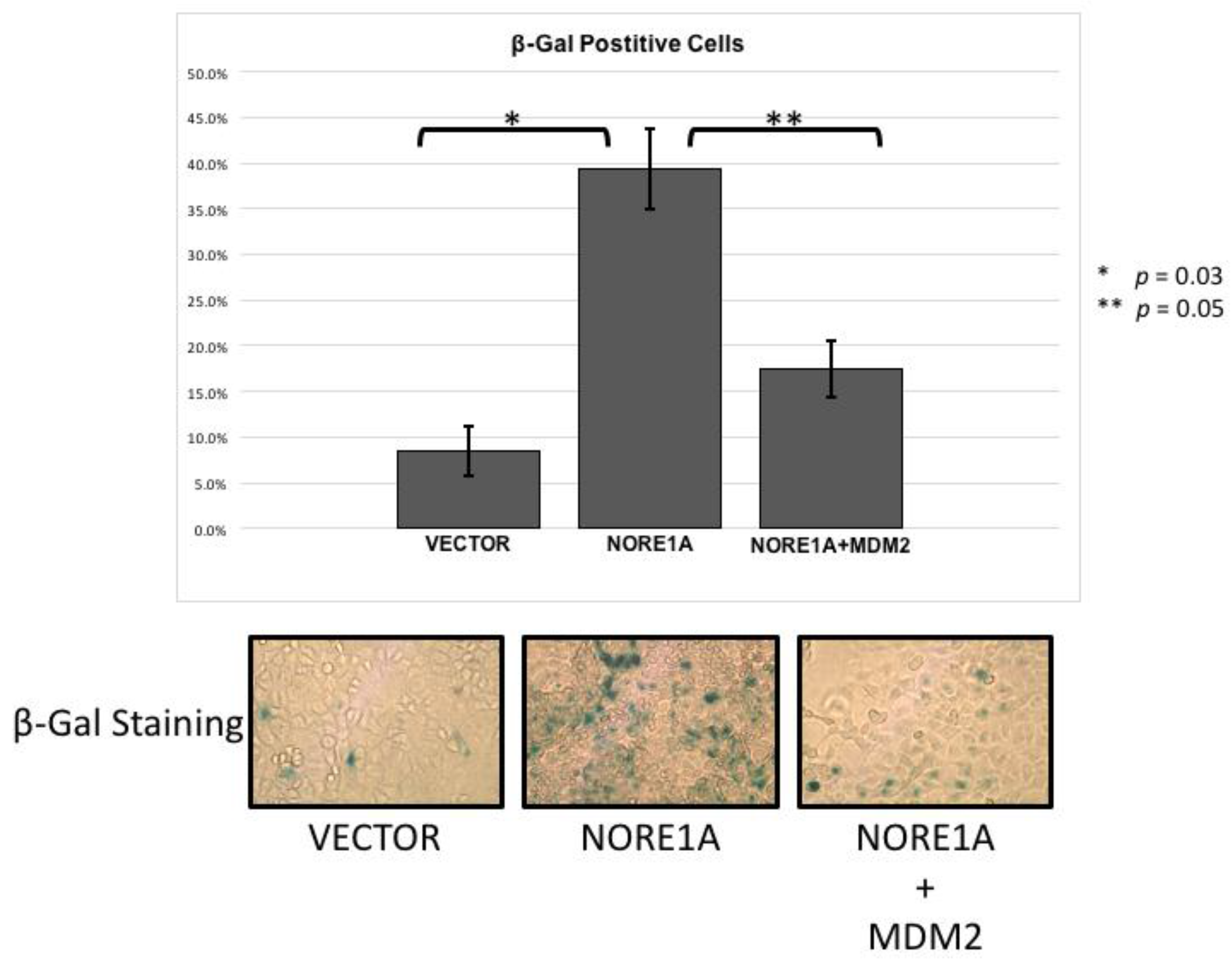
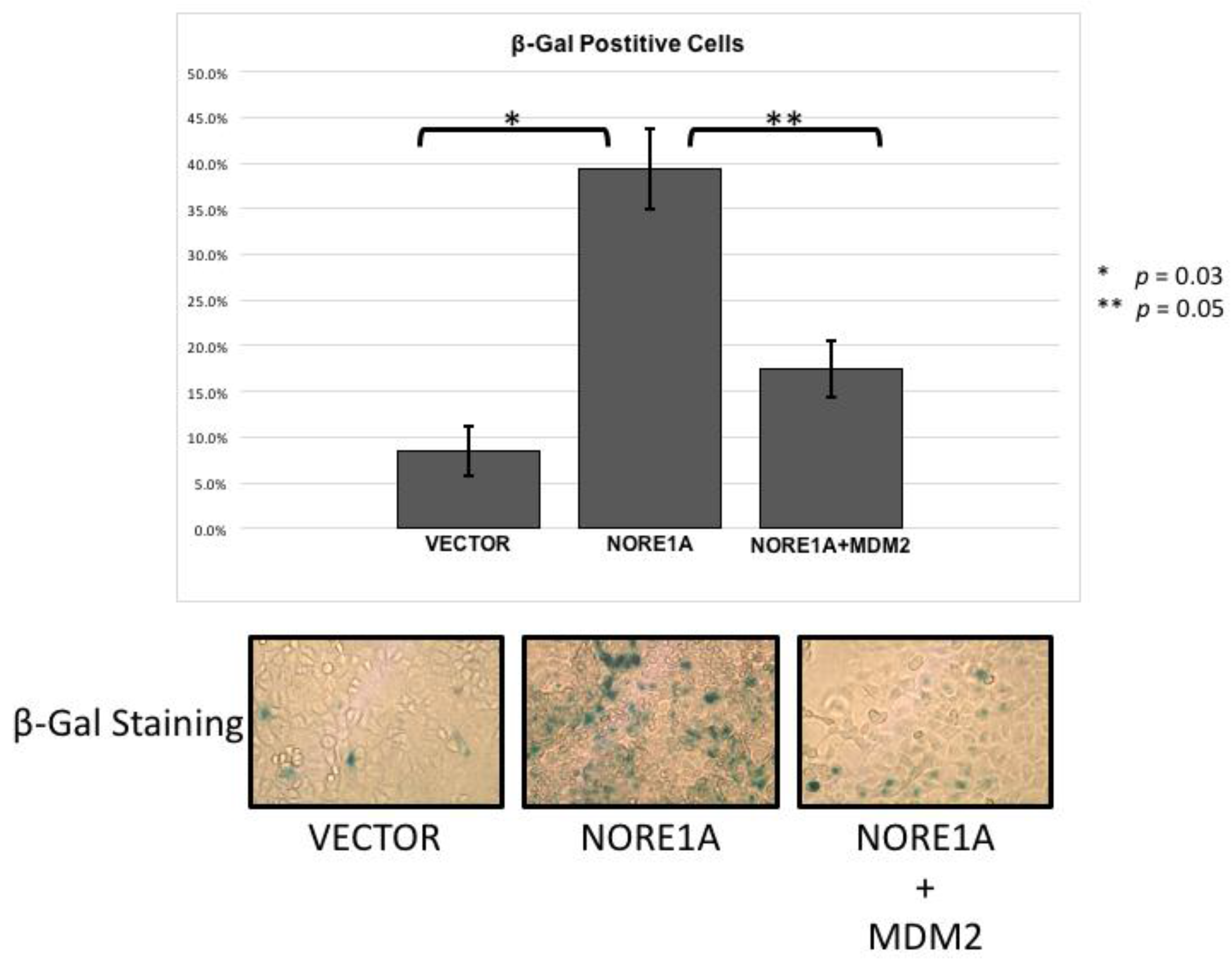
2.4. MDM2 Suppresses NORE1A Mediated Senescence
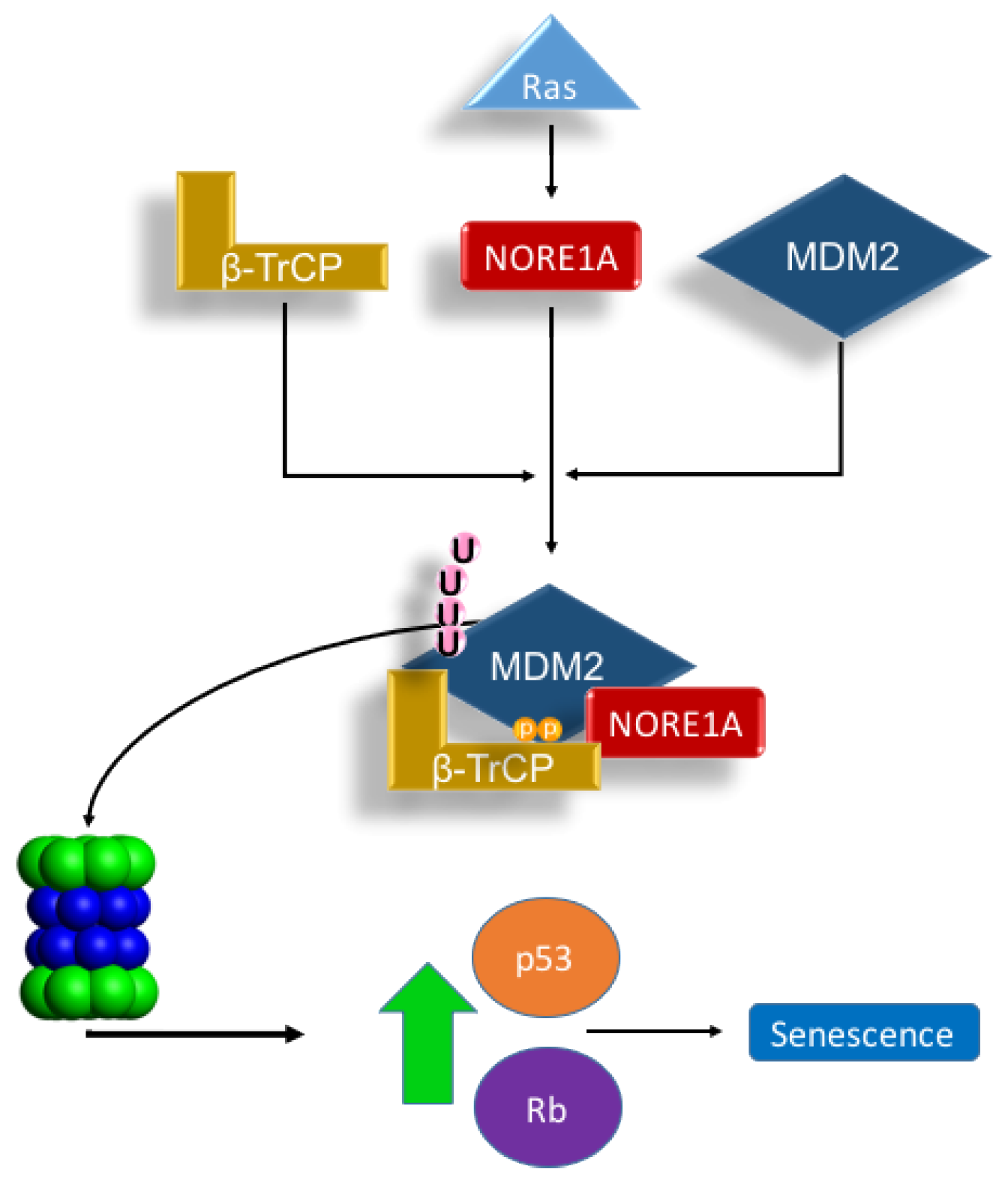
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids
4.3. Reagents, Western Blotting, and Antibodies
4.4. Senescence Assay
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Urso, L.; Calabrese, F.; Favaretto, A.; Conte, P.; Pasello, G. Critical review about MDM2 in cancer: Possible role in malignant mesothelioma and implications for treatment. Crit. Rev. Oncol. Hematol. 2016, 97, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Sasaki, M.; Maki, C.G. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J. Biol. Chem. 2007, 282, 14616–14625. [Google Scholar] [CrossRef] [PubMed]
- Levav-Cohen, Y.; Goldberg, Z.; Tan, K.H.; Alsheich-Bartok, O.; Zuckerman, V.; Haupt, S.; Haupt, Y. The p53-MDM2 loop: A critical juncture of stress response. Subcell. Biochem. 2014, 85, 161–186. [Google Scholar] [PubMed]
- Meng, X.; Franklin, D.A.; Dong, J.; Zhang, Y. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014, 74, 7161–7167. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; Cichowski, K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. The senescent side of tumor suppression. Cell cycle 2005, 4, 1722–1724. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.; Phillips, A.; Darley, M.; Blaydes, J.P. MEK-ERK signaling controls Hdm2 oncoprotein expression by regulating hdm2 mRNA export to the cytoplasm. J. Biol. Chem. 2005, 280, 16651–16658. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.; Biederer, C.; Woods, D.; Shifman, O.; Shirasawa, S.; Sasazuki, T.; McMahon, M.; Oren, M.; McCormick, F. Opposing effects of Ras on p53: Transcriptional activation of mdm2 and induction of p19ARF. Cell 2000, 103, 321–330. [Google Scholar] [CrossRef]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell. Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.D.; Martinez, A.; Ellis, C.A.; Vallecorsa, T.; Clark, G.J. The pro-apoptotic Ras effector Nore1 may serve as a Ras-regulated tumor suppressor in the lung. J. Biol. Chem. 2003, 278, 21938–21943. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Calvisi, D.F.; Barnoud, T.; Clark, J.; Schmidt, M.L.; Vos, M.D.; Clark, G.J. NORE1A is a Ras senescence effector that controls the apoptotic/senescent balance of p53 via HIPK2. J. Cell Biol. 2015, 208, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Donninger, H.; Vos, M.D.; Birrer, M.J.; Gordon, L.; Leaner, V.; Clark, G.J. NORE1A tumor suppressor candidate modulates p21CIP1 via p53. Cancer Res. 2009, 69, 4629–4637. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.L.; Donninger, H.; Clark, G.J. Ras regulates SCF(β-TrCP) protein activity and specificity via its effector protein NORE1A. J. Biol. Chem. 2014, 289, 31102–31110. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Park, S.J.; Sung, K.S.; Park, J.; Lee, S.B.; Park, S.Y.; Lee, H.J.; Ahn, J.W.; Choi, S.J.; Lee, S.G.; et al. Mdm2 associates with Ras effector NORE1 to induce the degradation of oncoprotein HIPK1. EMBO Rep. 2012, 13, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Tseng, A.; Gao, D.; Zhai, B.; Zhang, Q.; Shaik, S.; Wan, L.; Ang, X.L.; Mock, C.; Yin, H.; et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell 2010, 18, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Sdek, P.; Ying, H.; Chang, D.L.; Qiu, W.; Zheng, H.; Touitou, R.; Allday, M.J.; Xiao, Z.X. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 2005, 20, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.D.; Tsai, K.Y.; Jacks, T. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol. 2000, 2, 563–568. [Google Scholar] [PubMed]
- Song, M.S.; Song, S.J.; Kim, S.Y.; Oh, H.J.; Lim, D.S. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 2008, 27, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Thaler, S.; Hahnel, P.S.; Schad, A.; Dammann, R.; Schuler, M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer Res. 2009, 69, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, A.W.; Ram, R.; Shivakumar, L.; Gao, B.; Minna, J.D.; White, M.A. The RASSF1A tumor suppressor restrains anaphase-promoting complex/cyclosome activity during the G1/S phase transition to promote cell cycle progression in human epithelial cells. Mol. Cell. Biol. 2008, 28, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- Barnoud, T.; Donninger, H.; Clark, G.J. Ras regulates Rb via NORE1A. J. Biol. Chem. 2016, 291, 3114–3123. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, M.L.; Calvisi, D.F.; Clark, G.J. NORE1A Regulates MDM2 Via β-TrCP. Cancers 2016, 8, 39. https://doi.org/10.3390/cancers8040039
Schmidt ML, Calvisi DF, Clark GJ. NORE1A Regulates MDM2 Via β-TrCP. Cancers. 2016; 8(4):39. https://doi.org/10.3390/cancers8040039
Chicago/Turabian StyleSchmidt, M. Lee, Diego F. Calvisi, and Geoffrey J. Clark. 2016. "NORE1A Regulates MDM2 Via β-TrCP" Cancers 8, no. 4: 39. https://doi.org/10.3390/cancers8040039
APA StyleSchmidt, M. L., Calvisi, D. F., & Clark, G. J. (2016). NORE1A Regulates MDM2 Via β-TrCP. Cancers, 8(4), 39. https://doi.org/10.3390/cancers8040039