
Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response

Abstract
:1. Introduction
2. Expression of TAMs
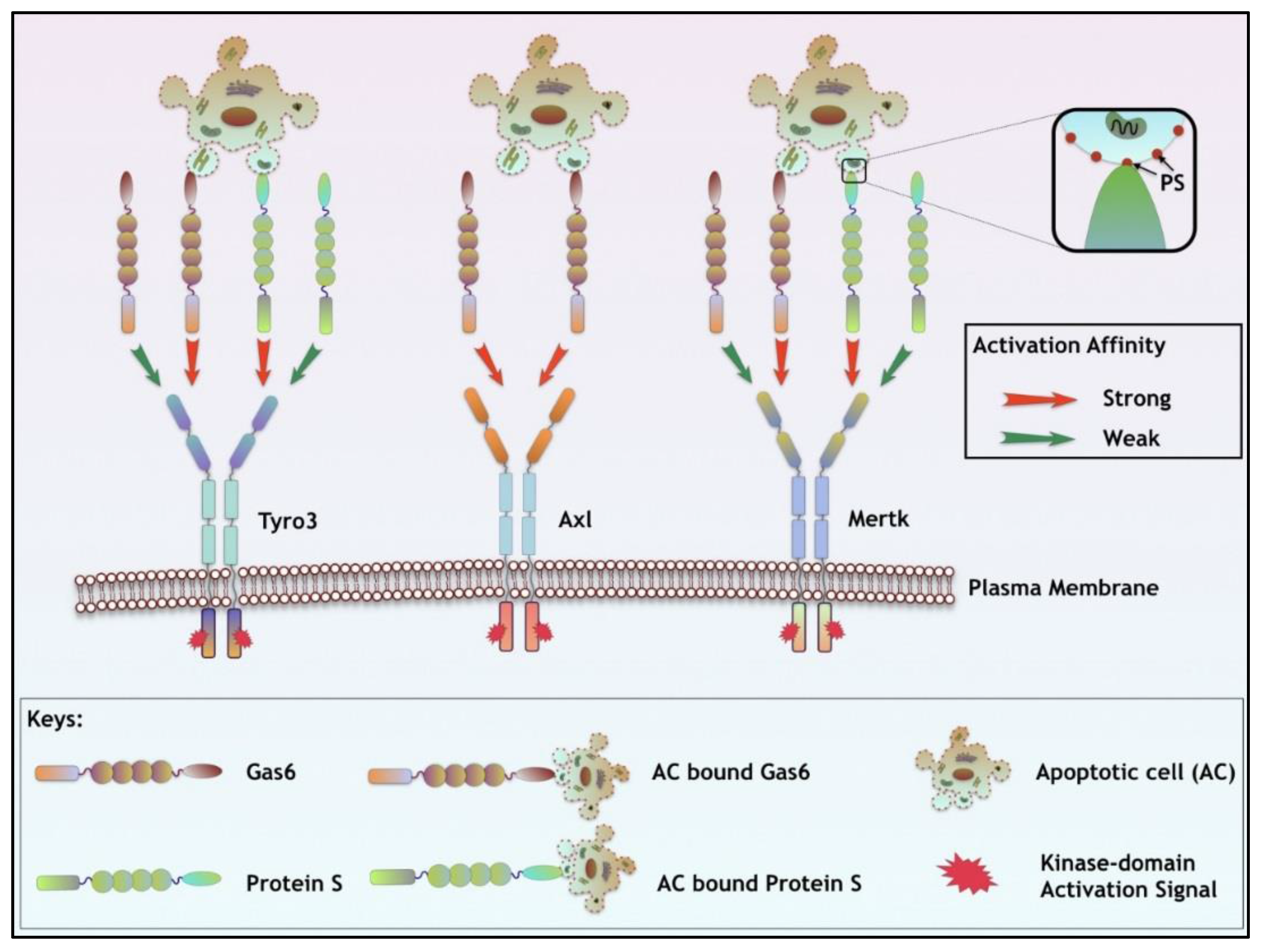
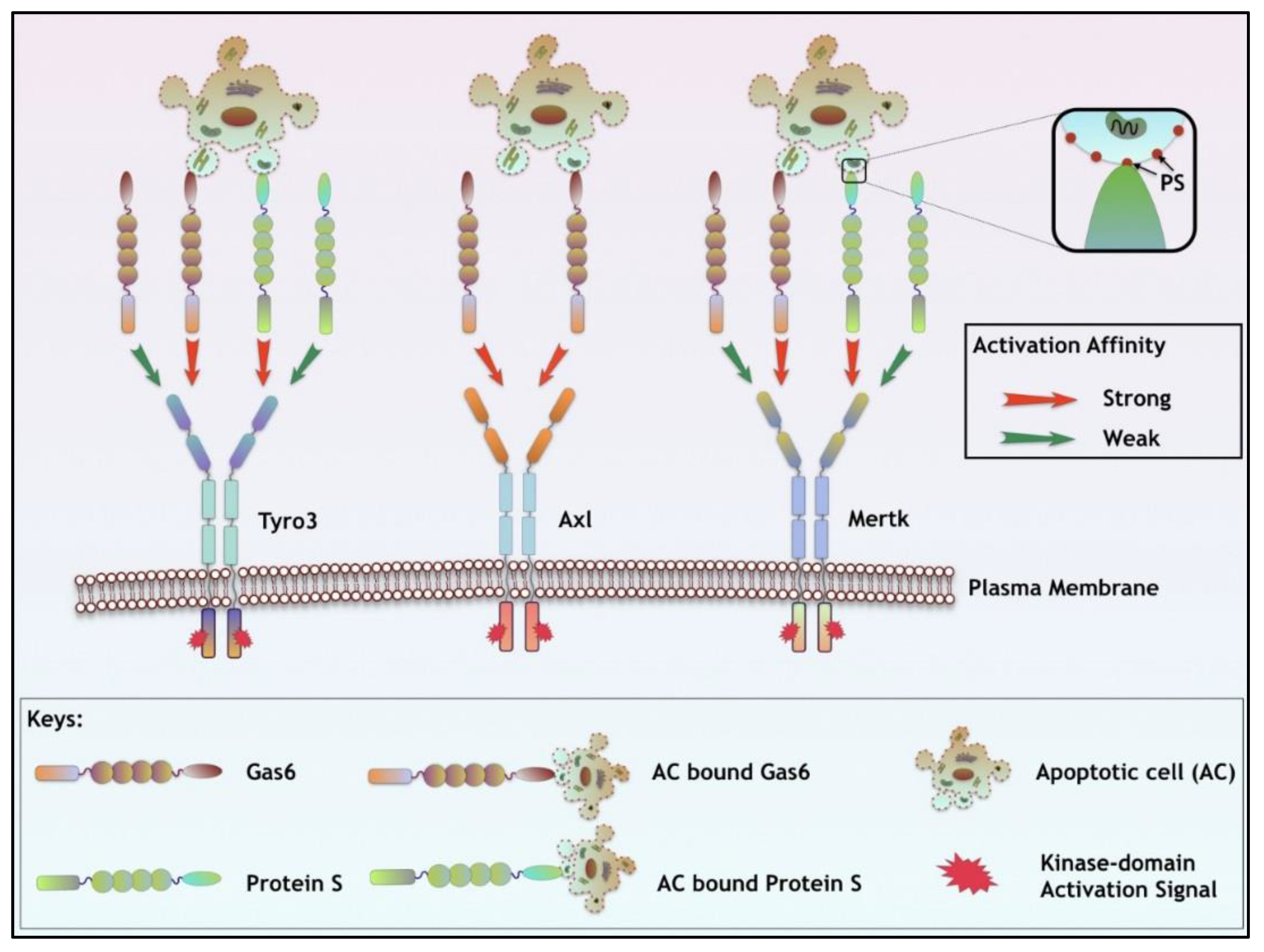
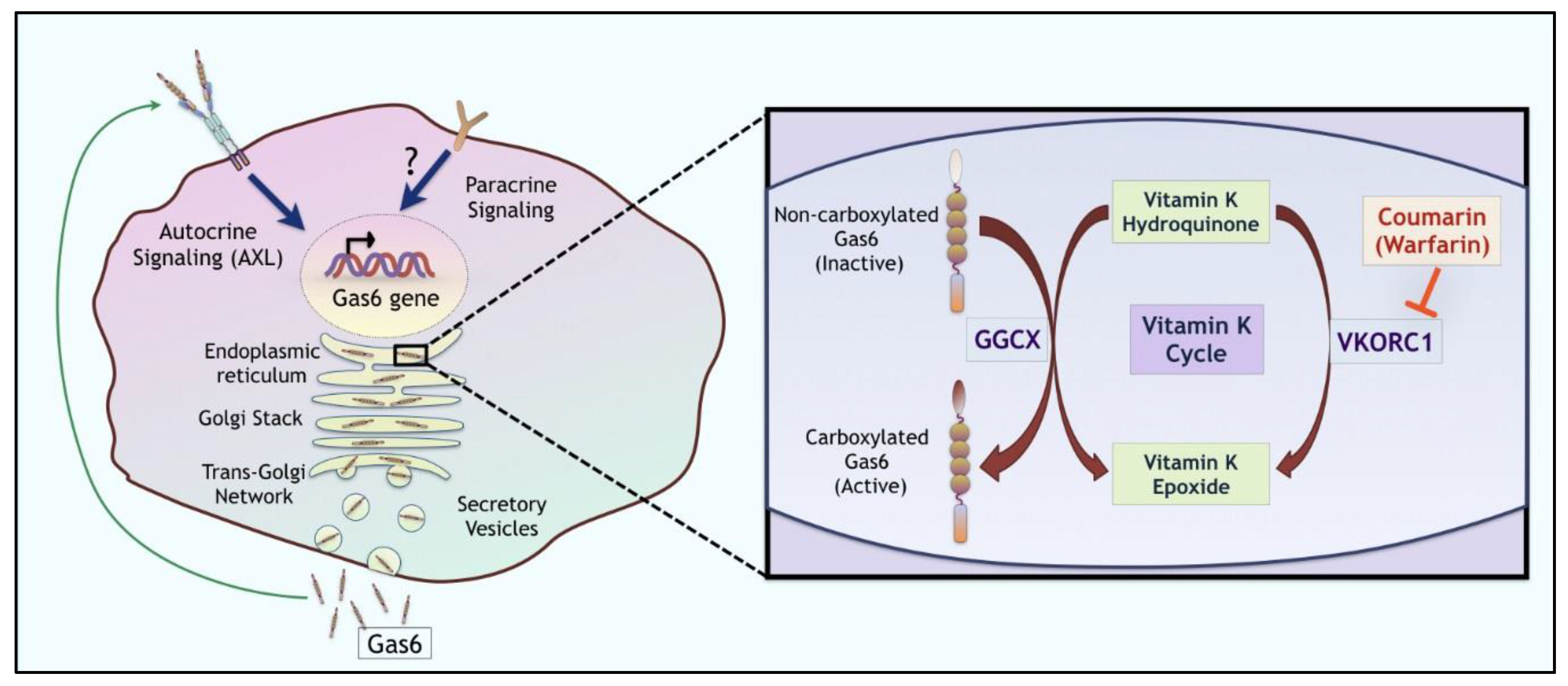
3. TAM Ligands
4. Targeting Ligand/TAMs for Anti-Tumor Therapeutic Response
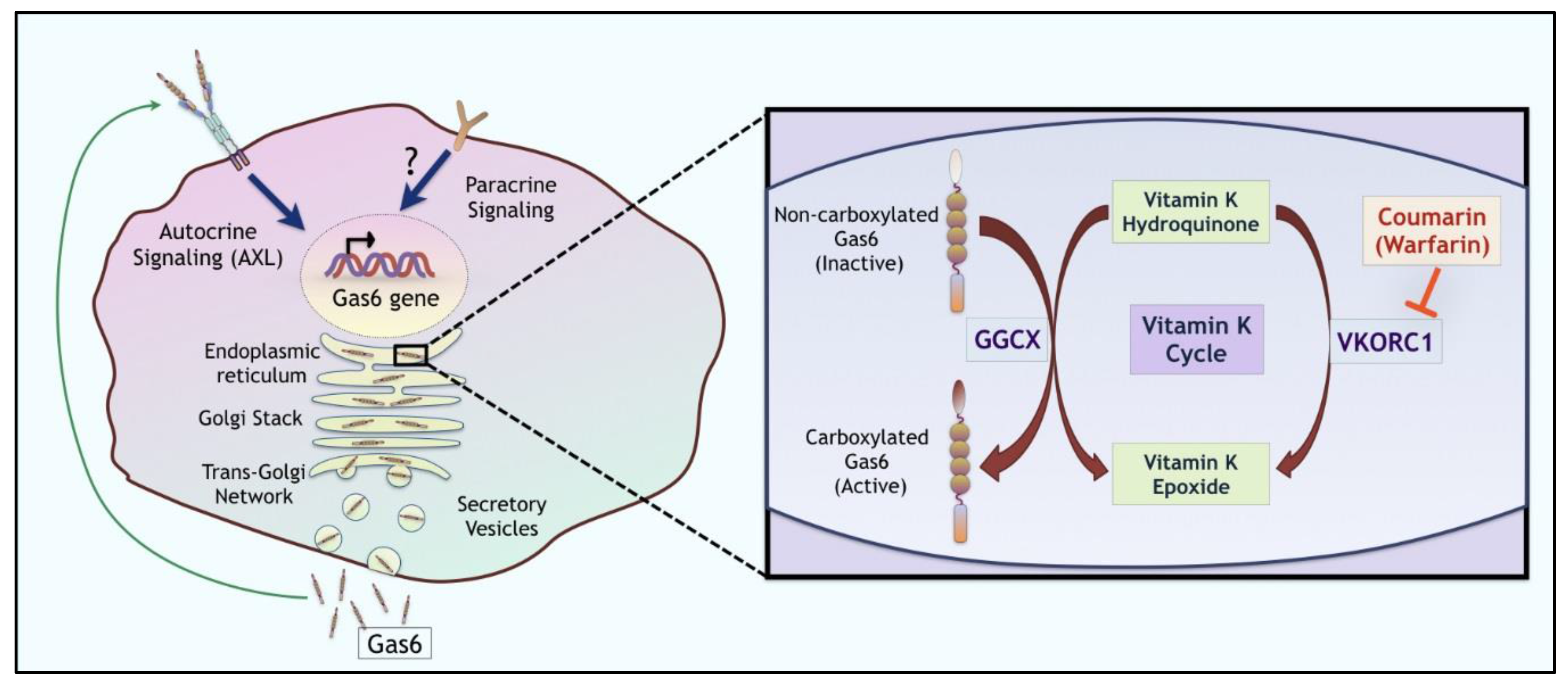
5. Targeting Gas6 and General Ligand Inhibitors
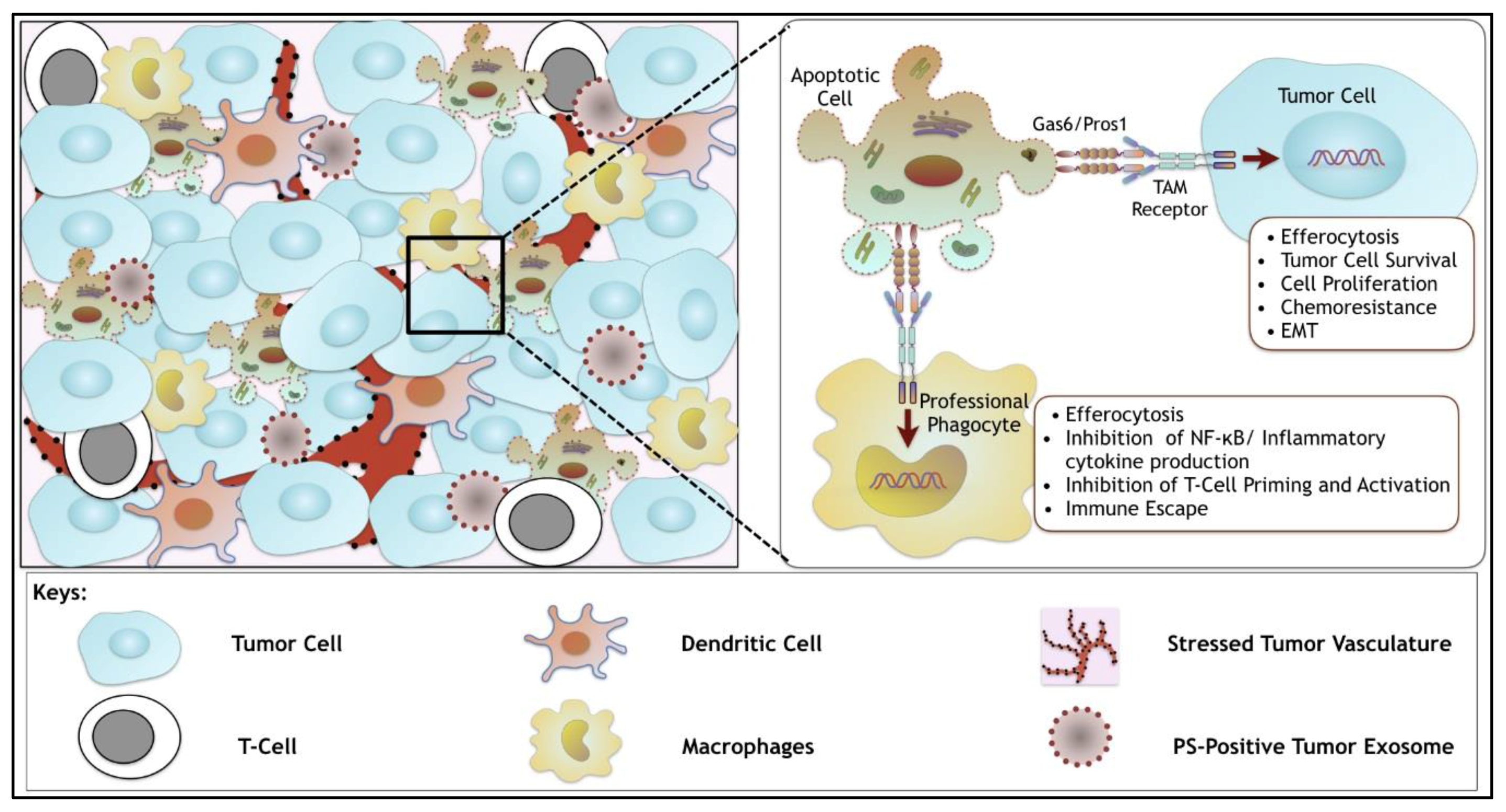
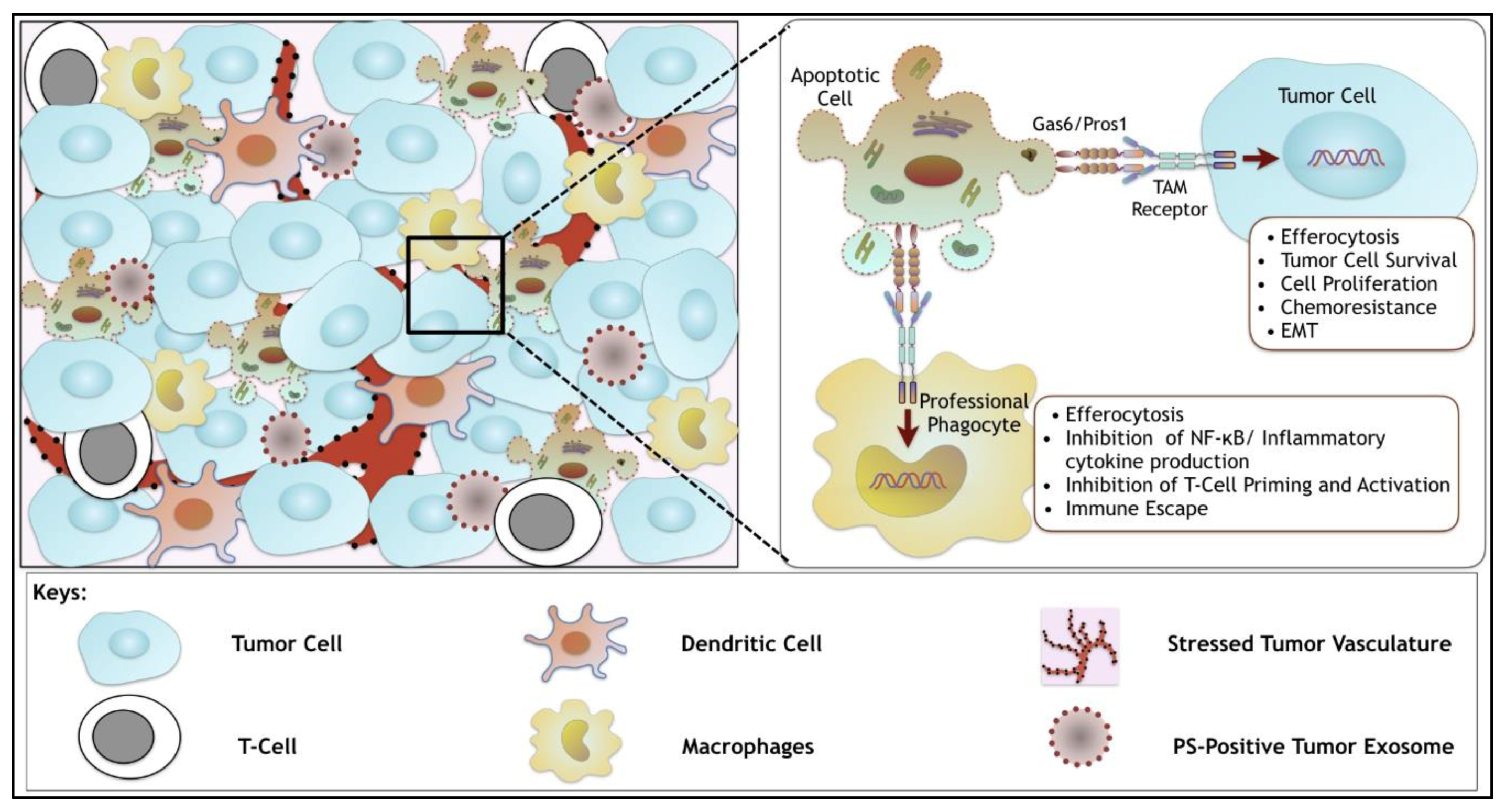
6. Synergistic Role of TAMs and of Dys-Regulated PS in the Tumor Microenvironment
7. Conclusions
- Are TAM receptors and their ligands (Gas6 and Pros1) up-regulated in poorly immunogenic tumors?
- Can TAM receptors and TAM ligand antagonist be used in combination with other check point inhibitors such as anti-PD1 and anti-CTLA-4?
- Can warfarin or non-γ-carboxylated Gas6 proteins be considered as adjuvant cancer therapeutics?
- Does over-expression of Ggcx and Vkorc1 contribute to tumorigenicity of the tumor microenvironment?
- What is the molecular mechanism(s) by which TAM post-receptor signaling drive anti-inflammatory and immune inhibitory signals?
Acknowledgments
Conflicts of Interest
References
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter-Hufford, A.; Ravichandran, K.S. Clearing the dead: Apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, a008748. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [PubMed]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin k-dependent proteins related to Protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein s and its relative, gas6, are ligands for the tyro 3/axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [PubMed]
- Lemke, G.; Lu, Q. Macrophage regulation by tyro 3 family receptors. Curr. Opin. Immunol. 2003, 15, 31–36. [Google Scholar] [CrossRef]
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the tyro 3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by mer. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, S.; Wang, H.; Wu, H.; Lu, Q.; Han, D. TAM receptors and the regulation of erythropoiesis in mice. Haematologica 2009, 94, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Lemke, G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron 1991, 6, 691–704. [Google Scholar] [CrossRef]
- Lapraz, F.; Rottinger, E.; Duboc, V.; Range, R.; Duloquin, L.; Walton, K.; Wu, S.Y.; Bradham, C.; Loza, M.A.; Hibino, T.; et al. Rtk and TGF-β signaling pathways genes in the sea urchin genome. Dev. Biol. 2006, 300, 132–152. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, H.; Pienta, K.J. Axl as a mediator of cellular growth and survival. Oncotarget 2014, 5, 8818–8852. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 2007, 178, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Zahuczky, G.; Kristof, E.; Majai, G.; Fesus, L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of mertk in enhanced phagocytosis. PLoS ONE 2011, 6, e21349. [Google Scholar] [CrossRef] [PubMed]
- McColl, A.; Bournazos, S.; Franz, S.; Perretti, M.; Morgan, B.P.; Haslett, C.; Dransfield, I. Glucocorticoids induce protein s-dependent phagocytosis of apoptotic neutrophils by human macrophages. J. Immunol. 2009, 183, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. Elife 2014. [Google Scholar] [CrossRef] [PubMed]
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, M.; Huppi, K.; Pitt, J.J.; Dorsey, T.H.; Ambs, S.; Caplen, N.J. Identification of the receptor tyrosine kinase AXL in breast cancer as a target for the human miR-34a microRNA. Breast Cancer Res. Treat. 2011, 130, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Bellosta, P.; Basilico, C. Cleavage and release of a soluble form of the receptor tyrosine kinase ark in vitro and in vivo. J. Cell. Physiol. 1996, 168, 737–744. [Google Scholar] [CrossRef]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A soluble form of the mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Valverde, P. Effects of Gas6 and hydrogen peroxide in Axl ubiquitination and downregulation. Biochem. Biophys. Res. Commun. 2005, 333, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Crosier, P.S.; Hall, L.R.; Vitas, M.R.; Lewis, P.M.; Crosier, K.E. Identification of a novel receptor tyrosine kinase expressed in acute myeloid leukemic blasts. Leuk. Lymphoma 1995, 18, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sherick, A.B.; Menachof, K.; Eisenman, K.M.; McGranahan, A.; McGary, C.; Hunsucker, S.A.; Schlegel, J.; Armistead, P.M.; Liang, X.; Kireev, D.; et al. Mer receptor tyrosine kinase is a potential therapeutic target in acute myeloid leukemia. Blood 2012, 120, 1317. [Google Scholar]
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of Axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [PubMed]
- Knubel, K.H.; Pernu, B.M.; Sufit, A.; Nelson, S.; Pierce, A.M.; Keating, A.K. Mertk inhibition is a novel therapeutic approach for glioblastoma multiforme. Oncotarget 2014, 5, 1338–1351. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, J.; Sambade, M.J.; Sather, S.; Moschos, S.J.; Tan, A.C.; Winges, A.; DeRyckere, D.; Carson, C.C.; Trembath, D.G.; Tentler, J.J.; et al. Mertk receptor tyrosine kinase is a therapeutic target in melanoma. J. Clin. Investig. 2013, 123, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.P.; Whang, Y.E.; Mohler, J.L.; Earp, H.S. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: Role of ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005, 65, 10514–10523. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Robinson, D.R.; Kung, H.J. Signal pathways in up-regulation of chemokines by tyrosine kinase Mer/Nyk in prostate cancer cells. Cancer Res. 2004, 64, 7311–7320. [Google Scholar] [CrossRef] [PubMed]
- Meric, F.; Lee, W.P.; Sahin, A.; Zhang, H.; Kung, H.J.; Hung, M.C. Expression profile of tyrosine kinases in breast cancer. Clin. Cancer Res. 2002, 8, 361–367. [Google Scholar] [PubMed]
- Kimani, S.G.; Kumar, S.; Davra, V.; Chang, Y.J.; Kasikara, C.; Geng, K.; Tsou, W.I.; Wang, S.; Hoque, M.; Bohac, A.; et al. Normalization of TAM post-receptor signaling reveals a cell invasive signature for Axl tyrosine kinase. Cell Commun. Signal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Craven, R.J.; Xu, L.H.; Weiner, T.M.; Fridell, Y.W.; Dent, G.A.; Srivastava, S.; Varnum, B.; Liu, E.T.; Cance, W.G. Receptor tyrosine kinases expressed in metastatic colon cancer. Int. J. Cancer 1995, 60, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Bosurgi, L.; Bernink, J.H.; Delgado Cuevas, V.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef] [PubMed]
- Sawabu, T.; Seno, H.; Kawashima, T.; Fukuda, A.; Uenoyama, Y.; Kawada, M.; Kanda, N.; Sekikawa, A.; Fukui, H.; Yanagita, M.; et al. Growth arrest-specific gene 6 and Axl signaling enhances gastric cancer cell survival via Akt pathway. Mol. Carcinog. 2007, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Ye, X.; Pham, T.; Lin, E.; Chan, S.; McNamara, E.; Neve, R.M.; Belmont, L.; Koeppen, H.; Yauch, R.L.; et al. Axl inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014, 74, 5878–5890. [Google Scholar] [CrossRef] [PubMed]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of Gas6/Axl signaling by HIF promotes renal metastasis through SCR and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Baron, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [PubMed]
- Suarez, R.M.; Chevot, F.; Cavagnino, A.; Saettel, N.; Radvanyi, F.; Piguel, S.; Bernard-Pierrot, I.; Stoven, V.; Legraverend, M. Inhibitors of the TAM subfamily of tyrosine kinases: Synthesis and biological evaluation. Eur. J. Med. Chem. 2013, 61, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. Mertk inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Carrera Silva, E.A.; Chan, P.Y.; Joannas, L.; Errasti, A.E.; Gagliani, N.; Bosurgi, L.; Jabbour, M.; Perry, A.; Smith-Chakmakova, F.; Mucida, D.; et al. T cell-derived Protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 2013, 39, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Mark, M.R.; Chen, J.; Hammonds, R.G.; Sadick, M.; Godowsk, P.J. Characterization of Gas6, a member of the superfamily of g domain-containing proteins, as a ligand for rse and Axl. J. Biol. Chem. 1996, 271, 9785–9789. [Google Scholar] [PubMed]
- Nakano, T.; Kishino, J.; Arita, H. Characterization of a high-affinity and specific binding site for Gas6. FEBS Lett. 1996, 387, 75–77. [Google Scholar] [CrossRef]
- Chen, J.; Carey, K.; Godowski, P.J. Identification of gas6 as a ligand for mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene 1997, 14, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.A.; Maylock, C.A.; Williams, J.A.; Paweletz, C.P.; Shu, H.; Shacter, E. Serum-derived Protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat. Immunol. 2003, 4, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Rezende, S.M.; Simmonds, R.E.; Lane, D.A. Coagulation, inflammation, and apoptosis: Different roles for proteins and the protein S-C4b binding protein complex. Blood 2004, 103, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, Tyro3, Axl, and Mer, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [PubMed]
- Kirane, A.; Ludwig, K.F.; Sorrelle, N.; Haaland, G.; Sandal, T.; Ranaweera, R.; Toombs, J.E.; Wang, M.; Dineen, S.P.; Micklem, D.; et al. Warfarin blocks Gas6-mediated Axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. 2015, 75, 3699–3705. [Google Scholar] [CrossRef] [PubMed]
- Hasanbasic, I.; Rajotte, I.; Blostein, M. The role of gamma-carboxylation in the anti-apoptotic function of Gas6. J. Thromb. Haemost. 2005, 3, 2790–2797. [Google Scholar] [CrossRef] [PubMed]
- Rajotte, I.; Hasanbasic, I.; Blostein, M. Gas6-mediated signaling is dependent on the engagement of its gamma-carboxyglutamic acid domain with phosphatidylserine. Biochem. Biophys. Res. Commun. 2008, 376, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.H.; Hao, G.; Best, A.M.; Sun, X.; Thorpe, P.E. Highly specific pet imaging of prostate tumors in mice with an iodine-124-labeled antibody fragment that targets phosphatidylserine. PLoS ONE 2013, 8, e84864. [Google Scholar] [CrossRef] [PubMed]
- Kimani, S.G.; Geng, K.; Kasikara, C.; Kumar, S.; Sriram, G.; Wu, Y.; Birge, R.B. Contribution of defective PS recognition and efferocytosis to chronic inflammation and autoimmunity. Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [PubMed]
- Balogh, I.; Hafizi, S.; Stenhoff, J.; Hansson, K.; Dahlback, B. Analysis of Gas6 in human platelets and plasma. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1280–1286. [Google Scholar] [CrossRef] [PubMed]
- Heeb, M.J. Role of the pros1 gene in thrombosis: Lessons and controversies. Expert Rev. Hematol. 2008, 1, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, K.; Nakabayashi, T.; Naitoh, S.; Takeda, M.; Tarumi, T.; Mizoguchi, I.; Ieko, M.; Koike, T. One novel and one recurrent mutation in the PROS1 gene cause type I protein S deficiency in patients with pulmonary embolism associated with deep vein thrombosis. Am. J. Hematol. 2006, 81, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Cosemans, J.M.; van Kruchten, R.; Olieslagers, S.; Schurgers, L.J.; Verheyen, F.K.; Munnix, I.C.; Waltenberger, J.; Angelillo-Scherrer, A.; Hoylaerts, M.F.; Carmeliet, P.; et al. Potentiating role of Gas6 and Tyro3, Axl and Mer (TAM) receptors in human and murine platelet activation and thrombus stabilization. J. Thromb. Haemost. 2010, 8, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Christoph, S.; Deryckere, D.; Schlegel, J.; Frazer, J.K.; Batchelor, L.A.; Trakhimets, A.Y.; Sather, S.; Hunter, D.M.; Cummings, C.T.; Liu, J.; et al. UNC569, a novel small-molecule mer inhibitor with efficacy against acute lymphoblastic leukemia in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sherick, A.B.; Zhang, W.; Menachof, K.K.; Hill, A.A.; Rinella, S.; Kirkpatrick, G.; Page, L.S.; Stashko, M.A.; Jordan, C.T.; Wei, Q.; et al. Efficacy of a Mer and Flt3 tyrosine kinase small molecule inhibitor, UNC1666, in acute myeloid leukemia. Oncotarget 2015, 6, 6722–6736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; DeRyckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. Unc2025, a potent and orally bioavailable Mer/Flt3 dual inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Adjei, A.A. Met: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.M.; An, Y.; Cai, Z.W.; Chen, X.T.; Clark, C.; Cornelius, L.A.; Dai, J.; Gullo-Brown, J.; Gupta, A.; Henley, B.; et al. Discovery of n-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl )-2-oxo-1,2-dihydropyridine-3-carboxamide (bms-777607), a selective and orally efficacious inhibitor of the met kinase superfamily. J. Med. Chem. 2009, 52, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S., II; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered AXL "decoy receptor" effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Weinger, J.G.; Omari, K.M.; Marsden, K.; Raine, C.S.; Shafit-Zagardo, B. Up-regulation of soluble Axl and mer receptor tyrosine kinases negatively correlates with gas6 in established multiple sclerosis lesions. Am. J. Pathol. 2009, 175, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Vaisar, T.; Subramanian, M.; Mautner, L.; Blobel, C.; Tabas, I. Shedding of the mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase cdelta, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 2011, 286, 33335–33344. [Google Scholar] [CrossRef] [PubMed]
- Law, A.L.; Parinot, C.; Chatagnon, J.; Gravez, B.; Sahel, J.A.; Bhattacharya, S.S.; Nandrot, E.F. Cleavage of mer tyrosine kinase (MerTK) from the cell surface contributes to the regulation of retinal phagocytosis. J. Biol. Chem. 2015, 290, 4941–4952. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.S.; Spite, M.; Fredman, G.; Tabas, I. Mertk cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [PubMed]
- Santulli-Marotto, S.; Gervais, A.; Fisher, J.; Strake, B.; Ogden, C.A.; Riveley, C.; Giles-Komar, J. Discovering molecules that regulate efferocytosis using primary human macrophages and high content imaging. PLoS ONE 2015, 10, e0145078. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Schmidt, T.; Tjwa, M.; van Geyte, K.; Lievens, D.; Lutgens, E.; Vanhoutte, D.; Borgel, D.; Plaisance, S.; Hoylaerts, M.; et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood 2010, 115, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Moody, G.; Belmontes, B.; Masterman, S.; Wang, W.; King, C.; Murawsky, C.; Tsuruda, T.; Liu, S.; Radinsky, R.; Beltran, P.J. Antibody-mediated neutralization of autocrine Gas6 inhibits the growth of pancreatic ductal adenocarcinoma tumors in vivo. Int. J. Cancer 2016, 139, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Maquoi, E.; Voros, G.; Carmeliet, P.; Collen, D.; Lijnen, H.R. Role of gas-6 in adipogenesis and nutritionally induced adipose tissue development in mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Robins, R.S.; Lemarie, C.A.; Laurance, S.; Aghourian, M.N.; Wu, J.; Blostein, M.D. Vascular Gas6 contributes to thrombogenesis and promotes tissue factor up-regulation after vessel injury in mice. Blood 2013, 121, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Avanzi, G.C.; Gallicchio, M.; Cavalloni, G.; Gammaitoni, L.; Leone, F.; Rosina, A.; Boldorini, R.; Monga, G.; Pegoraro, L.; Varnum, B.; et al. Gas6, the ligand of Axl and Rse receptors, is expressed in hematopoietic tissue but lacks mitogenic activity. Exp. Hematol. 1997, 25, 1219–1226. [Google Scholar] [PubMed]
- Foley, J.H.; Conway, E.M. Gas6 gains entry into the coagulation cascade. Blood 2013, 121, 570–571. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Arai, H.; Yanagita, M.; Matsubara, T.; Kanamori, H.; Nakano, T.; Iehara, N.; Fukatsu, A.; Kita, T.; Doi, T. Growth arrest-specific gene 6 is involved in glomerular hypertrophy in the early stage of diabetic nephropathy. J. Biol. Chem. 2003, 278, 18229–18234. [Google Scholar] [CrossRef] [PubMed]
- Akitake-Kawano, R.; Seno, H.; Nakatsuji, M.; Kimura, Y.; Nakanishi, Y.; Yoshioka, T.; Kanda, K.; Kawada, M.; Kawada, K.; Sakai, Y.; et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis 2013, 34, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Ekman, C.; Linder, A.; Akesson, P.; Dahlback, B. Plasma concentrations of Gas6 (growth arrest specific protein 6) and its soluble tyrosine kinase receptor Saxl in sepsis and systemic inflammatory response syndromes. Crit. Care 2010, 14, R158. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; King, R.M.; Philipson, L. Genes specifically expressed at growth arrest of mammalian cells. Cell 1988, 54, 787–793. [Google Scholar] [CrossRef]
- Park, I.K.; Giovenzana, C.; Hughes, T.L.; Yu, J.; Trotta, R.; Caligiuri, M.A. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood 2009, 113, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Llacuna, L.; Barcena, C.; Bellido-Martin, L.; Fernandez, L.; Stefanovic, M.; Mari, M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Garcia de Frutos, P.; Morales, A. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 2010, 52, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Sica, A. Macrophages give Gas(6) to cancer. Blood 2010, 115, 2122–2123. [Google Scholar] [CrossRef] [PubMed]
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in Ccl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Spector, S.A. Development and characterization of a human microglia cell model of Hiv-1 infection. J. Neurovirol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique Tgf-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, J.; Hammonds, G.; Phillips, H.; Armanini, M.; Wood, P.; Bunge, R.; Godowski, P.J.; Sliwkowski, M.X.; Mather, J.P. Identification of Gas6 as a growth factor for human schwann cells. J. Neurosci. 1996, 16, 2012–2019. [Google Scholar] [PubMed]
- Borgel, D.; Clauser, S.; Bornstain, C.; Bieche, I.; Bissery, A.; Remones, V.; Fagon, J.Y.; Aiach, M.; Diehl, J.L. Elevated growth-arrest-specific protein 6 plasma levels in patients with severe sepsis. Crit. Care Med. 2006, 34, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Massin, F.; Cravoisy, A.; Dupays, R.; Barraud, D.; Nace, L.; Bollaert, P.E. Growth arrest-specific protein 6 plasma concentrations during septic shock. Crit. Care 2007, 11, R8. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Higashino, K.; Kikuchi, N.; Kishino, J.; Nomura, K.; Fujita, H.; Ohara, O.; Arita, H. Vascular smooth muscle cell-derived, gla-containing growth-potentiating factor for Ca2+-mobilizing growth factors. J. Biol. Chem. 1995, 270, 5702–5705. [Google Scholar] [PubMed]
- Azuma, K.; Tsukui, T.; Ikeda, K.; Shiba, S.; Nakagawa, K.; Okano, T.; Urano, T.; Horie-Inoue, K.; Ouchi, Y.; Ikawa, M.; et al. Liver-specific gamma-glutamyl carboxylase-deficient mice display bleeding diathesis and short life span. PLoS ONE 2014, 9, e88643. [Google Scholar] [CrossRef] [PubMed]
- Ran, S.; Downes, A.; Thorpe, P.E. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002, 62, 6132–6140. [Google Scholar] [PubMed]
- Stafford, J.H.; Thorpe, P.E. Increased exposure of phosphatidylethanolamine on the surface of tumor vascular endothelium. Neoplasia 2011, 13, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Birge, R.B. Efferocytosis. Curr. Biol. 2016, 26, R558–R559. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.C.; Young, C.; Hicks, D.; Owens, P.; Williams, A.; Vaught, D.B.; Morrison, M.M.; Lim, J.; Williams, M.; Brantley-Sieders, D.M.; et al. Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Investig. 2014, 124, 4737–4752. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new mertk-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Maiguel, D.; Kim, Y.; Li, W. Identification of tubby and tubby-like protein 1 as eat-me signals by phage display. Exp. Cell Res. 2010, 316, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new mertk ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Ligand Properties | Gas6 | Protein S |
|---|---|---|
| TAM Specificity | Axl >> Tyro3 > Mertk | Tyro3 > Mertk; Axl (ND) |
| TAM Specificity (In the presence of PS) | Tyro3 = Mertk >> Axl | Tyro3 = Mertk; Axl (ND) |
| Vit. K dependency | √ | √ |
| Plasma Concentration | 0.16 to 0.28 nM/L | 0.30 μM/L (~1000 times) |
| Cell Type | Reference |
|---|---|
| Adipose Tissue | [79] |
| Bone Marrow cells | [77,80,81,82] |
| Endothelial Cells | [6,79,80,82,83,84] |
| Epithelial Cells | [85] |
| Fibroblast | [6,79,86,87] |
| Hematopoietic cells | [10,19,81] |
| Hematopoietic stem cells | [88] |
| Hepatocytes | [89] |
| Macrophage | [77,90,91] |
| Mesangial cells (kidney) | [84] |
| Microglia Cells | [92,93] |
| Neuron | [94] |
| Pericytes | [83] |
| Plasma | [61,86,95,96] |
| Platelets | [64,80,82] |
| Stromal Cells | [85] |
| Vascular smooth muscle cell | [80,82,83,97] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davra, V.; Kimani, S.G.; Calianese, D.; Birge, R.B. Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response. Cancers 2016, 8, 107. https://doi.org/10.3390/cancers8120107
Davra V, Kimani SG, Calianese D, Birge RB. Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response. Cancers. 2016; 8(12):107. https://doi.org/10.3390/cancers8120107
Chicago/Turabian StyleDavra, Viralkumar, Stanley G. Kimani, David Calianese, and Raymond B. Birge. 2016. "Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response" Cancers 8, no. 12: 107. https://doi.org/10.3390/cancers8120107
APA StyleDavra, V., Kimani, S. G., Calianese, D., & Birge, R. B. (2016). Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response. Cancers, 8(12), 107. https://doi.org/10.3390/cancers8120107