
Identifying Cancer Driver Genes Using Replication-Incompetent Retroviral Vectors

Abstract
:1. Introduction
2. Identifying Cancer Genes by High-Throughput Sequencing Is Challenging
3. Insertional Mutagenesis Screens
3.1. Transposon Based Insertional Mutagenesis Screens
3.2. Insertional Mutagenesis Using Replication-Competent Retroviruses
3.3. Replication-Incompetent Vectors Can Be Pseudotyped to Allow Mutagenesis of Essentially Any Mammalian Cell
3.4. Replication-Incompetent Vectors Can Cause Cancer via Insertional Mutagenesis
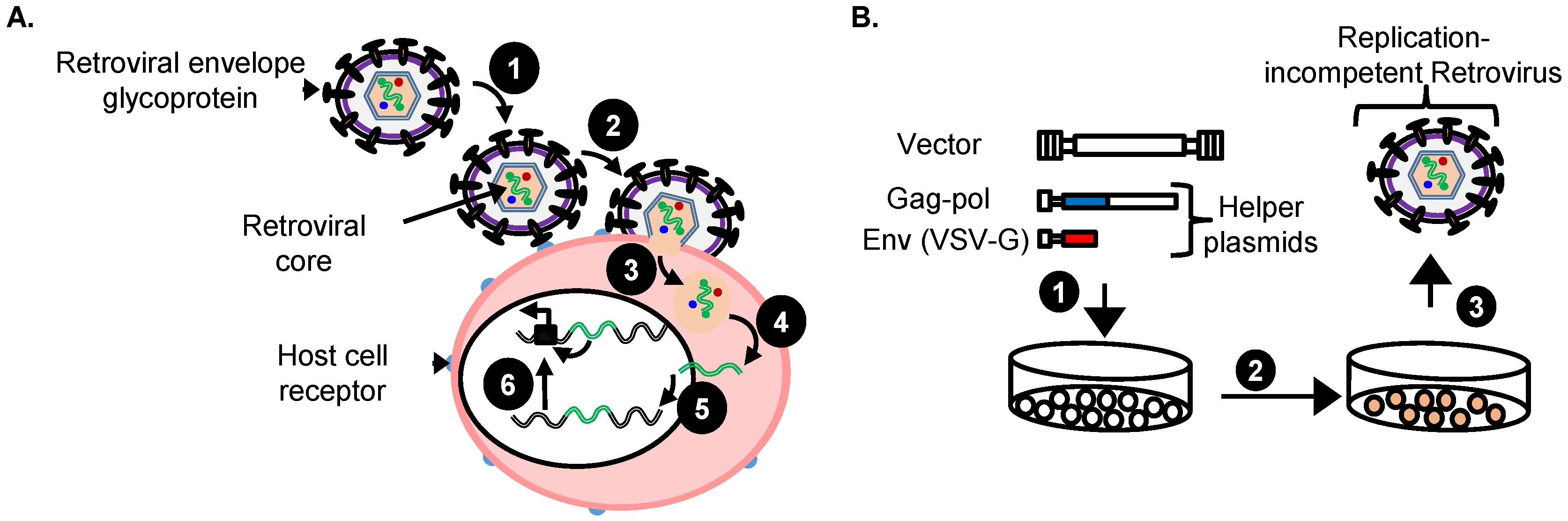
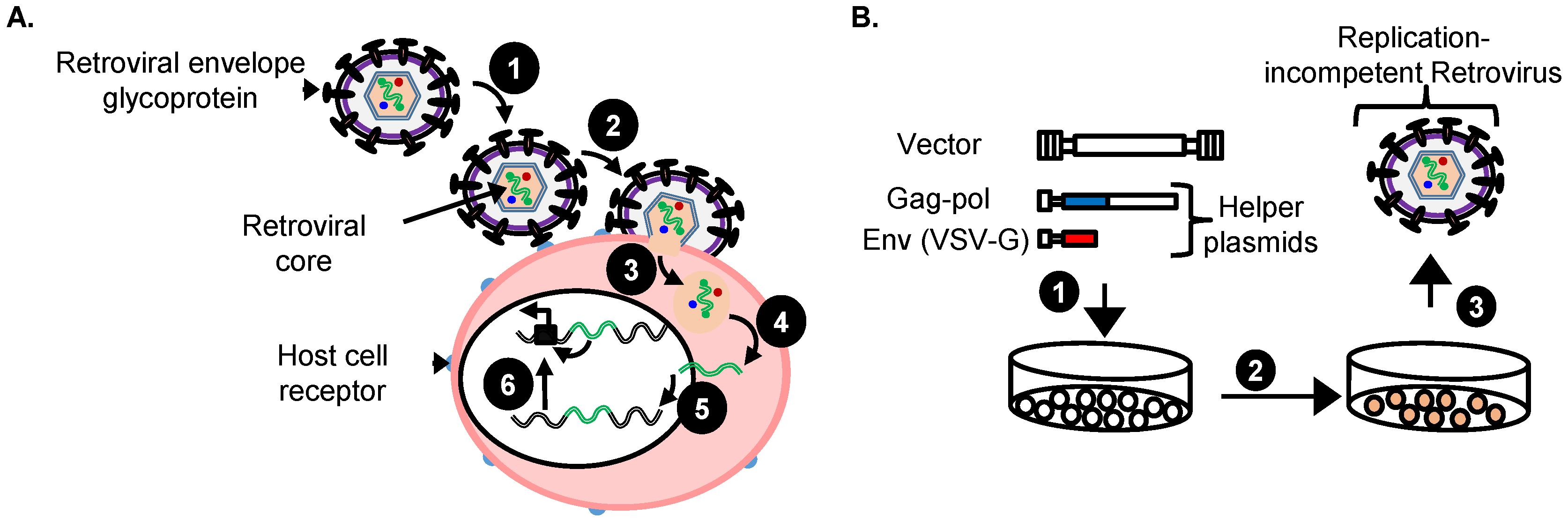
4. Replication-Incompetent Retroviral Vector Design and Use
4.1. Choice of Replication-Incompetent Retroviral Vector Type
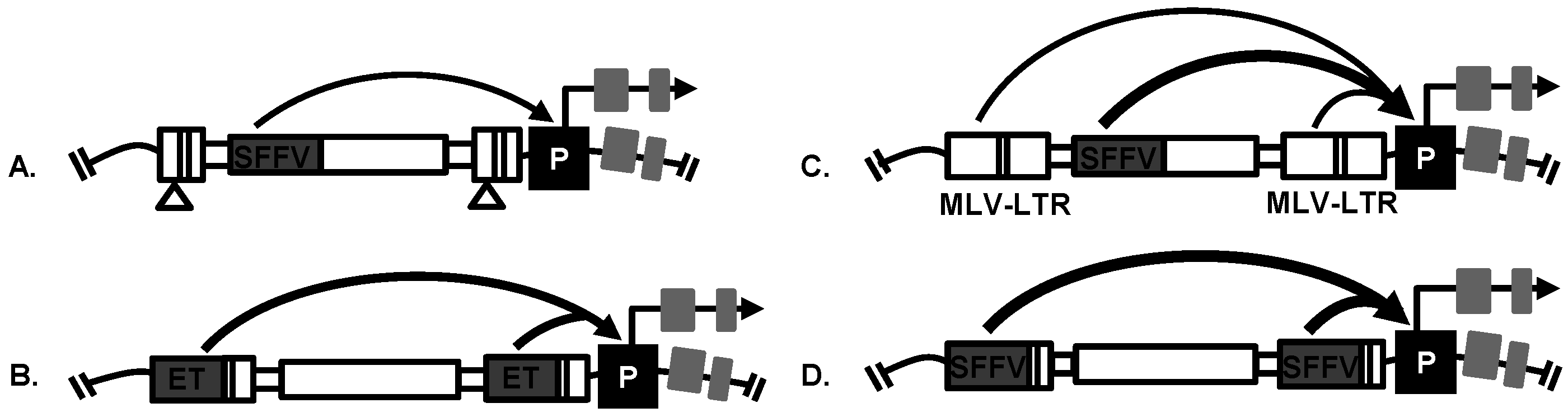
4.2. Replication-Incompetent Retroviral Vector Design
4.3. Production of Replication-Incompetent Retroviral Vectors
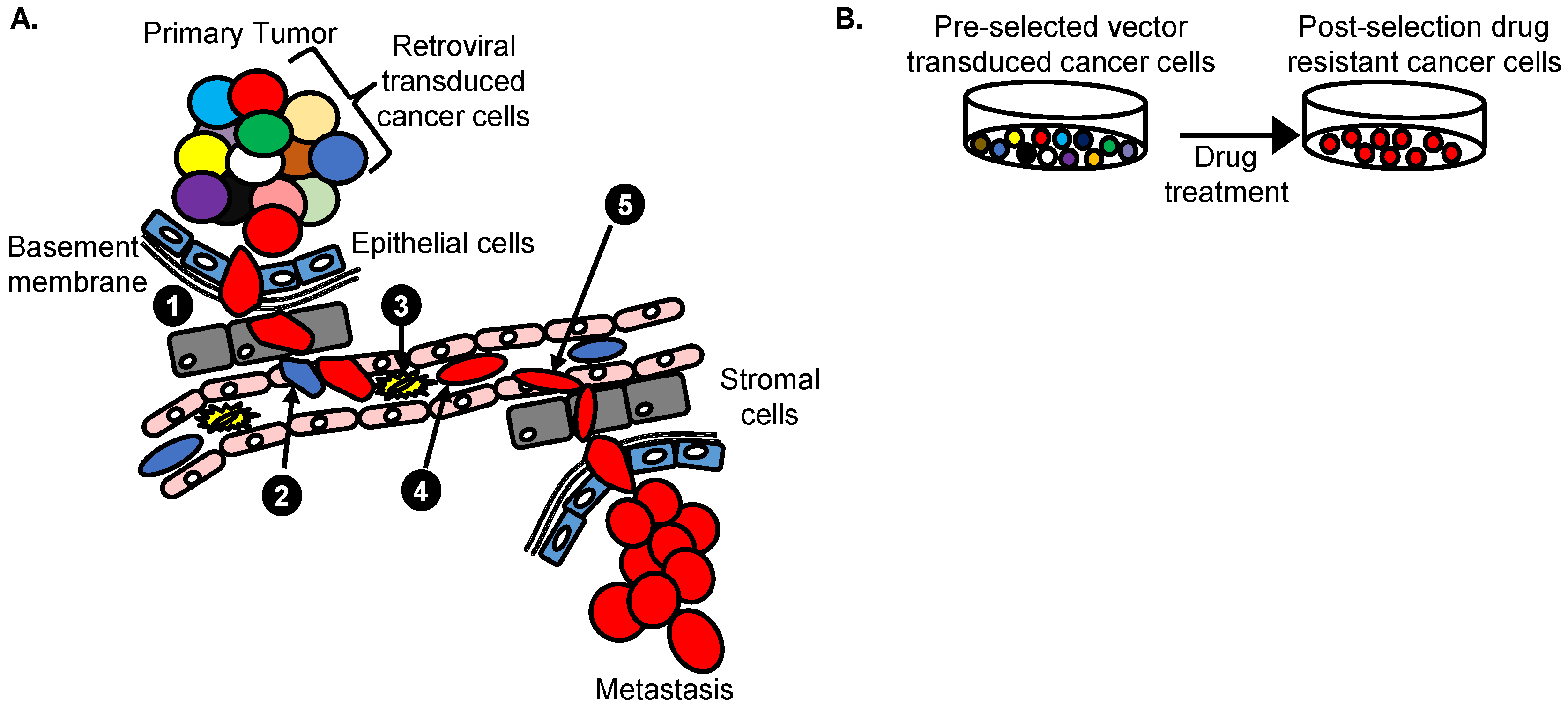
4.4. Vectors with a Drug Resistance Gene Can Be Used at a Low MOI to Reduce Passenger Mutations
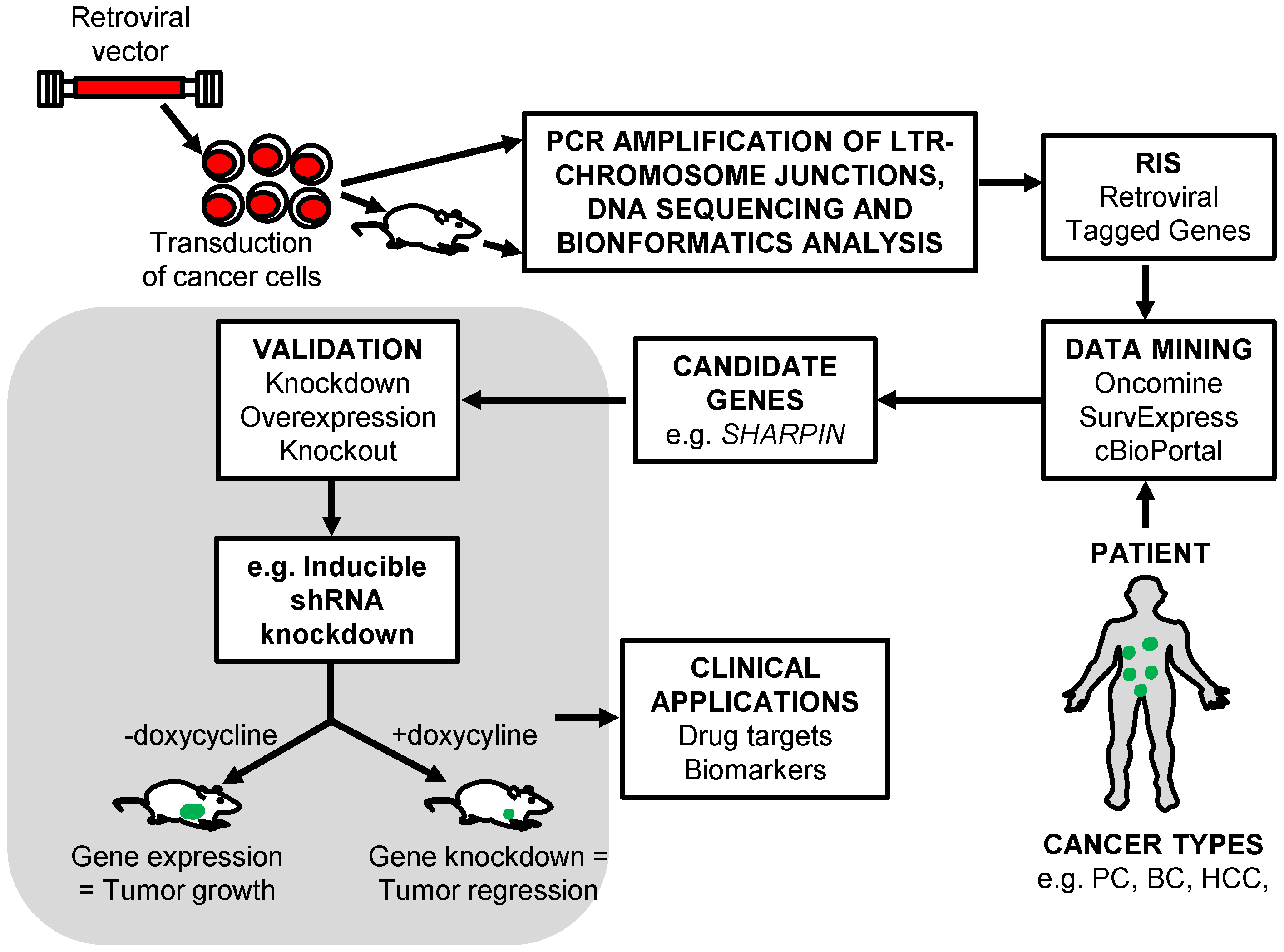
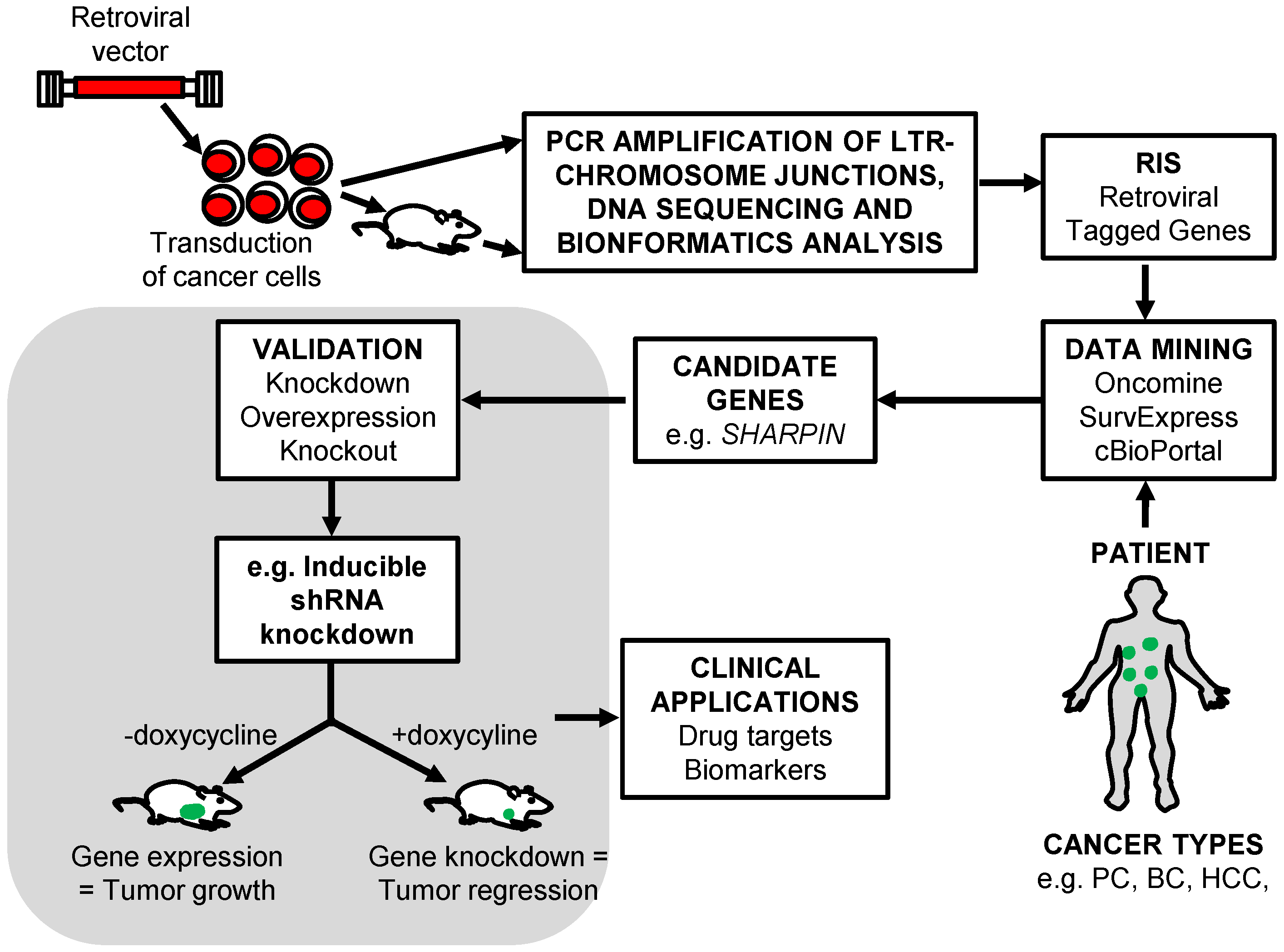
5. The Identification of RIS That Tag Cancer Driver Genes
5.1. Shuttle Vector Rescue Approach
5.2. LAM-PCR
5.3. MGS-PCR
6. Retroviral Insertional Mutagenesis Screens to Identify Cancer Driver Genes
6.1. Prostate Cancer
6.2. Breast Cancer
6.3. Hepatocellular Carcinoma
7. Retroviral Vector Screens to Identify Cancer Drug Resistance Genes
7.1. HER 2+ Breast Cancer
7.2. Pancreatic Adenocarcinoma
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of alk as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [PubMed]
- Zhang, J.; Liu, J.; Sun, J.; Chen, C.; Foltz, G.; Lin, B. Identifying driver mutations from sequencing data of heterogeneous tumors in the era of personalized genome sequencing. Brief. Bioinform. 2014, 15, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Uren, A.G.; Kool, J.; Berns, A.; van Lohuizen, M. Retroviral insertional mutagenesis: Past, present and future. Oncogene 2005, 24, 7656–7672. [Google Scholar] [CrossRef] [PubMed]
- Collier, L.S.; Carlson, C.M.; Ravimohan, S.; Dupuy, A.J.; Largaespada, D.A. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 2005, 436, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.J.; Akagi, K.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 2005, 436, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Jenkins, N.A. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer 2010, 10, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Moriarity, B.S.; Largaespada, D.A. Sleeping Beauty transposon insertional mutagenesis based mouse models for cancer gene discovery. Curr. Opin. Genet. Dev. 2015, 30, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.J.; Fritz, S.; Largaespada, D.A. Transposition and gene disruption in the male germline of the mouse. Genesis 2001, 30, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.M.; Dupuy, A.J.; Fritz, S.; Roberg-Perez, K.J.; Fletcher, C.F.; Largaespada, D.A. Transposon mutagenesis of the mouse germline. Genetics 2003, 165, 243–256. [Google Scholar] [PubMed]
- Rad, R.; Rad, L.; Wang, W.; Strong, A.; Ponstingl, H.; Bronner, I.F.; Mayho, M.; Steiger, K.; Weber, J.; Hieber, M.; et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat. Genet. 2015, 47, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Landrette, S.F.; Xu, T. Somatic genetics empowers the mouse for modeling and interrogating developmental and disease processes. PLoS Genet. 2011, 7, e1002110. [Google Scholar] [CrossRef] [PubMed]
- Bii, V.M.; Rae, D.T.; Trobridge, G.D. A novel gammaretroviral shuttle vector insertional mutagenesis screen identifies sharpin as a breast cancer metastasis gene and prognostic biomarker. Oncotarget 2015, 6, 39507–39520. [Google Scholar] [PubMed]
- Ranzani, M.; Cesana, D.; Bartholomae, C.C.; Sanvito, F.; Pala, M.; Benedicenti, F.; Gallina, P.; Sergi, L.S.; Merella, S.; Bulfone, A.; et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat. Methods 2013, 10, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Nalla, A.K.; Williams, T.F.; Collins, C.P.; Rae, D.T.; Trobridge, G.D. Lentiviral vector-mediated insertional mutagenesis screen identifies genes that influence androgen independent prostate cancer progression and predict clinical outcome. Mol. Carcinog. 2015, 55, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Schinke, E.N.; Bii, V.; Nalla, A.; Rae, D.T.; Tedrick, L.; Meadows, G.G.; Trobridge, G.D. A novel approach to identify driver genes involved in androgen-independent prostate cancer. Mol. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Ranzani, M.; Annunziato, S.; Calabria, A.; Brasca, S.; Benedicenti, F.; Gallina, P.; Naldini, L.; Montini, E. Lentiviral vector-based insertional mutagenesis identifies genes involved in the resistance to targeted anti-cancer therapies. Mol. Ther. 2014, 22, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D. Genotoxicity of retroviral hematopoietic stem cell gene therapy. Expert Opin. Biol. Ther. 2011, 11, 581–593. [Google Scholar] [CrossRef] [PubMed]
- West, M.; Blanchette, C.; Dressman, H.; Huang, E.; Ishida, S.; Spang, R.; Zuzan, H.; Olson, J.A.; Marks, J.R.; Nevins, J.R. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 11462–11467. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.B.; Harpole, D.; Lancaster, J.M.; Berchuck, A.; et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Gray, J.W. Translating insights from the cancer genome into clinical practice. Nature 2008, 452, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to raf inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Big science: The cancer genome challenge. Nature 2010, 464, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Vandin, F.; Upfal, E.; Raphael, B.J. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012, 22, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hao, J.; Jiang, W.; He, T.; Zhang, X.; Jiang, T.; Jiang, R. Identifying potential cancer driver genes by genomic data integration. Sci. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular reconstruction of sleeping beauty, a tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Mirzaei, H.; Sahebkar, A.; Jaafari, M.R.; Hadjati, J.; Javanmard, S.H.; Mirzaei, H.R.; Salehi, R. Piggybac as a novel vector in cancer gene therapy: Current perspective. Cancer Gene Ther. 2016, 23, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Kaufman, C.D.; Zayed, H.; Miskey, C.; Walisko, O.; Izsvák, Z. The sleeping beauty transposable element: Evolution, regulation and genetic applications. Curr. Issues Mol. Biol. 2004, 6, 43–55. [Google Scholar] [PubMed]
- Chao, M.C.; Abel, S.; Davis, B.M.; Waldor, M.K. The design and analysis of transposon insertion sequencing experiments. Nat. Rev. Microbiol. 2016, 14, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Geurts, A.M.; Collier, L.S.; Geurts, J.L.; Oseth, L.L.; Bell, M.L.; Mu, D.; Lucito, R.; Godbout, S.A.; Green, L.E.; Lowe, S.W.; et al. Gene mutations and genomic rearrangements in the mouse as a result of transposon mobilization from chromosomal concatemers. PLoS Genet. 2006, 2, e156. [Google Scholar] [CrossRef] [PubMed]
- Vigdal, T.J.; Kaufman, C.D.; Izsvák, Z.; Voytas, D.F.; Ivics, Z. Common physical properties of DNA affecting target site selection of Sleeping Beauty and other tc1/mariner transposable elements. J. Mol. Biol. 2002, 323, 441–452. [Google Scholar] [CrossRef]
- Ikeda, R.; Kokubu, C.; Yusa, K.; Keng, V.W.; Horie, K.; Takeda, J. Sleeping Beauty transposase has an affinity for heterochromatin conformation. Mol. Cell Biol. 2007, 27, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Takeda, J.; Horie, K. Enhancement of Sleeping Beauty transposition by CpG methylation: Possible role of heterochromatin formation. Mol. Cell Biol. 2004, 24, 4004–4018. [Google Scholar] [CrossRef] [PubMed]
- Zayed, H.; Izsvák, Z.; Khare, D.; Heinemann, U.; Ivics, Z. The DNA-bending protein hmgb1 is a cellular cofactor of sleeping beauty transposition. Nucleic Acids Res. 2003, 31, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.H.; Coates, C.J.; George, A.L. PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mayhew, D.; Chen, X.; Johnston, M.; Mitra, R.D. “Calling cards” For DNA-binding proteins in mammalian cells. Genetics 2012, 190, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, C.; Lu, D.; Ning, Z.; Cox, T.; Melvin, D.; Wang, X.; Bradley, A.; Liu, P. Chromosomal transposition of piggyBac in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.M.; Skapura, D.; Justice, M.J. Genetic profile of insertion mutations in mouse leukemias and lymphomas. Genome Res. 2000, 10, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.W.; Chattopadhyay, S.K.; Lander, M.R.; Taddesse-Heath, L.; Naghashfar, Z.; Morse, H.C.; Fredrickson, T.N. Accelerated appearance of multiple B cell lymphoma types in NFS/N mice congenic for ecotropic murine leukemia viruses. Lab. Investig. 2000, 80, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, V.; Kimm, M.A.; Boer, M.; Wessels, L.; Theelen, W.; Jonkers, J.; Hilkens, J. Mmtv insertional mutagenesis identifies genes, gene families and pathways involved in mammary cancer. Nat. Genet. 2007, 39, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.H.; Turner, G.; Trubetskoy, A.; Verhoeven, E.; Wientjens, E.; Hulsman, D.; Russell, R.; DePinho, R.A.; Lenz, J.; van Lohuizen, M. Genome-wide retroviral insertional tagging of genes involved in cancer in cdkn2a-deficient mice. Nat. Genet. 2002, 32, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Wielgosz, M.M.; Hargrove, P.; Kepes, S.; Gray, J.; Persons, D.A.; Nienhuis, A.W. Transduction of human primitive repopulating hematopoietic cells with lentiviral vectors pseudotyped with various envelope proteins. Mol. Ther 2010, 18, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Wu, R.A.; Hansen, M.; Ironside, C.; Watts, K.L.; Olsen, P.; Beard, B.C.; Kiem, H.P. Cocal-pseudotyped lentiviral vectors resist inactivation by human serum and efficiently transduce primate hematopoietic repopulating cells. Mol. Ther. 2010, 18, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Slavin, S.; Aker, M.; Ficara, F.; Deola, S.; Mortellaro, A.; Morecki, S.; Andolfi, G.; Tabucchi, A.; Carlucci, F.; et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 2002, 296, 2410–2413. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Vai, S.; Mortellaro, A.; Casorati, G.; Ficara, F.; Andolfi, G.; Ferrari, G.; Tabucchi, A.; Carlucci, F.; Ochs, H.D.; et al. Immune reconstitution in ADA-SCID after pbl gene therapy and discontinuation of enzyme replacement. Nat. Med. 2002, 8, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Bougnères, P.; Schmidt, M.; Kalle, C.V.; Fischer, A.; Cavazzana-Calvo, M.; Aubourg, P. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012, 507, 187–198. [Google Scholar] [PubMed]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Diez, I.A.; Dewey, R.A.; Bohm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Sadelain, M.; Glorioso, J.C. Occurrence of leukaemia following gene therapy of X-linked SCID. Nat. Rev. Cancer 2003, 3, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Check, E. A tragic setback. Nature 2002, 420, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-Associated clonal t cell proliferation in two patients after gene therapy for scid-x1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Sgualdino, J.; Rudilosso, L.; Merella, S.; Naldini, L.; Montini, E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J. Clin. Investig. 2012, 122, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Dustin, R.T.; Grant, T.D. Retroviral genotoxicity. In Gene Therapy—Tools and Potential Applications; Molina, F.M., Ed.; InTech: Rijeka, Croatia, 2013; pp. 399–427. [Google Scholar]
- Cavazza, A.; Moiani, A.; Mavilio, F. Mechanisms of retroviral integration and mutagenesis. Hum. Gene Ther. 2013, 24, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral dna integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Beard, B.C.; Dickerson, D.; Beebe, K.; Gooch, C.; Fletcher, J.; Okbinoglu, T.; Miller, D.G.; Jacobs, M.A.; Kaul, R.; Kiem, H.P.; et al. Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells. Mol. Ther. 2007, 15, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi Sergi, L.; Benedicenti, F.; Ambrosi, A.; Di Serio, C.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, I.; Woods, N.B.; Panagopoulos, I.; Andersson, E.; Mikkola, H.; Fahlman, C.; Zufferey, R.; Carlsson, L.; Trono, D.; Karlsson, S. Lentivirus vector gene expression during es cell-derived hematopoietic development in vitro. J. Virol. 2000, 74, 10778–10784. [Google Scholar] [CrossRef] [PubMed]
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of Murine Leukemia Virus DNA depends on mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar] [PubMed]
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar] [PubMed]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular stomatitis virus g glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [PubMed]
- Zielske, S.P.; Gerson, S.L. Lentiviral transduction of P140K mgmt into human CD34(+) hematopoietic progenitors at low multiplicity of infection confers significant resistance to BG/BCNU and allows selection in vitro. Mol. Ther. 2002, 5, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.S.; Wahlers, A.; Kuhlcke, K.; Stahle, B.; Zander, A.R.; Baum, C.; Fehse, B. Dose finding with retroviral vectors: Correlation of retroviral vector copy numbers in single cells with gene transfer efficiency in a cell population. Blood 2003, 102, 3934–3937. [Google Scholar] [CrossRef] [PubMed]
- Kraunus, J.; Schaumann, D.H.; Meyer, J.; Modlich, U.; Fehse, B.; Brandenburg, G.; von Laer, D.; Klump, H.; Schambach, A.; Bohne, J.; et al. Self-inactivating retroviral vectors with improved rna processing. Gene Ther. 2004, 11, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Wahlers, A.; Schwieger, M.; Li, Z.; Meier-Tackmann, D.; Lindemann, C.; Eckert, H.G.; von Laer, D.; Baum, C. Influence of multiplicity of infection and protein stability on retroviral vector-mediated gene expression in hematopoietic cells. Gene Ther. 2001, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Sellers, S.; Gomes, T.J.; Larochelle, A.; Lopez, R.; Adler, R.; Krouse, A.; Donahue, R.E.; Childs, R.W.; Dunbar, C.E. Ex vivo expansion of retrovirally transduced primate cd34+ cells results in overrepresentation of clones with MDS1/EVI1 insertion sites in the myeloid lineage after transplantation. Mol. Ther 2010, 18, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. Blat--the blast-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Beard, B.C.; Adair, J.E.; Trobridge, G.D.; Kiem, H.P. High-throughput genomic mapping of vector integration sites in gene therapy studies. Methods Mol. Biol. 2014, 1185, 321–344. [Google Scholar] [PubMed]
- Rae, D.T.; Collins, C.P.; Hocum, J.D.; Browning, D.L.; Trobridge, G.D. Modified genomic sequencing PCR using the miseq platform to identify retroviral integration sites. Hum. Gene Ther. Methods 2015, 26, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Hocum, J.D.; Battrell, L.R.; Maynard, R.; Adair, J.E.; Beard, B.C.; Rawlings, D.J.; Kiem, H.P.; Miller, D.G.; Trobridge, G.D. Visa-vector integration site analysis server: A web-based server to rapidly identify retroviral integration sites from next-generation sequencing. BMC Bioinform. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2014, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Milstein, M.; Mooser, C.K.; Hu, H.; Fejzo, M.; Slamon, D.; Goodglick, L.; Dry, S.; Colicelli, J. RIN1 is a breast tumor suppressor gene. Cancer Res. 2007, 67, 11510–11516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, H.; Zhou, H.; Du, T.; Zeng, L.; Cao, Y.; Chen, J.; Lai, Y.; Li, J.; Wang, G.; et al. Activation of Nuclear Factor kappa B pathway and downstream targets survivin and livin by SHARPIN contributes to the progression and metastasis of prostate cancer. Cancer 2014, 120, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- De Melo, J.; Tang, D. Elevation of SIPL1 (SHARPIN) increases breast cancer risk. PLoS ONE 2015, 10, e0127546. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. Survexpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Park, E.S.; Lee, J.S.; Lee, Y.H.; Ishikawa, T.; Kim, Y.J.; Thorgeirsson, S.S. Identification of potential driver genes in human liver carcinoma by genomewide screening. Cancer Res. 2009, 69, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses her2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of aromatase inhibitor resistance. Nat. Rev. Cancer 2015, 15, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Keng, V.W.; Villanueva, A.; Chiang, D.Y.; Dupuy, A.J.; Ryan, B.J.; Matise, I.; Silverstein, K.A.; Sarver, A.; Starr, T.K.; Akagi, K.; et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat. Biotechnol. 2009, 27, 264–274. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer a | Retroviral Vector b | Screen c | Identified Gene d | Reference e |
|---|---|---|---|---|
| Androgen-independent prostate cancer | Self-inactivating LV | In vivo | PTRF, GCOM1, MEX3D | [24] |
| In vitro | ATPAF1, TRPM4 | [24] | ||
| Breast cancer | MLV-LTR γRV | In vivo | SHARPIN, WWTR1, MAF1, RIN1 | [21] |
| Androgen-independent prostate cancer | Self-inactivating LV | In vivo | GLYATL1, FLNA, OBSCN, STRA13, WHSC1, ARFGAP3, KDM2A, FAM83H, CLDN7, CNOT6, B3GNT9 | [23] |
| Hepatocellular carcinoma | LV-LTR | In vivo | SOS1, BRAF, FIGN, RTL1 | [22] |
| HER 2+ breast cancer | Self-inactivating LV | In vitro | PIK3CA, PIK3CB, MAP4K3, CADM2, SOS1 | [25] |
| Pancreatic adenocarcinoma | Self-inactivating LV | In vitro | SOS1, MROH1, LOC100128338 | [25] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bii, V.M.; Trobridge, G.D. Identifying Cancer Driver Genes Using Replication-Incompetent Retroviral Vectors. Cancers 2016, 8, 99. https://doi.org/10.3390/cancers8110099
Bii VM, Trobridge GD. Identifying Cancer Driver Genes Using Replication-Incompetent Retroviral Vectors. Cancers. 2016; 8(11):99. https://doi.org/10.3390/cancers8110099
Chicago/Turabian StyleBii, Victor M., and Grant D. Trobridge. 2016. "Identifying Cancer Driver Genes Using Replication-Incompetent Retroviral Vectors" Cancers 8, no. 11: 99. https://doi.org/10.3390/cancers8110099
APA StyleBii, V. M., & Trobridge, G. D. (2016). Identifying Cancer Driver Genes Using Replication-Incompetent Retroviral Vectors. Cancers, 8(11), 99. https://doi.org/10.3390/cancers8110099