
The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy

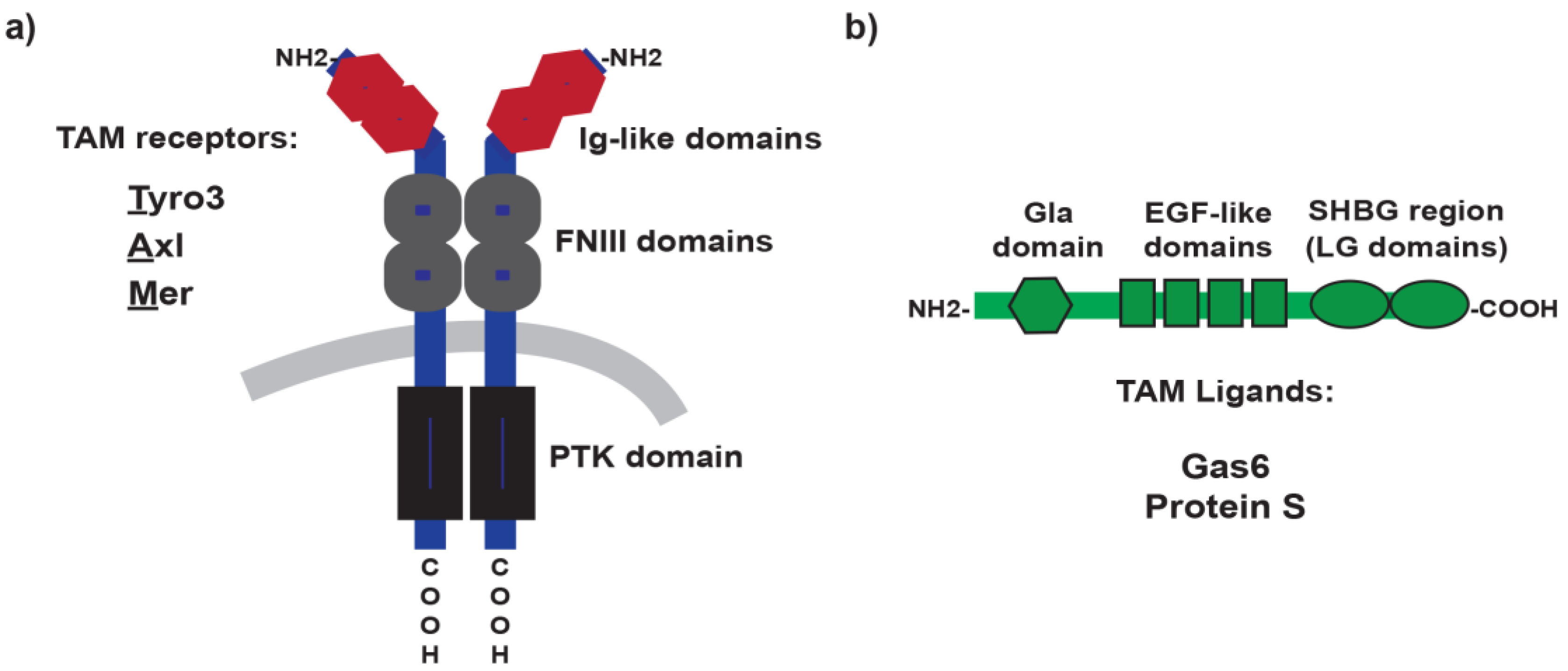{kind=link}
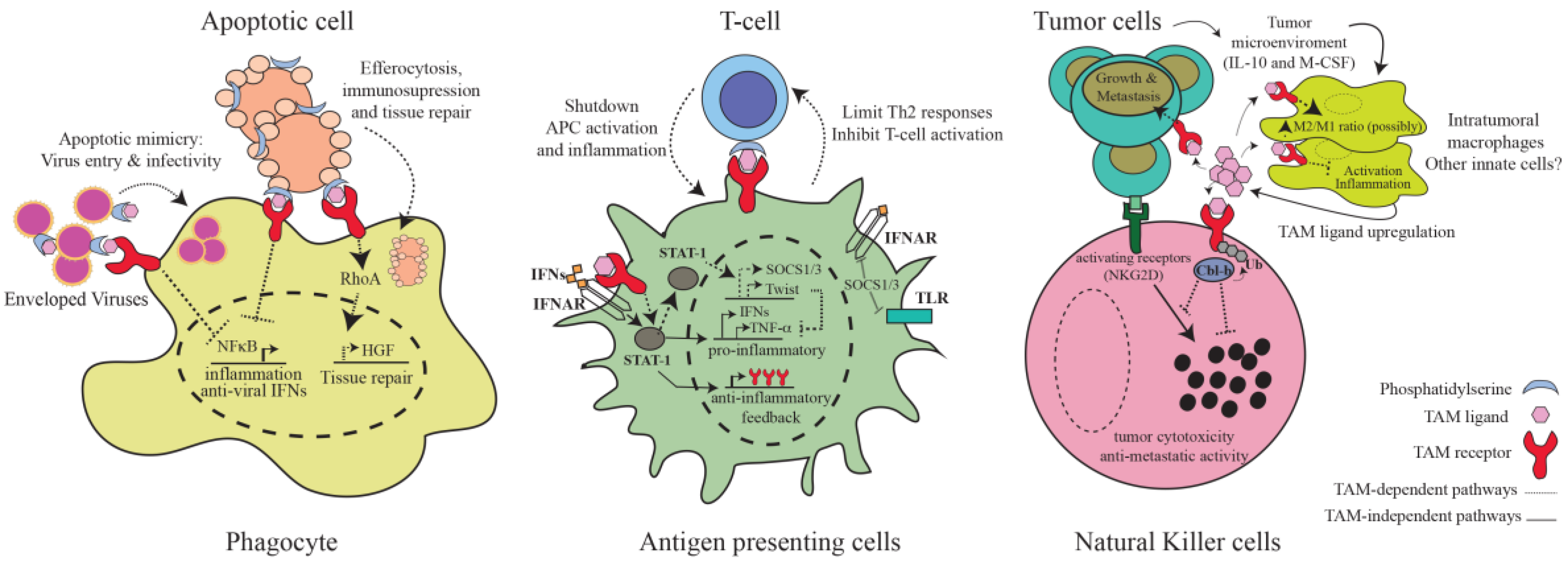{kind=link}
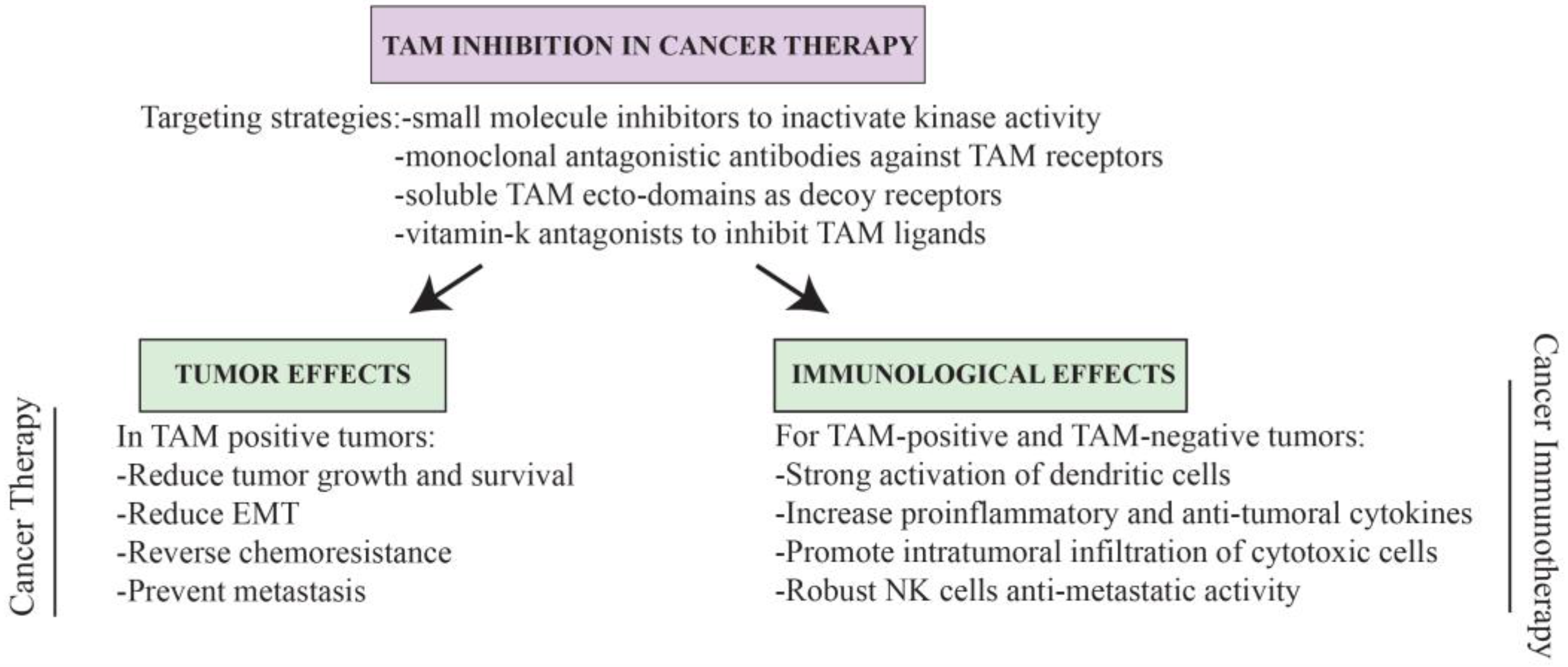{kind=link}
Abstract
:1. Introduction
2. TAM Receptors and Ligands: A Brief Overview
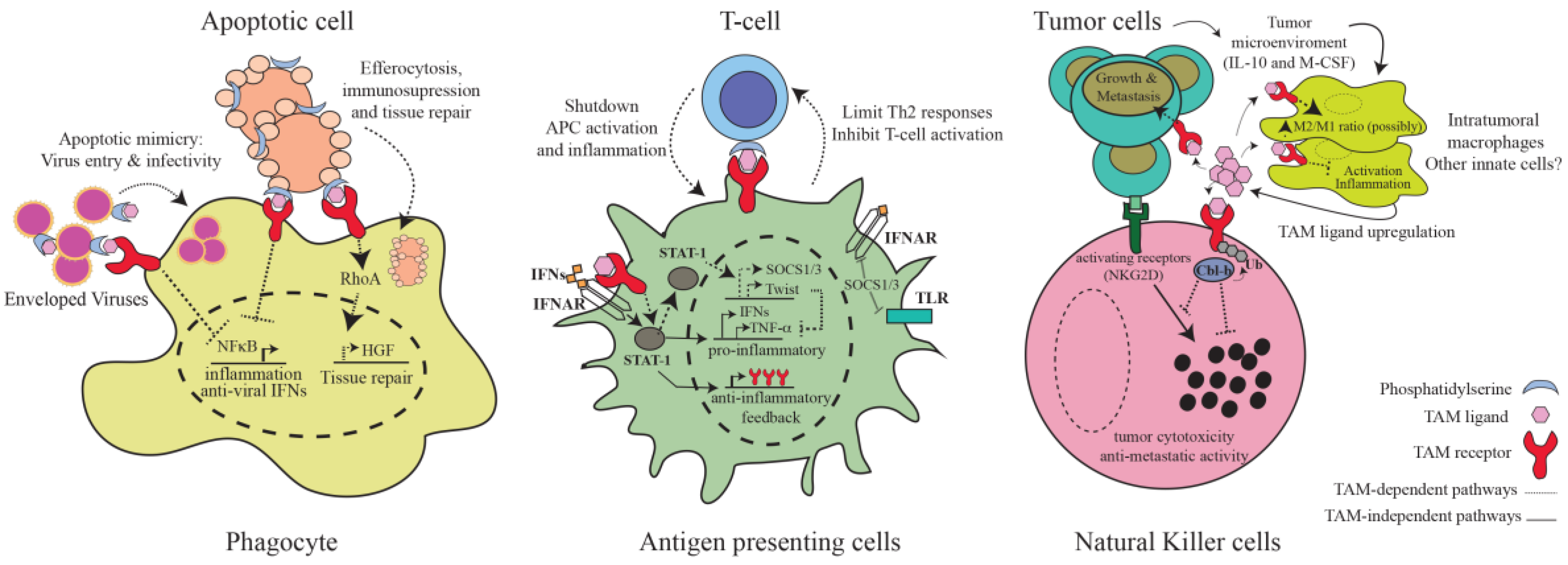
3. TAM-Mediated Regulation of Immunity
3.1. Inhibition of Inflammatory Pathways in Antigen-Presenting Cells
3.2. Phagocytosis of Apoptotic Cells
3.3. TAM Receptors as Integrators of Innate and Adaptive Immunity
3.4. NK and NKT Cells
3.5. Virus Entry and Infectivity
4. Functional Diversification for TAM Receptors
5. TAM Signaling in Autoimmunity
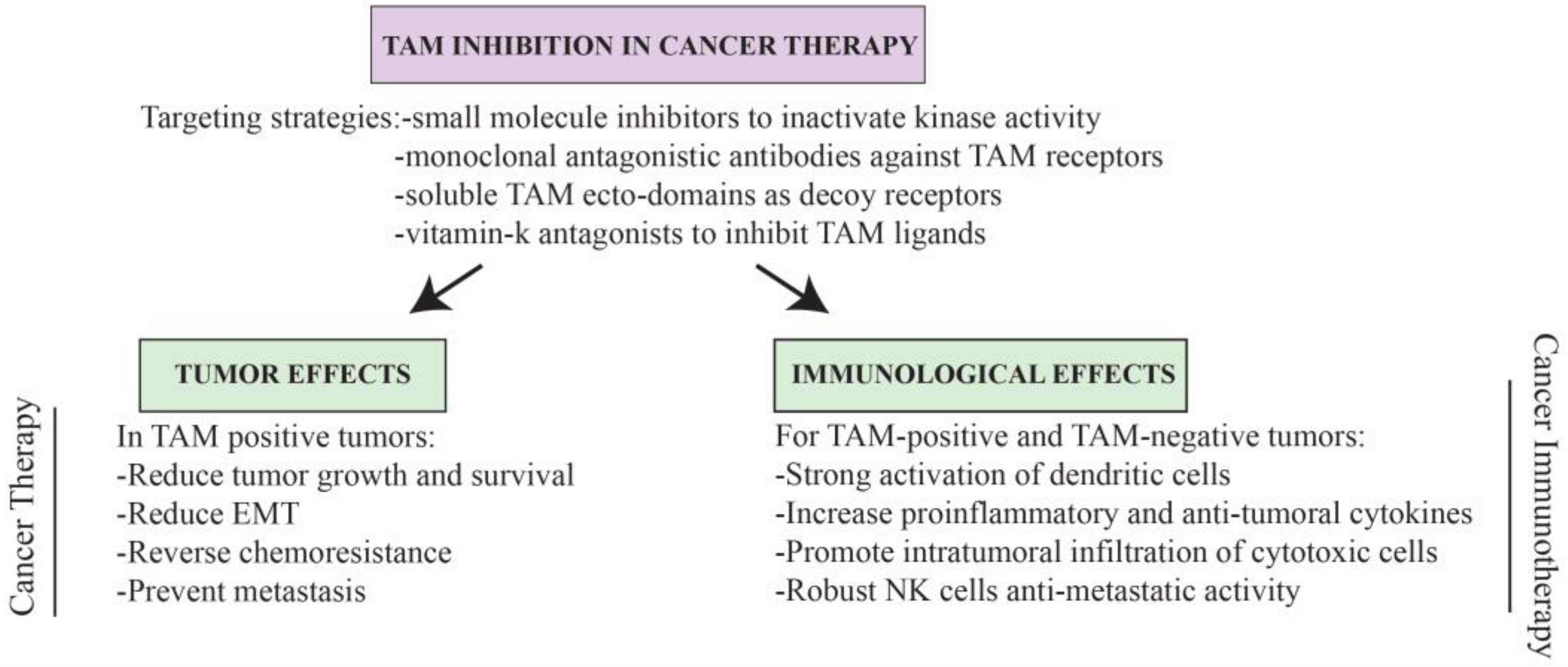
6. TAMing Anti-Tumor Immunity
7. Conclusions and Perspectives for TAM-Based Cancer Therapy
Acknowledgments
Conflicts of Interest
References
- Lai, C.; Lemke, G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron 1991, 6, 691–704. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.-M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. ERole of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Burnier, L.; Lambrechts, D.; Fish, R.J.; Tjwa, M.; Plaisance, S.; Sugamele, R.; DeMol, M.; Martinez-Soria, E.; Maxwell, P.H.; et al. Role of Gas6 in erythropoiesis and anemia in mice. J. Clin. Investig. 2008, 118, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Melaragno, M.G.; Cavet, M.E.; Yan, C.; Tai, L.-K.; Jin, Z.-G.; Haendeler, J.; Berk, B.C. Gas6 inhibits apoptosis in vascular smooth muscle: Role of Axl kinase and Akt. J. Mol. Cell. Cardiol. 2004, 37, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Happonen, K.E.; Tran, S.; Mörgelin, M.; Prince, R.; Calzavarini, S.; Angelillo-Scherrer, A.; Dahlbäck, B. The Gas6-Axl protein interaction mediates endothelial uptake of platelet microparticles. J. Biol. Chem. 2016, 291, 10586–10601. [Google Scholar] [CrossRef] [PubMed]
- Healy, A.M.; Schwartz, J.J.; Zhu, X.; Herrick, B.E.; Varnum, B.; Farber, H.W. Gas 6 promotes Axl-mediated survival in pulmonary endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1273–L1281. [Google Scholar] [PubMed]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [PubMed]
- Feng, W.; Yasumura, D.; Matthes, M.T.; LaVail, M.M.; Vollrath, D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J. Biol. Chem. 2002, 277, 17016–17022. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, M.; Hunter, D.M.; Strunk, K.E.; Earp, H.S.; Cook, R.S. Epithelial cell-directed efferocytosis in the post-partum mammary gland is necessary for tissue homeostasis and future lactation. BMC Dev. Biol. 2010, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.S.; Hakeda, Y.; Takakura, N.; Kameda, T.; Hamaguchi, I.; Miyamoto, T.; Kakudo, S.; Nakano, T.; Kumegawa, M.; Suda, T. Tyro3 Receptor tyrosine kinase and its ligand, Gas6, stimulate the function of osteoclasts. Stem Cells 1998, 16, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Wang, X.; Li, M.; Lin, P.H.; Yao, Q.; Chen, C. Human protein S inhibits the uptake of AcLDL and expression of SR-A through Mer receptor tyrosine kinase in human macrophages. Blood 2009, 113, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Meng, L.; Jiang, X.; Cvm, N.K.; Ding, J.; Li, Q.; Lu, Q. TAM receptors support neural stem cell survival, proliferation and neuronal differentiation. PLoS ONE 2014, 9, e115140. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Daniels, B.P.; Shrestha, B.; Proenca-Modena, J.L.; Lew, E.D.; Lazear, H.M.; Gorman, M.J.; Lemke, G.; Klein, R.S.; Diamond, M.S. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat. Med. 2015, 21, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, J.H.; van der Poll, T.; van’t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-A.; Jo, S.-Y.; Lee, H.-Y.; Lee, C. Inhibition of IL-6/STAT3 axis and targeting Axl and Tyro3 receptor tyrosine kinases by apigenin circumvent taxol resistance in ovarian cancer cells. Int. J. Oncol. 2015, 46, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Chowdhury, S.; Stebbing, J. TAMing resistance to multi-targeted kinase inhibitors through Axl and Met inhibition. Oncogene 2016, 35, 2684–2686. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.A.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2008; Volume 100, pp. 35–83. [Google Scholar]
- Baladi, T.; Abet, V.; Piguel, S. State-of-the-art of small molecule inhibitors of the TAM family: The point of view of the chemist. Eur. J. Med. Chem. 2015, 105, 220–237. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Schmidt, T.; Tjwa, M.; van Geyte, K.; Lievens, D.; Lutgens, E.; Vanhoutte, D.; Borgel, D.; Plaisance, S.; Hoylaerts, M.; et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood 2010, 115, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Gore, M.; Lemke, G. Structure, expression, and activity of Tyro3, a neural adhesion-related receptor tyrosine kinase. Oncogene 1994, 9, 2567–2578. [Google Scholar] [PubMed]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R.; Beau, M.M.L.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.K.; Dawson, T.L.; Mullaney, D.L.; Snodgrass, H.R.; Earp, H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994, 5, 647–657. [Google Scholar] [PubMed]
- Stitt, T.N.; Conn, G.; Goret, M.; Lai, C.; Bruno, J.; Radzlejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [PubMed]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (Gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagórska, A.; Través, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor–ligand–phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.-L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 2007, 178, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Uehara, H.; Shacter, E. Auto-oxidation and oligomerization of protein S on the apoptotic cell surface is required for Mer tyrosine kinase-mediated phagocytosis of apoptotic cells. J. Immunol. 2008, 180, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Vaisar, T.; Subramanian, M.; Mautner, L.; Blobel, C.; Tabas, I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 2011, 286, 33335–33344. [Google Scholar] [CrossRef] [PubMed]
- O’Bryan, J.P.; Fridell, Y.W.; Koski, R.; Varnum, B.; Liu, E.T. The transforming receptor tyrosine kinase, Axl, is post-translationally regulated by proteolytic cleavage. J. Biol. Chem. 1995, 270, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.-I.; Nguyen, K.-Q.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, Tyro3, Axl, and Mer, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Rigby, A.C.; Morelli, X.; Grant, M.A.; Huang, G.; Furie, B.; Seaton, B.; Furie, B.C. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 2003, 10, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Dalen, J.E.; Anderson, D.R.; Poller, L.; Bussey, H.; Ansell, J.; Deykin, D. Oral anticoagulants: Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001, 119, 8S–21S. [Google Scholar] [CrossRef] [PubMed]
- Kirane, A.; Ludwig, K.F.; Sorrelle, N.; Haaland, G.; Sandal, T.; Ranaweera, R.; Toombs, J.E.; Wang, M.; Dineen, S.P.; Micklem, D.; et al. Warfarin blocks Gas6-mediated Axl activation required for pancreatic cancer epithelial plasticity and metastasis. Cancer Res. 2015, 75, 3699–3705. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Arai, H.; Yanagita, M.; Matsubara, T.; Kanamori, H.; Nakano, T.; Iehara, N.; Fukatsu, A.; Kita, T.; Doi, T. Growth arrest-specific gene 6 is involved in glomerular hypertrophy in the early stage of diabetic nephropathy. J. Biol. Chem. 2003, 278, 18229–18234. [Google Scholar] [CrossRef] [PubMed]
- Waizenegger, J.S.; Ben-Batalla, I.; Weinhold, N.; Meissner, T.; Wroblewski, M.; Janning, M.; Riecken, K.; Binder, M.; Atanackovic, D.; Taipaleenmaeki, H.; et al. Role of growth arrest-specific gene 6-Mer axis in multiple myeloma. Leukemia 2015, 29, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Kurohara, M.; Yasuda, H.; Moriyama, H.; Nakayama, M.; Sakata, M.; Yamada, K.; Kotani, R.; Hara, K.; Yokono, K.; Nagata, M. Low-dose warfarin functions as an immunomodulator to prevent cyclophosphamide-induced NOD diabetes. Kobe J. Med. Sci. 2008, 54, E1–E13. [Google Scholar] [PubMed]
- Shao, W.-H.; Zhen, Y.; Eisenberg, R.A.; Cohen, P.L. The Mer receptor tyrosine kinase is expressed on discrete macrophage subpopulations and mainly uses Gas6 as its ligand for uptake of apoptotic cells. Clin. Immunol. (Orlando, Fla.) 2009, 133, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Immunological genome consortium gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Wallet, M.A.; Sen, P.; Flores, R.R.; Wang, Y.; Yi, Z.; Huang, Y.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Wang, B.; et al. MerTK is required for apoptotic cell-induced T cell tolerance. J. Exp. Med. 2008, 205, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, B.A.; Zizzo, G.; Ulas, M.; Linan, M.K.; Schreiter, J.; Cohen, P.L. Increased expression of Mer tyrosine kinase in circulating dendritic cells and monocytes of lupus patients: Correlations with plasma interferon activity and steroid therapy. Arthritis Res. Ther. 2014, 16, R76. [Google Scholar] [CrossRef] [PubMed]
- Gould, W.R.; Baxi, S.M.; Schroeder, R.; Peng, Y.W.; Leadley, R.J.; Peterson, J.T.; Perrin, L.A. Gas6 receptors Axl, Sky and Mer enhance platelet activation and regulate thrombotic responses. J. Thromb. Haemost. 2005, 3, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Gadue, P.; Gong, S.; Garrett, S.; Stein, P.L.; Cohen, P.L. The mer receptor tyrosine kinase: Expression and function suggest a role in innate immunity. Eur. J. Immunol. 2003, 33, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Carrera Silva, E.A.; Chan, P.Y.; Joannas, L.; Errasti, A.E.; Gagliani, N.; Bosurgi, L.; Jabbour, M.; Perry, A.; Smith-Chakmakova, F.; Mucida, D.; et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 2013, 39, 160–170. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Harkes, I.C.; Dougherty, L.; Wicks, I.P. Expression of receptor tyrosine kinase Axl and its ligand Gas6 in rheumatoid arthritis. Am. J. Pathol. 1999, 154, 1171–1180. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Koller, B.H.; Earp, H.S.; Matsushima, G.K. A novel receptor tyrosine kinase, Mer, inhibits TNF-α production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 1999, 162, 3498–3503. [Google Scholar] [PubMed]
- Sharif, M.N.; Šošić, D.; Rothlin, C.V.; Kelly, E.; Lemke, G.; Olson, E.N.; Ivashkiv, L.B. Twist mediates suppression of inflammation by type I IFNs and Axl. J. Exp. Med. 2006, 203, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Burstyn-Cohen, T. TAM receptors and the clearance of apoptotic cells: TAM signaling and apoptotic cell clearance. Ann. N. Y. Acad. Sci. 2010, 1209, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Peters, K.N.; Behar, S.M. Macrophages clean up: Efferocytosis and microbial control. Curr. Opin. Microbiol. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter-Hufford, A.; Ravichandran, K.S. Clearing the dead: Apoptotic Cell sensing, recognition, engulfment, and digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, a008748. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.P.; Earp, H.S. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: A mechanism for localizing guanine nucleotide-exchange factor action. J. Biol. Chem. 2003, 278, 42596–42603. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Singh, S.; Georgescu, M.-M.; Birge, R.B. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 2005, 118, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Bosurgi, L.; Bernink, J.H.; Delgado Cuevas, V.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Wallet, M.A.; Yi, Z.; Huang, Y.; Henderson, M.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Baldwin, A.S.; Tisch, R.M. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-κB activation in dendritic cells. Blood 2007, 109, 653–660. [Google Scholar] [CrossRef] [PubMed]
- A-Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Díaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Baen, J.-Y.; Lee, Y.-J.; Choi, Y.-H.; Kang, J.L. The TAM-family receptor Mer mediates production of HGF through the RhoA-dependent pathway in response to apoptotic cells. Mol. Biol. Cell 2012, 23, 3254–3265. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; Boyer, S.N.; Heeb, M.J.; Griffin, J.H.; Grusby, M.J. Protein S is inducible by interleukin 4 in T cells and inhibits lymphoid cell procoagulant activity. Proc. Natl. Acad. Sci. USA 1997, 94, 11484–11489. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; Stitt, T.N.; Grusby, M.J. Cross-linking of protein S bound to lymphocytes promotes aggregation and inhibits proliferation. Cell. Immunol. 1997, 181, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Cabezon, R.; Carrera-Silva, E.A.; Florez-Grau, G.; Errasti, A.E.; Calderon-Gomez, E.; Lozano, J.J.; Espana, C.; Ricart, E.; Panes, J.; Rothlin, C.V.; et al. MERTK as negative regulator of human T cell activation. J. Leukoc. Biol. 2015, 97, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.Y.; Silva, E.A.C.; De Kouchkovsky, D.; Joannas, L.D.; Hao, L.; Hu, D.; Huntsman, S.; Eng, C.; Licona-Limon, P.; Weinstein, J.S.; et al. The TAM family receptor tyrosine kinase Tyro3 is a negative regulator of type 2 immunity. Science 2016, 352, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Walzer, T.; Vivier, E. NK cell development: Gas matters. Nat. Immunol. 2006, 7, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Caraux, A.; Lu, Q.; Fernandez, N.; Riou, S.; Di Santo, J.P.; Raulet, D.H.; Lemke, G.; Roth, C. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat. Immunol. 2006, 7, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-K.; Giovenzana, C.; Hughes, T.L.; Yu, J.; Trotta, R.; Caligiuri, M.A. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood 2009, 113, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J. Viral apoptotic mimicry party: P.S. bring your own Gas6. Cell Host Microbe 2011, 9, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Drayman, N.; Glick, Y.; Ben-nun-Shaul, O.; Zer, H.; Zlotnick, A.; Gerber, D.; Schueler-Furman, O.; Oppenheim, A. Pathogens use structural mimicry of native host ligands as a mechanism for host receptor engagement. Cell Host Microbe 2013, 14, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Zagórska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A.T. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Grabiec, A.M.; Kaur, M.; Bell, T.J.; Fujino, N.; Cook, P.C.; Svedberg, F.R.; MacDonald, A.S.; Maciewicz, R.A.; Singh, D.; et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. 2015, 8, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of ebola and marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika virus infection in human skin cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.T.; Pang, I.K.; Carrera Silva, E.A.; Bosurgi, L.; Miner, J.J.; Diamond, M.S.; Iwasaki, A.; Rothlin, C.V. AXL receptor tyrosine kinase is required for T cell priming and antiviral immunity. eLife 2016, 5, e12414. [Google Scholar] [CrossRef] [PubMed]
- Zagórska, A.; Través, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed]
- McColl, A.; Bournazos, S.; Franz, S.; Perretti, M.; Morgan, B.P.; Haslett, C.; Dransfield, I. Glucocorticoids induce protein S-dependent Phagocytosis of apoptotic neutrophils by human macrophages. J. Immunol. 2009, 183, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Qi, N.; Shang, T.; Wu, H.; Deng, T.; Han, D. Sertoli cell-initiated testicular innate immune response through toll-like receptor-3 activation is negatively regulated by Tyro3, Axl, and mer receptors. Endocrinology 2010, 151, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Chen, Q.; Yan, K.; Liu, Z.; Zhang, X.; Liu, P.; Chen, Y.; Han, D. Breakdown of immune homeostasis in the testis of mice lacking Tyro3, Axl and Mer receptor tyrosine kinases. Immunol. Cell Biol. 2013, 91, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Y.; Ge, Y.; Ma, P.; Ma, Q.; Ma, J.; Wang, H.; Xue, S.; Han, D. Immunoexpression of Tyro3 family receptors—Tyro3, Axl, and Mer—And their ligand Gas6 in postnatal developing mouse testis. J. Histochem. Cytochem. 2005, 53, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Chen, Y.; Wang, H.; Wang, H.; Wu, H.; Lu, Q.; Han, D. Gas6 and the Tyro3 receptor tyrosine kinase subfamily regulate the phagocytic function of Sertoli cells. Reproduction (Camb. Engl.) 2008, 135, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Recarte-Pelz, P.; Tàssies, D.; Espinosa, G.; Hurtado, B.; Sala, N.; Cervera, R.; Reverter, J.C.; de Frutos, P.G. Vitamin K-dependent proteins GAS6 and Protein S and TAM receptors in patients of systemic lupus erythematosus: Correlation with common genetic variants and disease activity. Arthritis Res. Ther. 2013, 15, R41. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, L.; Zhang, D.; Yu, H.; Tan, H.; Zhang, J.; Deng, B.; Kijlstra, A.; Yang, P. Analysis of receptor tyrosine kinase genetics identifies two novel risk loci in GAS6 and PROS1 in Behçet’s disease. Sci. Rep. 2016, 6, 26662. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Chu, N.-F.; Shieh, Y.-S.; Hung, Y.-J. The growth arrest-specific 6 (Gas6) gene polymorphism c.834+7G>A is associated with type 2 diabetes. Diabetes Res. Clin. Pract. 2012, 95, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Sun, X.; Zhu, L.; Hu, F.; Shi, L.; Fan, C.; Li, Z.; Su, Y. Different expression patterns and clinical significance of mAxl and sAxl in systemic lupus erythematosus. Lupus 2014, 23, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Guermazi, S.; Hamza, M.; Dellagi, K. Protein S deficiency and antibodies to protein S in patients with Behçet’s disease. Thromb. Res. 1997, 86, 197–204. [Google Scholar] [CrossRef]
- Szász, A.; Strifler, G.; Vörös, A.; Váczi, B.; Tubak, V.; Puskás, L.G.; Belső, N.; Kemény, L.; Nagy, I. The expression of TAM receptors and their ligand Gas6 is downregulated in psoriasis. J. Dermatol. Sci. 2013, 71, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Cakal, B.; Gokmen, A.; Yalinkilic, M.; Cakal, E.; Ayaz, S.; Nadir, I.; Ozin, Y.; Dagli, U.; Ulker, A. Natural anticoagulant protein levels in Turkish patients with inflammatory bowel disease. Blood Coagul. Fibrinolysis. 2010, 21, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Aadland, E.; Odegaard, O.R.; Røseth, A.; Try, K. Free protein S deficiency in patients with chronic inflammatory bowel disease. Scand. J. Gastroenterol. 1992, 27, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Ballantine, L.; Midgley, A.; Harris, D.; Richards, E.; Burgess, S.; Beresford, M.W. Increased soluble phagocytic receptors sMer, sTyro3 and sAxl and reduced phagocytosis in Juvenile-onset Systemic Lupus Erythematosus. Pediatr. Rheumatol. Online J. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Wang, J.; Ma, N.; Yang, M.; Fu, H.; Liang, Y.; Huang, F.; Yang, Z.; Zhong, R. The association of Tyro3/Axl/Mer signaling with inflammatory response, disease activity in patients with primary Sjögren’s syndrome. Jt. Bone Spine Rev. Rhum. 2015, 82, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ekman, C.; Jönsen, A.; Sturfelt, G.; Bengtsson, A.A.; Gottsäter, A.; Lindblad, B.; Lindqvist, E.; Saxne, T.; Dahlbäck, B. Increased plasma levels of the soluble Mer tyrosine kinase receptor in systemic lupus erythematosus relate to disease activity and nephritis. Arthritis Res. Ther. 2011, 13, R62. [Google Scholar] [CrossRef] [PubMed]
- Gheita, T.A.; Bassyouni, I.H.; Bassyouni, R.H. Plasma concentrations of growth arrest specific protein 6 and the soluble form of its tyrosine kinase receptor Axl in patients with systemic lupus erythematosus and Behçets disease. J. Clin. Immunol. 2012, 32, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Qi, N.; Liu, P.; Zhang, Y.; Wu, H.; Chen, Y.; Han, D. Development of a spontaneous liver disease resembling autoimmune hepatitis in mice lacking Tyro3, Axl and Mer receptor tyrosine kinases. PLoS ONE 2013, 8, e66604. [Google Scholar] [CrossRef] [PubMed]
- Waterborg, C.E.J.; Través, P.G.; Zagórska, A.; Lemke, G.; Beermann, S.; van de Loo, F.A. The TAM receptors Axl and Mer play a joint-specific protective role in experimental arthritis. J. Immunol. 2016, 196, 117. [Google Scholar]
- Weinger, J.G.; Brosnan, C.F.; Loudig, O.; Goldberg, M.F.; Macian, F.; Arnett, H.A.; Prieto, A.L.; Tsiperson, V.; Shafit-Zagardo, B. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during Experimental Autoimmune Encephalomyelitis. J. Neuroinflamm. 2011, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Van den Brand, B.T.; Abdollahi-Roodsaz, S.; Vermeij, E.A.; Bennink, M.B.; Arntz, O.J.; Rothlin, C.V.; van den Berg, W.B.; van de Loo, F.A.J. Therapeutic efficacy of Tyro3, Axl, and Mer tyrosine kinase agonists in collagen-induced arthritis. Arthritis Rheum. 2013, 65, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Tsiperson, V.; Li, X.; Schwartz, G.J.; Raine, C.S.; Shafit-Zagardo, B. GAS6 enhances repair following cuprizone-induced demyelination. PLoS ONE 2010, 5, e15748. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.S.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.N.; Wong, E.B.; Soni, C.; Rahman, Z.S.M. Prolonged apoptotic cell accumulation in germinal centers of Mer-deficient mice causes elevated B cell and CD4+ Th cell responses leading to autoantibody production. J. Immunol. 2013, 190, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-Y.; Suh, C.-H. Incomplete clearance of apoptotic cells in systemic lupus erythematosus: Pathogenic role and potential biomarker. Int. J. Rheum. Dis. 2015, 18, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Warner, S.L.; Vankayalapati, H.; Bearss, D.J.; Sharma, S. Targeting Axl and Mer kinases in cancer. Mol. Cancer Ther. 2011, 10, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.P.; Kohlschmidt, J.; Maharry, K.; Volinia, S.; Mrózek, K.; Nicolet, D.; Schwind, S.; Becker, H.; Metzeler, K.H.; Mendler, J.H.; et al. GAS6 expression identifies high-risk adult AML patients: Potential implications for therapy. Leukemia 2014, 28, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, K.; Yan, Z.; Xia, Y.; Li, J.; Shi, L.; Zou, Q.; Wan, X.; Jiao, B.; Wang, H.; et al. Axl expression stratifies patients with poor prognosis after hepatectomy for hepatocellular carcinoma. PLoS ONE 2016, 11, e0154767. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suda, K.; Shimizu, S.; Sakai, K.; Mizuuchi, H.; Tomizawa, K.; Takemoto, T.; Nishio, K.; Mitsudomi, T. Clinical, pathological, and molecular features of lung adenocarcinomas with AXL expression. PLoS ONE 2016, 11, e0154186. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-S.; Yang, P.-W.; Wong, L.-F.; Lee, J.-M. The AXL receptor tyrosine kinase is associated with adverse prognosis and distant metastasis in esophageal squamous cell carcinoma. Oncotarget 2016, 7, 36956–36970. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.; Pfeiffer, C.; Strube, S.; Alsadeq, A.; Fedders, H.; Vokuhl, C.; Loges, S.; Waizenegger, J.; Ben-Batalla, I.; Cario, G.; et al. Mer tyrosine kinase promotes the survival of t(1;19)-positive acute lymphoblastic leukemia (ALL) in the central nervous system (CNS). Blood 2015, 125, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Ammoun, S.; Provenzano, L.; Zhou, L.; Barczyk, M.; Evans, K.; Hilton, D.A.; Hafizi, S.; Hanemann, C.O. Axl/Gas6/NFκB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene 2014, 33, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.-W.; Hou, P.-C.; Wu, H.-C.; Chang, Y.-L.; Lin, S.-C.; Lin, S.-C.; Lin, B.-W.; Lee, J.-C.; Chang, Y.-J.; Sun, H.S.; et al. Targeting Tyro3 inhibits epithelial–mesenchymal transition and increases drug sensitivity in colon cancer. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Knubel, K.H.; Pernu, B.M.; Sufit, A.; Nelson, S.; Pierce, A.M.; Keating, A.K. MerTK inhibition is a novel therapeutic approach for glioblastoma multiforme. Oncotarget 2014, 5, 1338–1351. [Google Scholar] [CrossRef] [PubMed]
- Moody, G.; Belmontes, B.; Masterman, S.; Wang, W.; King, C.; Murawsky, C.; Tsuruda, T.; Liu, S.; Radinsky, R.; Beltran, P.J. Antibody-mediated neutralization of autocrine Gas6 inhibits the growth of pancreatic ductal adenocarcinoma tumors in vivo: Anti-tumor activity of a fully human Gas6 neutralizing antibody in vitro and in vivo. Int. J. Cancer 2016, 139, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; An, Q.; Birt, M.; Pilkington, G.J.; Hafizi, S. Small molecule inhibition of Axl receptor tyrosine kinase potently suppresses multiple malignant properties of glioma cells. Oncotarget 2015, 6, 16183–16197. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Valls, A.F.; Yerbes, R.; von Richter, S.; Kahlert, C.; Loges, S.; Weitz, J.; Schneider, M.; de Almodovar, C.R.; Ulrich, A.; et al. TAM receptors Tyro3 and Mer as novel targets in colorectal cancer. Oncotarget 2016, 7, 56355–56370. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.J.; Gardner, J.; Vendel, M.C.; Ailor, E.; Szak, S.; Huang, F.; Doern, A.; Tan, X.; Yang, W.; Grueneberg, D.A.; et al. Evaluation of Tyro3 expression, Gas6-mediated Akt phosphorylation, and the impact of anti-Tyro3 antibodies in melanoma cell lines. Biochemistry (Mosc.) 2013, 52, 3102–3118. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.A.; Kohrt, J.T.; Filipski, K.J.; Kaufman, M.; Sheehan, D.; Edmunds, J.E.; Delaney, A.; Wang, Y.; Bourbonais, F.; Lee, D.-Y.; et al. Novel and selective spiroindoline-based inhibitors of Sky kinase. Bioorg. Med. Chem. Lett. 2012, 22, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Traoré, T.; Cavagnino, A.; Saettel, N.; Radvanyi, F.; Piguel, S.; Bernard-Pierrot, I.; Stoven, V.; Legraverend, M. New aminopyrimidine derivatives as inhibitors of the TAM family. Eur. J. Med. Chem. 2013, 70, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. First Axl inhibitor enters clinical trials. Nat. Biotechnol. 2013, 31, 775–776. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in cancer: A medicinal chemistry perspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, X.; Koul, S.; Lee, C.Y.; Zhang, Z.; Halmos, B. AXL kinase as a novel target for cancer therapy. Oncotarget 2014, 5, 9546–9563. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O. Negative signalling by inhibitory receptors: The NK cell paradigm. Immunol. Rev. 2008, 224, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Lacour, F.; Oberling, C.; Guerin, M. Effect of dicoumarol on the development of metastases of the T8 epithelioma in the rat; new research. Bull. Assoc. Fr. Pour Létude Cancer 1957, 44, 88–91. [Google Scholar]
- Ryan, J.J.; Ketcham, A.S.; Wexler, H. Reduced incidence of spontaneous metastases with long-term Coumadin therapy. Ann. Surg. 1968, 168, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Ketcham, A.S.; Wexler, H. Warfarin therapy as an adjunct to the surgical treatment of malignant tumors in mice. Cancer Res. 1969, 29, 2191–2194. [Google Scholar] [PubMed]
- McCulloch, P.; George, W.D. Warfarin inhibits metastasis of Mtln3 rat mammary carcinoma without affecting primary tumour growth. Br. J. Cancer 1989, 59, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. A study of the mechanism by which anticoagulation with warfarin inhibits blood-borne metastases. Cancer Res. 1973, 33, 1217–1224. [Google Scholar] [PubMed]
- Schulman, S.; Lindmarker, P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. N. Engl. J. Med. 2000, 342, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Tagalakis, V.; Tamim, H.; Blostein, M.; Collet, J.-P.; Hanley, J.A.; Kahn, S.R. Use of warfarin and risk of urogenital cancer: A population-based, nested case-control study. Lancet Oncol. 2007, 8, 395–402. [Google Scholar] [CrossRef]
- Pengo, V.; Noventa, F.; Denas, G.; Pengo, M.F.; Gallo, U.; Grion, A.M.; Iliceto, S.; Prandoni, P. Long-term use of vitamin K antagonists and incidence of cancer: A population-based study. Blood 2011, 117, 1707–1709. [Google Scholar] [CrossRef] [PubMed]
- Pottegård, A.; Friis, S.; Hallas, J. Cancer risk in long-term users of vitamin K antagonists: A population-based case-control study. Int. J. Cancer 2013, 132, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Henderson, W.G.; Rickles, F.R.; Forman, W.B.; Cornell, C.J.; Forcier, R.J.; Edwards, R.L.; Headley, E.; Kim, S.H.; O’Donnell, J.F. Effect of warfarin anticoagulation on survival in carcinoma of the lung, colon, head and neck, and prostate. Final report of VA Cooperative Study #75. Cancer 1984, 53, 2046–2052. [Google Scholar] [PubMed]
- Aisner, J.; Goutsou, M.; Maurer, L.H.; Cooper, R.; Chahinian, P.; Carey, R.; Skarin, A.; Slawson, R.; Perry, M.C.; Green, M.R. Intensive combination chemotherapy, concurrent chest irradiation, and warfarin for the treatment of limited-disease small-cell lung cancer: A Cancer and Leukemia Group B pilot study. J. Clin. Oncol. 1992, 10, 1230–1236. [Google Scholar] [PubMed]
- Chahinian, A.P.; Propert, K.J.; Ware, J.H.; Zimmer, B.; Perry, M.C.; Hirsh, V.; Skarin, A.; Kopel, S.; Holland, J.F.; Comis, R.L. A randomized trial of anticoagulation with warfarin and of alternating chemotherapy in extensive small-cell lung cancer by the Cancer and Leukemia Group B. J. Clin. Oncol. 1989, 7, 993–1002. [Google Scholar] [PubMed]
- Akl, E.A.; Kamath, G.; Kim, S.Y.; Yosuico, V.; Barba, M.; Terrenato, I.; Sperati, F.; Schünemann, H.J. Oral anticoagulation may prolong survival of a subgroup of patients with cancer: A cochrane systematic review. J. Exp. Clin. Cancer Res. CR 2007, 26, 175–184. [Google Scholar] [PubMed]
- Yanagita, M.; Arai, H.; Ishii, K.; Nakano, T.; Ohashi, K.; Mizuno, K.; Varnum, B.; Fukatsu, A.; Doi, T.; Kita, T. Gas6 regulates mesangial cell proliferation through Axl in experimental glomerulonephritis. Am. J. Pathol. 2001, 158, 1423–1432. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Leighton, J.A.; Ghosh, S. Tyro3, Axl, and Mertk receptor signaling in inflammatory bowel disease and colitis-associated cancer. Inflamm. Bowel Dis. 2014, 20, 1472–1480. [Google Scholar] [CrossRef] [PubMed]
- Akitake-Kawano, R.; Seno, H.; Nakatsuji, M.; Kimura, Y.; Nakanishi, Y.; Yoshioka, T.; Kanda, K.; Kawada, M.; Kawada, K.; Sakai, Y.; et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis 2013, 34, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Dougan, M.; Dranoff, G. Immune therapy for cancer. Annu. Rev. Immunol. 2009, 27, 83–117. [Google Scholar] [CrossRef] [PubMed]
- Hoos, A. Development of immuno-oncology drugs—From CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Scutera, S.; Fraone, T.; Musso, T.; Cappello, P.; Rossi, S.; Pierobon, D.; Orinska, Z.; Paus, R.; Bulfone-Paus, S.; Giovarelli, M. Survival and migration of human dendritic cells are regulated by an IFN-α-inducible Axl/Gas6 pathway. J. Immunol. 2009, 183, 3004–3013. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Hedl, M.; Abraham, C. TAM receptor-dependent regulation of SOCS3 and MAPKs contributes to proinflammatory cytokine downregulation following chronic NOD2 stimulation of human macrophages. J. Immunol. 2015, 194, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Burstyn-Cohen, T.; Heeb, M.J.; Lemke, G. Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J. Clin. Investig. 2009, 119, 2942–2953. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolino, M.; Penninger, J.M. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers 2016, 8, 97. https://doi.org/10.3390/cancers8100097
Paolino M, Penninger JM. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers. 2016; 8(10):97. https://doi.org/10.3390/cancers8100097
Chicago/Turabian StylePaolino, Magdalena, and Josef M. Penninger. 2016. "The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy" Cancers 8, no. 10: 97. https://doi.org/10.3390/cancers8100097
APA StylePaolino, M., & Penninger, J. M. (2016). The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers, 8(10), 97. https://doi.org/10.3390/cancers8100097