1. Introduction
Over the last several decades it has become increasingly clear that the immune system plays an important role in cancer prevention through a process called immune surveillance [
1]. In turn, the selective pressure exerted by the immune system causes tumors to develop multiple mechanisms to escape immune detection, including the induction of immune tolerance, local suppression of the immune response, and systematic disruption of T-cell signaling [
2]. Recently, there has been intense focus on immunotherapy, which the National Cancer Institute defines as “biologic therapy that uses substances to stimulate or suppress the immune system to help the body fight cancer, infection, and other diseases”. However, the idea that the immune system can be used to treat or cure cancer is not new; in fact, the first studies using immunomodulation for cancer therapy were conducted over a century ago [
3]. Despite this long history, modern cancer immunotherapies including monoclonal antibodies, tumor vaccines, oncolytic viruses, and proinflammatory molecules, have only met significant clinical success over the last decade [
4].
One cancer that may benefit from immunotherapy is head and neck squamous cell carcinoma (HNSCC). Worldwide, HNSCC represents the sixth most common cancer, resulting in approximately 550,000 diagnoses and 300,000 deaths per year [
5]. The relatively poor overall survival (OS) for HNSCC patients despite advances in surgical techniques, chemotherapy, and radiation therapy results, in part, from an inability to further intensify therapy due to unacceptable morbidity, and partly from the profound immunosuppressive effects of the tumor itself [
6]. Therefore, significant efforts have been directed towards stimulating the immune response against HNSCC to improve survival and reduce morbidity associated with the disease. In this review, we outline immunomodulatory approaches to HNSCC treatment that are currently under investigation as Phase I, II, or III clinical trials.
2. Identification of Clinical Trials
Current clinical trials for HNSCC immunotherapies were identified by querying the ClinicalTrials.gov database for “HNSCC” + “immune” or “squamous cell carcinoma” + “head and neck” + “immunotherapy”. At the time of the search in April 2016, this approach yielded 53 trials, which were manually reviewed for inclusion. To focus on current clinical trials, studies that were withdrawn without results were excluded, while trials that were terminated or completed without posted results were included. In total, 33 trials covering 22 different immunotherapeutic approaches were retained for discussion (
Table 1). For clarity, the immunotherapeutic agents are divided into four broad categories based on the class of therapy, with further sub-classification of different agents based on the actual or proposed mechanism of action (
Table 1). The four major classes of immunotherapy covered by this review are vaccine therapies, monoclonal antibodies, oncolytic viruses and active immunotherapeutics, and immunomodulators (
Table 1).
3. Vaccine Therapy
Anticancer vaccine therapies involve generating an antitumor immune response by presenting a tumor-associated antigen (TAA) plus an immunostimulatory adjuvant, resulting in immune sensitization to tumor antigens. To date, several vaccination strategies have been applied, including the transfection of TAA expression plasmids into patient tissues (DNA vaccines), the administration of TAA peptides (peptide vaccines), and the use of cultured human or microbial cells to generate an antitumor immune response.
3.1. DNA Vaccines
3.1.1. INO-3112
INO-3112 is a combination of two previously developed DNA vaccines, VGX-3100 and INO-9012, originally developed for the treatment of cervical cancer. The expression plasmids contained within the vaccine produce E6 and E7 proteins from human papillomavirus (HPV)16 and HPV18, respectively, resulting in an HPV-specific CD8+ T cell response. Given the selectivity for HPV proteins, this vaccine is only appropriate for HPV-positive HNSCC. The vaccine is administered as an intramuscular injection of plasmid DNA once every three weeks to a total of four doses. Because the plasmid-encoded antigens must be expressed to generate an immune response, each injection is accompanied by electroporation with the CELLECTRA
® device, which causes surrounding myocytes to incorporate and express the vaccine plasmids. Interim results from Phase I trials of INO-3112 including 19 patients showed that 80% (4/5) of patients tested had elevated anti-E6/E7 antibody titers for HPV16 and HPV18 and that 87.5% (7/8) of patients tested demonstrated elevated CD8+ T cell responses following vaccination [
7,
8].
Adverse effects in the study group were generally mild and included injection site pain (58%), local erythema (21%), hematoma (13%), and swelling (13%). All adverse events were Grade 2 or lower. Efficacy endpoints have yet to be reported for INO-3112, however a phase II trial of VGX-3100, which is included in INO-3112, demonstrated similar immune responses and resulted in regression of cervical intraepithelial neoplasia grade II or III lesions in 48.2% of patients in the experimental group compared to disease regression in only 30% of patients receiving placebo [
9]. INO-3112 is currently being tested in a Phase I/II trial for HNSCC with results expected in 2017.
3.1.2. Allovectin-7
The Allovectin-7 vaccine is a DNA/lipid complex containing plasmids encoding the Human Leukocyte Antigen-B7 (HLA-B7) heavy chain and β
2 microglobulin, resulting in the expression of the complete major histocompatibility complex type-I (MHC-I) molecules on the surface of tumor cells. Many tumor types evade immune recognition by reducing MHC-I expression, thereby preventing the presentation of tumor antigens on the cell surface [
10,
11]. Accordingly, increasing MHC-I may facilitate immune recognition of tumor antigens as foreign, promoting antitumor immunity. In addition, because the HLA-B7 molecule expressed by Allovectin-7 is frequently allogenic to the recipient, expression of HLA-B7 itself may promote a strong inflammatory response. In contrast to INO-3112, which requires electroporation for plasmid expression, the DNA/lipid complex used by Allovectin-7 allows for direct uptake of plasmid DNA by the myocytes surrounding the injection site. Phase I and II trials of Allovectin-7 in HNSCC patients demonstrated that the vaccine is safe and well-tolerated, with 33% of patients exhibiting partial or complete tumor response [
12]. However, no HLA-B7-positive patients demonstrated a clinical response to the vaccine therapy, suggesting that the antitumor effects of the vaccine may be due to the immune response to HLA-B7 itself, and not tumor antigens [
12]. Additionally, a phase III trial of Allovectin-7 in metastatic malignant melanoma failed to reach its primary or secondary endpoints of tumor response and improved OS compared to chemotherapy [
13], which resulted in the termination of the Allovectin program in 2013. At the time, a Phase II/III trial for the use of Allovectin-7 in HNSCC was underway, although no results were reported before the program was cancelled.
3.2. Peptide Vaccines
3.2.1. MAGE-A3/HPV16
The MAGE-A3 vaccine was originally developed for treatment of non-small cell lung carcinoma, although a phase III trial of >2000 patients failed to demonstrate efficacy over placebo for this patient population [
14]. The vaccine targets MAGE-A3, a human protein that is highly expressed in a variety of tumors and during embryogenesis, but is only found in the placenta and testis of adults [
15]. The MAGE-A3/HPV16 combines MAGE-A3 peptides with furin-sensitive linkers and the GL-0810 vaccine containing an HPV16-specific peptide. Importantly, peptides from both vaccines contain the human immunodeficiency virus-1 (HIV-1) Tat membrane translocation sequence, which allows the peptides to efficiently cross cell membranes and be cleaved into short peptides for presentation on MHC class I complexes [
16]. To improve the immune response, MAGE-A3 has been tested with various immunostimulants, of which AS15 has produced the best clinical response in Phase II trials [
17]. However, a Phase I dose escalation trial of MAGE-A3/HPV16 found that, despite good tolerability and the development of robust T cell and antibody responses, all 16 patients experienced disease progression [
18]. Phase III trials of the MAGE-A3 vaccine in NSCLC and melanoma were also terminated prematurely due to futility, suggesting that MAGE-A3 vaccination is unlikely to be an optimal immunotherapeutic strategy [
17,
19]. Phase I trials testing MAGE-A3/HPV16 in HNSCC were scheduled for completion in 2012, however the current status and results from these trials is not known.
3.2.2. Mucin-1
Mucin-1 (MUC1) is a heavily glycosylated protein that undergoes proteolytic cleavage to form heterodimeric complexes on the cell surface, where it helps to provide a protective barrier between the cell and the external environment [
20]. Accordingly, MUC1 is expressed by most epithelial cells and is overexpressed in multiple tumor types. In tumor cells MUC1 promotes tumor growth, metastasis, and drug resistance, and the C-terminal tail may serve as an oncogenic signaling molecule [
21]. Tumor-associated MUC1 is characterized by altered glycosylation patterns, which enable differential targeting of tumor MUC1 for vaccine therapy [
22].
Indeed, in 2009 MUC1 was listed as the second-highest priority tumor antigen for vaccine therapy [
23], and currently there are nearly 30 ongoing clinical trials involving MUC1, some of which are exploring the exciting possibility of using MUC1 vaccination for cancer prevention, in addition to cancer treatment. In general, MUC1 vaccines are well-tolerated, generate tumor-specific T cell responses, and have shown evidence of efficacy in a variety of tumor types. However, despite the large number of early-phase trials, only a few MUC1 vaccine programs have reached stage III clinical trials, and therefore the long-term utility of MUC1 vaccination for cancer treatment or prevention remains to be determined. A Phase I/II MUC1 vaccine trial for HNSCC is currently underway with a predicted completion date in 2021.
3.2.3. AlloVax
The two-part AlloVax vaccine is comprised of chaperone protein-enriched tumor cell lysate from the patient’s own tumor and the AlloStim adjuvant, an intentionally mismatched allogenic stem cell transplant. This combination produces a host-versus-graft response called the “mirror effect” [
24], which stimulates an eventual host-versus-tumor response. This approach has yielded T cell responses in vitro, however further clinical trials are required to determine the efficacy and safety profile of this vaccination strategy. Although AlloVax has not been subjected to extensive trials, Phase I and II trials are underway for HNSCC with estimated completion dates in 2016 and 2018, respectively.
3.2.4. ISA101 and ISA201 (HESPECTA)
The ISA101 vaccine is a mixture of 13 overlapping 25–35 amino acid synthetic peptides derived from the HPV16 E6 and E7 proteins [
25]. The peptide mixture (nine E6 peptides and four E7 peptides) is combined with the Montanide ISA-51 adjuvant, which contains all potential T cell epitopes and therefore produces T cell activation irrespective of the HLA type of the patient. Although results from trials of ISA101 in head and neck cancer patients have yet to be reported (
Table 1), promising results have been observed in patients with HPV16-induced non-invasive vulvar and vaginal lesions. One trial of 20 patients with vulvar intraepithelial neoplasia demonstrated complete or partial response in 12 of 20 patients at 3 months and 15 of 19 patients at 12 months [
26]. Furthermore, in a trial of 43 patients, HPV16-positive high-grade vulvar and vaginal intraepithelial neoplasia treated with ISA101 plus imiquimod or ISA101 alone, clinical response was observed at 3 months in 18 of 34 patients completing the vaccine series and in 15 of 29 patients evaluated at 12 months [
27]. Imiquimod had no impact on response; however, in both trials clinical response was correlated with stronger vaccine-specific immune responses [
26,
27]. Trials of ISA101 for patients with HPV-positive head and neck cancers are currently underway.
ISA201 (HESPECTA) is a second generation vaccine based on ISA101 in which two HPV16 E6 peptides are conjugated to Amplivant, a synthetic Toll-like receptor-2 (TLR2) agonist [
28]. A Phase I trial of HESPECTA in patients with non-metastatic HPV16-positive head and neck cancer is currently underway (
Table 1). Results are expected in December of 2016.
3.3. Biologic Vaccines
3.3.1. ADXS11-001
The ADXS11-001 vaccine consists of a live, attenuated strain of
Listeria monocytogenes engineered to secrete the HPV-E7 protein fused to the
Listeria listeriolysin O protein [
29].
Listeria monocytogenes infects antigen-presenting cells, resulting in antigen presentation and activation of CD4+ and CD8+ T cells targeting HPV-infected tumor cells. A Phase I trial in 15 patients with metastatic, refractory, or recurrent cervical cancer treated with two vaccinations of 1 × 10
9 colony forming units (CFUs), 3.3 × 10
9 CFUs, or 1 × 10
10 CFUs demonstrated median survival of 347 days, 1-year survival of 53%, and reduced tumor volume in 31% of patients. These results are particularly exciting when compared to historical controls, which have a median survival of 6 months and 1-year survival of 5% [
29,
30].
Similarly, preliminary results from the GOG-0265 trial demonstrated a 1-year survival rate of 38.5% for 26 women with persistent or recurrent metastatic cervical cancer treated with three doses of ADXS11-001 at 1 × 10
9 CFU, compared to a 20% 1-year survival rate for historical controls [
31]. In both trials, adverse events were manageable and primarily consisted of “flu-like” symptoms including fatigue, chills, fever, and headache. However, clinically-relevant hypotension has been reported, including dose-limiting diastolic hypotension that responded to intravenous (IV) fluid bolus in patients receiving 1 × 10
10 CFU doses [
29,
31]. Phase II clinical trials to establish the efficacy of ADXS11-001 for HPV-positive oropharyngeal HNSCC are currently underway.
3.3.2. Semi-Allogenic Human Fibroblast Vaccine
The theory underlying semi-allogenic cell transfer is that weakly immunogenic tumor-associated antigens can be converted to highly immunogenic antigens when presented by allogenic fibroblasts. To this end, fibroblast cell lines are transfected with DNA isolated from extirpated tumor cells, grown in culture, and then lethally irradiated before being injected into the patient from whom the tumor DNA was derived [
32]. To our knowledge this approach has yet to be applied to human subjects, although studies in murine cancer models have produced promising results. In one study, LM murine fibroblast cells, genetically engineered to express interleukin-2 (IL-2), were transfected with genomic DNA isolated from a sporadic breast cancer in a C3H/He mouse before being injected into tumor-bearing C3H/He mice [
33]. The mice that received fibroblast injections survived significantly longer than control animals, likely due to a robust CD8+ T cell response [
33]. Similar approaches have been used in murine squamous cell carcinoma models [
34], and the recipient fibroblast cell lines have been modified to optimize the immune response [
35]. A current two-stage Phase I trial aims to expand this approach to human subjects with HNSCC, using the MRC-5 human fibroblast cell line [
36]. MRC-5 cells will be transfected with genomic DNA isolated from extirpated HNSCC, grown in culture, and then lethally irradiated prior to being injected into the patient from whom the tumor DNA was isolated. Enrollment is anticipated to begin in 2016, with reporting of preliminary results by 2018.
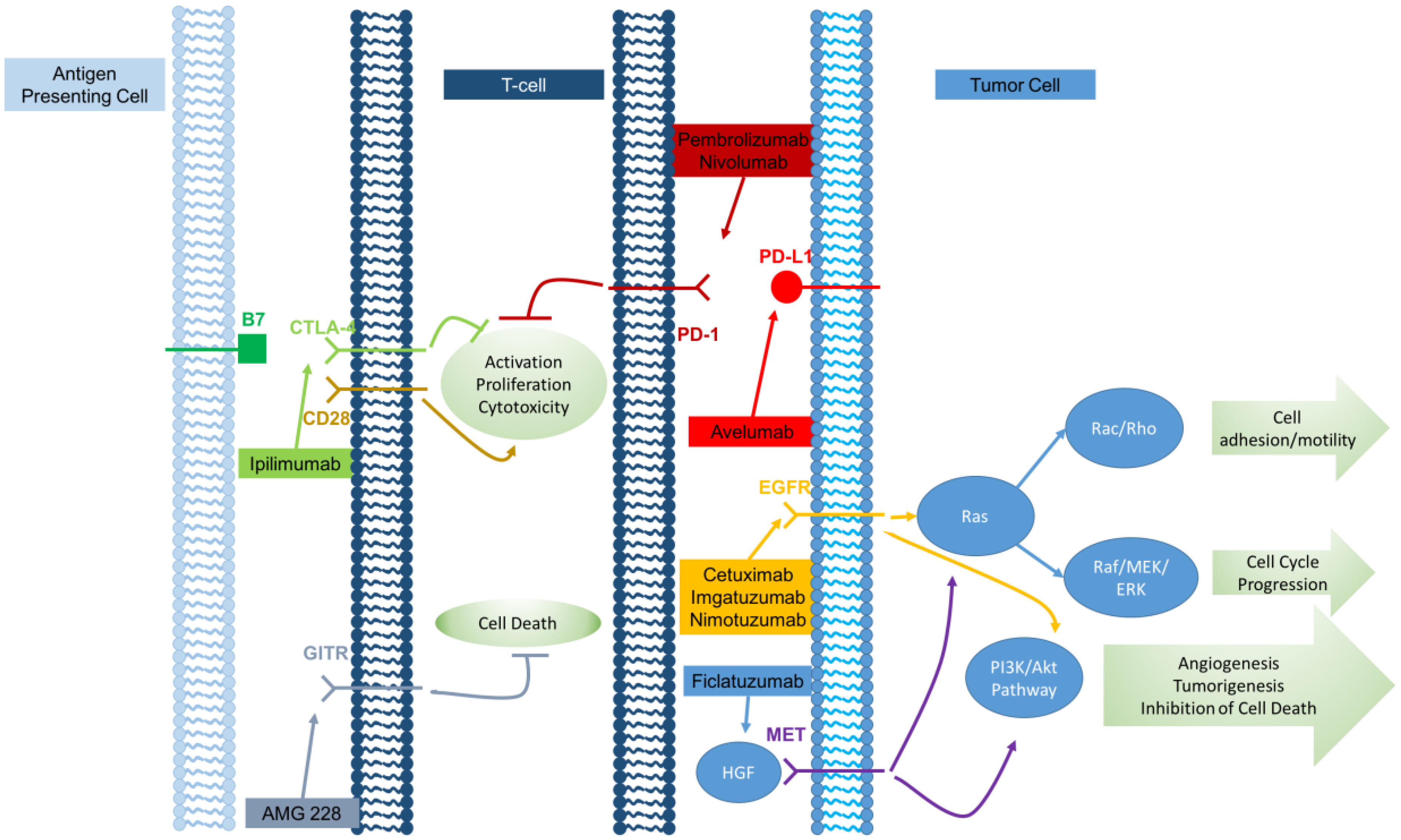
4. Monoclonal Antibodies
Over recent years, an increasingly diverse group of monoclonal antibodies have been applied to cancer therapy, representing a significant advance in cancer treatment. Although these antibodies target a range of molecules, many antibodies mediate their antitumor effects through similar mechanisms including the targeting of tumor cells for antibody-dependent cell-mediated cytotoxicity (ADCC), the direct inhibition of tumor growth signals, and inhibiting signaling pathways involved in maintaining immune self-tolerance.
Figure 1 graphically depicts the various monoclonal antibodies and where they act along the immune pathway.
4.1. Receptor Tyrosine Kinase Inhibitors
4.1.1. Cetuximab
Cetuximab is a humanized monoclonal murine antibody targeting the epidermal growth factor receptor (EGFR). EGFR is an erbB family receptor tyrosine kinase expressed by a wide variety of tumor types. Binding of a ligand to EGFR results in receptor dimerization, autophosphorylation, and induction of signaling cascades leading to cell proliferation. Cetuximab works by inhibiting ligand binding to EGFR, thereby blocking cell growth signaling [
37]. Importantly, for colorectal cancer cetuximab is only effective for tumors expressing wild-type KRAS, as mutant KRAS provides growth signals that bypass EGFR inhibition. However, for HNSCC cetuximab appears to be effective for both KRAS-wild type and KRAS-mutant tumors [
38]. Cetuximab was approved for HNSCC in 2006 after early Phase III trials demonstrated that cetuximab improved response to chemotherapy and reduced the risk of death for patients who have cetuximab-related skin toxicity [
39].
Later studies showed that cetuximab plus platinum-based chemotherapy increases overall and progression-free survival (PFS), and that 5-year survival is improved for patients receiving chemoradiotherapy who developed skin rash of grade ≥2 severity in response to cetuximab [
40,
41]. However, a more recent Phase III study of >800 patients found that adding cetuximab to platinum-based chemoradiotherapy regimens did not improve PFS or OS, suggesting that routine cetuximab administration may not be beneficial to all patients [
42]. Therefore, further research is needed to identify patients who are most likely to benefit from cetuximab therapy. Currently, there are over 80 ongoing clinical trials examining the use of cetuximab for various cancers, including four trials for HNSCC.
4.1.2. Imgatuzumab (GA201)
Imgatuzumab is a first-in-class glycoengineered humanized anti-EGFR monoclonal antibody. The unique characteristics of imgatuzumab result from the addition of bisected afucosylated carbohydrate moieties to the Fc domain, which enhances the interaction with Fcγ receptors on immune effector cells and produces enhanced antibody-dependent cell-mediated cytotoxicity [
43]. Phase I/II trials in patients with colorectal carcinoma demonstrated an acceptable safety profile, with skin rash as the most common adverse effect. Moreover, therapeutic responses were observed in patients with KRAS-mutant tumors, indicating that imgatuzumab may be more beneficial than cetuximab for this patient population [
44,
45]. In addition, one trial of 25 patients on single-agent imgatuzumab therapy produced a median OS of 9.3 months, which was longer than previously reported OS of 4.5 months for single-agent cetuximab therapy [
45]. Although further data are required, these results, as well as preclinical data from mouse model systems, suggest that imgatuzumab may be more efficacious for the treatment of solid tumor than existing anti-EGFR therapies [
43]. A Phase I trial of imgatuzumab was started in 2009; however, the drug testing pipeline was cancelled in 2013 before results were released.
4.1.3. Nimotuzumab
Nimotuzumab is a humanized anti-EGFR monoclonal antibody notable for its unique safety profile, where antitumor activity has been observed in the absence of the severe skin, mucosal, renal, and gastrointestinal toxicity observed with cetuximab [
46,
47]. A phase IIb trial in 92 Indian patients with stage III or IV HNSCC treated with nimotuzumab plus chemoradiation therapy versus single modality radiation therapy demonstrated 30-month PFS of 57% in the nimotuzumab group versus 22% in the control group [
47]. Moreover, while median OS was not achieved by 30 months for the nimotuzumab group, OS was 22 months for the control group [
47]. A similar phase IIb study of 76 Indian patients comparing nimotuzumab plus chemoradiation therapy, nimotuzumab plus radiation therapy, chemoradiation therapy, and radiation therapy alone demonstrated improved survival in patients receiving nimotuzumab, with maximal OS in the nimotuzumab plus chemoradiation therapy group [
48]. While further evidence is required, these data together suggest that nimotuzumab is a promising therapeutic agent for HNSCC. A Phase III trial of nimotuzumab is currently recruiting HNSCC patients, with results expected in 2021.
4.1.4. Ficlatuzumab
In contrast to the previously discussed monoclonal antibodies, which all target EGFR, ficlatuzumab is a humanized monoclonal antibody targeting the hepatocyte growth factor (HGF). HGF is the only known ligand of the cellular MET tyrosine kinase receptor, which activates cellular signaling cascades including PI3K/Akt, Ras/Rac/Rho, and Ras/MAPK, leading to cancer growth, metastasis, angiogenesis, and drug resistance [
49]. Importantly, overexpression of HGF and cMET has been identified in HNSCC, suggesting that immunotherapies targeting this pathway may be beneficial for this patient population [
49,
50]. Preclinical studies of ficlatuzumab monotherapy for non-small cell lung cancer (NSCLC), which has a similar genetic profile to HNSCC [
51], resulted in reduced tumor growth and decreased levels of phospho-MET, phospho-ERK, and phospho-Akt, but a concurrent increase in phospho-EGFR.
A combination of ficlatuzumab plus cetuximab therapy resulted in complete tumor response in all animals, indicating a role for dual inhibition of the HGF and EGFR pathways [
52]. Unfortunately, a phase II trial of ficlatuzumab plus the EGFR inhibitor gefitinib versus gefitinib alone showed no difference in PFS, although biomarker analysis demonstrated a paradoxically increased response to ficlatuzumab therapy for patients with activating EGFR mutations and lower cMET expression [
53]. Therefore, ficlatuzumab may be best suited for highly-selected patients with specific mutational and gene expression profiles. Phase I trials for ficlatuzumab in HNSCC patients are currently underway, with projected completion dates in 2018.
4.2. Checkpoint Inhibitors
Immune checkpoints limit the inflammatory response, reducing damage to normal tissues and preventing autoimmunity. Therapies that inhibit or disable these checkpoints may therefore break immune tolerance to TAAs and induce a host antitumor immune response. Currently, five checkpoint inhibitors are commercially available or are undergoing clinical trials. Three checkpoint inhibitors target programmed death-1 (PD-1) pathway. PD-1 is a cell surface receptor expressed on activated B and T cells that binds the PD-L1 and PD-L2 ligands. Because PD-1 activation by ligand binding decreases the inflammatory response [
54], inhibiting PD-1 by direct antibody binding or by targeting PD-L1 or PD-L2 may relieve immune tolerance, promoting immune cell-mediated tumor lysis. Other target pathways include CTLA-4, which is expressed on the surface of activated T cells and limits T cell activity, and the glucocorticoid induced tumor necrosis factor receptor (GITR), a T cell costimulatory molecule. Importantly, effective checkpoint inhibition therapy requires the presence of inhibited CD8+ T cells at the tumor periphery [
55].
4.2.1. Pembrolizumab
Pembrolizumab is a humanized monoclonal antibody targeting PD-1 that is currently Food and Drug Administration (FDA) approved as monotherapy for metastatic melanoma and metastatic NSCLC. In addition, pembrolizumab has shown promise for HNSCC. Specifically, preliminary results from the KEYNOTE-012 trial, which was comprised of patients with recurrent HNSCC, demonstrated an overall response rate of 18.2% and a disease control rate of nearly 50% [
56,
57]. Furthermore, pembrolizumab was better tolerated than aggressive chemoradiation therapy, with the most common adverse effects being fatigue, hypothyroidism, decreased appetite, and rash [
57]. Survival will be assessed as part of ongoing phase II and III trials; however, these early results suggest that PD-1 inhibition may be an effective target for HNSCC.
4.2.2. Nivolumab
Similar to pembrolizumab, nivolumab is a humanized monoclonal anti-PD-1 antibody that is FDA approved for the treatment of renal cell carcinoma, NSCLC, and melanoma. For HNSCC, the phase III CheckMate-141 trial, which compared nivolumab to investigators’ choice of cetuximab, docetaxel, or methotrexate, was terminated early due to superiority of the nivolumab arm over the control arm [
58]. Although results from CheckMate-141 have yet to be published, studies in NSCLC have demonstrated a clear survival benefit for nivolumab versus docetaxel chemotherapy, particularly among patients whose tumors expressed PD-1L [
59]. Therefore, these data provide additional evidence that immune checkpoint inhibition may be a valuable therapeutic approach for HNSCC.
4.2.3. Avelumab
In contrast to many of the monoclonal antibodies discussed to this point, avelumab is a fully human monoclonal anti-PD-L1 antibody. Importantly, preclinical studies have indicated that avelumab is capable of inducing antibody-dependent cell-mediated cytotoxicity [
60,
61], which may play a critical role in cancer immunotherapy [
62]. An open-label phase Ib trial of avelumab in patients with NSCLC that progressed on platinum therapy demonstrated complete or partial response in 12% of patients and stable disease in an additional 38% [
63]. Although early trials have indicated that adverse effects are common with avelumab, the drug appears to have an acceptable safety profile, with the most common adverse effects being fatigue, nausea, infusion reaction, diarrhea, chills, decreased appetite, pyrexia, flu-like illness, and arthralgia [
63,
64]. While early results are promising, the ultimate utility of avelumab for HNSCC will be determined by ongoing phase I, II, and III trials.
4.2.4. Ipilimumab
Ipilimumab differs from the checkpoint inhibitors discussed thus far in that this humanized monoclonal antibody targets CTLA-4, an immunomodulatory protein expressed on the surface of activated T cells. CTLA-4 binds to B7, which prevents B7 from interacting with the co-stimulatory molecule CD28, limiting T cell proliferation and IL-2 production [
65]. Blocking CTLA-4 relieves this T cell inhibition, resulting in a host antitumor immune response. Phase III trials of ipilimumab in patients with metastatic melanoma demonstrated improved OS for patients receiving ipilimumab [
66], which lead to FDA approval of the drug for unresectable or metastatic melanoma in 2011. Unfortunately, trials of ipilimumab have produced less promising results for lung cancer. It is important to note that ipilimumab exhibits unique tumor response kinetics including response after initial progression and objective responses occurring up to 6–12 months following treatment [
65].
A large Phase II study of patients with NSCLC or small cell lung cancer (SCLC) found that while ipilimumab increased overall PFS and immune-related PFS, which accounts for the unique response characteristics of ipilimumab [
67], there was no increase in OS for either SCLC or NSCLC patients [
68,
69]. Additionally, the greatest increase in PFS was observed among patients who received ipilimumab after chemotherapy, indicating that ipilimumab is most efficacious following tumor antigen release [
68,
69]. Importantly, ipilimumab produces a unique side-effect profile due to immune system activation, including diarrhea, enterocolitis, intestinal perforation, and hepatotoxicity [
70]. Taken together, these data suggest that ipilimumab may have promise for HNSCC, but investigators should be aware of the unique characteristics of this medication when considering any patient for this therapy. Ipilimumab is currently undergoing a Phase I/II trial for virus-associated tumors, including HNSCC, with a projected completion date in 2018.
4.2.5. AMG 228
AMG 228 is a novel monoclonal antibody currently undergoing Phase I safety trials for a variety of solid tumors including HNSCC. This antibody targets GITR, which is expressed on the surface of CD25+ CD4+ regulatory T cells and acts as an effector T cell co-stimulatory molecule, possibly by inhibiting T cell death [
71]. In animal model systems, GITR stimulation results in decreased self-tolerance and the development of autoimmunity [
72]. In addition, administration of GITR agonist antibodies in murine melanoma model systems results in tumor immunity and rejection [
73,
74]. Accordingly, GITR modulation has been listed as one of the National Cancer Institute’s top 25 most promising research areas [
71]. Results from the initial human trials of AMG 228 in solid tumors are expected in 2017.
5. Oncolytic Viruses and Active Immunotherapeutics
Oncolytic viruses represent a novel approach to cancer therapy that uses recombinant or engineered viruses to selectively kill tumor cells while sparing normal tissues. In addition, tumor cell lysis releases TAAs into the surrounding environment along with viral antigens, which may facilitate the production of an immune antitumor response. Currently, two oncolytic viruses are undergoing clinical trials for HNSCC.
5.1. Pexa-Vec (JX-594)
Pexa-Vec is a recombinant vaccinia virus (family
Poxviridae) that is deleted for the viral thymidine kinase (vTK) gene and contains an exogenous Granulocyte Macrophage – Colony Stimulating Factor (GM-CSF) gene [
75]. The vTK deletion renders the virus replication-deficient in normal human cells that express constitutive levels of cellular thymidine kinase (cTK), but permits virus replication in tumor cells that overexpress cTK [
76,
77]. In addition, Pexa-Vec replication is stimulated by EGFR/Ras signaling and type-I interferon resistance, giving the virus both relative selectivity for tumor cells and the ability to replicate in tumors with diverse genetic alterations [
77]. Because vaccinia, like all poxviruses, is highly cytolytic, viral replication results in direct oncolysis in addition to stimulation of GM-CSF sensitive leukocytes, such as neutrophils [
75]. The virus was originally developed for use in hepatocellular carcinoma, where Phase II trials have demonstrated dose-related oncolysis, antitumor immunity, and increased OS with direct intratumor injection [
78]. Despite the administration of Pexa-Vec via intratumor injections in initial trials, the virus is stable for intravenous administration although relatively high doses may be required to achieve a systemic antitumor effect [
79]. Systemic Pexa-Vec administration has an acceptable safety profile with mild adverse effects including pyrexia, chills, headache, nausea, vomiting, and grade I papulopustular rash [
79]. Accordingly, Pexa-Vec may be an exciting option for the treatment of a variety of solid tumors including HNSCC. A Phase I trial of Pexa-Vec in HNSCC has been completed, although the results have not yet been published.
5.2. TRICOM
TRICOM is a recombinant avian fowlpox virus that infects mammalian cells but is replication-deficient in mammals [
80]. The recombinant virus used in fowlpox-TRICOM expresses three costimulatory transgenes, B7.1 (CD80), Intercellular Adhesion Molecule 1 (ICAM-1), and lymphocyte function-associated antigen 3 (LFA-3), plus one or more tumor-associated antigens, such as Carcinoembryonic antigen (CEA) and MUC-1 [
80]. In a Phase II trial where patients with metastatic colorectal carcinoma received dendritic cells infected with CEA/MUC-1 TRICOM (PANVAC), OS was not reached by 72 months of follow-up for patients receiving PANVAC, but was 44 months in contemporary controls, indicating that PANVAC therapy may improve survival for patients with solid tumors. In addition, pilot studies of systemic PANVAC therapy for patients with metastatic ovarian or breast carcinoma have suggested that this approach may provide clinical benefit to selected patients [
81]. The vaccine appears to be well-tolerated and the virus itself elicits only a limited immune response, allowing the virus to be administered repeatedly to achieve a more robust antitumor immune response [
80,
81]. Phase I studies of the PANVAC vaccine in head and neck cancer patients are nearing completion.
6. Immunomodulators
For the purposes of this review, we define immunomodulators as a diverse group of small molecule agonists and compounds designed to increase the host antitumor immune response. In general, these compounds result in generalized immune system stimulation, rather than a directed antitumor response.
6.1. Toll-Like Receptor Agonists
TLRs are a family of transmembrane signaling proteins that form an important component of the innate inflammatory response by detecting conserved microbe-associated molecular patterns (MAMPs) and endogenous products known as damage-associated molecular patterns (DAMPs) [
82]. Accumulating evidence indicates that TLR expression is altered in malignant cells and that DAMP release by dying cancer cells results in TLR activation, which may contribute to clinically relevant antitumor inflammatory responses [
82,
83]. Based on these observations, several TLR agonists have been developed for cancer immunotherapy applications.
6.1.1. Motolimod (VTX-2237)
Motolimod is a small molecule TLR8 agonist that enhances monocyte, dendritic cell, and natural killer (NK) cell activation and increased ADCC [
84]. Pre-clinical studies have indicated that motolimod stimulation of peripheral blood mononuclear cells (PBMCs) isolated from healthy donors increases cetuximab-mediated lysis of cultured HNSCC cells [
85]. Phase I trials have demonstrated that motolimod has an acceptable safety profile and increases NK cell activation in HNSCC patients, which may augment NK-mediated tumor lysis in response to other therapies, such as cetuximab [
86,
87]. However, further studies are needed to demonstrate the overall efficacy of motolimod for HNSCC patients. Results from a Phase Ib trial of motolimod in HNSCC are expected this year.
6.1.2. IMO-2055 (EMD 1201081)
Similar to motolimod, IMO-2055 is a synthetic TLR9 agonist that enhanced NK cell activation and stimulated a potent antitumor immune response in combination with monoclonal antibody therapy in preclinical trials [
88,
89]. However, a phase I study of IMO-2055 provided only limited evidence that IMO-2055 produces clinically-relevant antitumor effects in vivo [
90]. A further Phase II study of IMO-2055 plus cetuximab versus cetuximab monotherapy for HNSCC patients also failed to demonstrate clinical efficacy [
91], limiting potential applications of IMO-2055 for HNSCC. Importantly, evidence from other Phase I and III trials has indicated that IMO-2055 and other TLR9 agonists increase known myelosuppressive effects of platinum-based chemotherapy, which may lead to serious and potentially fatal adverse effects including hypotension, febrile neutropenia, and septicemia [
92,
93,
94]. Together, these data indicate that IMO-2055 likely has an unacceptable risk to efficacy ratio and may not be appropriate for clinical use. A Phase I study of IMO-2055 in HNSCC was terminated before results were released.
6.1.3. Picibanil (OK-432)
In contrast to the synthetic TLR agonists discussed above, picibanil is a lyophilized preparation of the
Streptococcus pyogenes su-strain that has been treated with benzylpenicillin [
95]. Originally developed and approved for the treatment of lymphangiomas [
96,
97], picibanil induces a substantial immune inflammatory response, likely via TLR4 signaling pathways [
95]. Notably, picibanil has been in clinical use for over 30 years, and therefore has a well-characterized safety and adverse effect profile. Strikingly, pre-clinical studies have indicated that active components of picibanil may induce direct lysis of head and neck cancer cells, suggesting that picibanil may induce HNSCC regression both by direct and immunostimulatory effects [
98]. Indeed, a Japanese study of 81 patients with oral squamous cell carcinoma who were treated with picibanil plus chemoradiotherapy versus chemoradiotherapy alone found significantly increased OS and PFS among patients who received picibanil [
99]. Accordingly, picibanil may be a useful adjuvant to standard therapeutic approaches for head and neck cancers. A Phase I trial of picibanil was completed in 2012, however results have not been published.
6.2. Exogenous Cytokines
Cytokines are a diverse family of small, non-structural signaling proteins that play diverse roles in modifying immune and inflammatory responses [
100]. Importantly, increasing evidence indicates that tumors modulate the local cytokine milieu to produce a pro-inflammatory environment while simultaneously facilitating tumor immune system evasion [
101]. Therefore, altering the cytokine response by providing exogenous cytokine mixtures may facilitate the immune antitumor response.
6.2.1. IL-12
One mechanism by which tumors evade immune recognition is through local immunosuppression, resulting in downregulation of the IL-12–interferon γ–HLA-DR axis [
101]. Therefore, increasing local IL-12 levels via an exogenous plasmid vector may facilitate tumor clearance by the immune system. In animal model systems, intratumoral electroporation of an IL-12 expression plasmid (pIL-12) resulted in significant intratumoral IL-12 expression, leading to disease stabilization and eventually complete regression without systemic toxicity [
102]. Phase I trials of intratumoral plasmid IL-12 electroporation in patients with metastatic melanoma resulted in a dose-dependent increase in tumor cell necrosis and lymphocyte infiltration, in addition to stabilization or regression of non-electroporated metastases in over 50% of treated patients [
103]. As in animal systems, intratumoral electroporation was associated with minimal systemic effects [
103], indicating that plasmid IL-12 preparations may be a promising therapy for advanced or metastatic disease. Current trials are examining the efficacy of IL-12 delivered as liposomal or plasmid preparations for HNSCC.
6.2.2. IRX-2
IRX-2 is a biologic immunotherapeutic containing a mixture of cytokines including IL-1β, IL-2, IL-6, IL-8, TNFα, GM-CSF, and IFN-γ [
104]. Preclinical studies have shown that this cytokine cocktail is active on multiple immune cell types, inducing dendritic cell maturation, T cell activation, and NK cell stimulation [
104,
105,
106,
107]. Phase I and II trials have demonstrated that IRX-2 is safe and generally well-tolerated, although some patients may develop potentially serious adverse effects including anemia and lymphopenia [
108,
109]. In addition, a Phase II study of 42 head and neck cancer patients treated with IRX-2 plus chemoradiotherapy demonstrated higher OS and longer time to recurrence compared to contemporary off-study controls [
108]. IRX-2 treatment was associated with symptomatic improvement in 57% of patients [
108], suggesting that IRX-2 may be an efficacious treatment to augment traditional chemoradiotherapy and surgery for head and neck cancers. A phase II trial examining the use of IRX-2 for HNSCC patients is currently underway, with an estimated completion in 2019.
7. Future Directions
One exciting potential application of immunotherapeutics, particularly vaccine therapies, is cancer prevention by vaccinating selected high-risk patients against TAAs. In the case of HPV-associated cancer, vaccination against high-risk HPV strains results in a clear reduction in the incidence of cervical cancer [
110], and has the potential to prevent >90% of HPV-positive head and neck cancers [
111]. However, the effect of HPV vaccination after the establishment of persistent HPV is not known. Moreover, it remains to be determined whether vaccination against HPV antigens, such as E6 or E7, will be an effective cancer prevention strategy for individuals with persistent HPV infection at high risk for developing HPV-associated oropharyngeal cancer. Identifying individuals with persistent oropharyngeal HPV infection also remains a barrier to cancer preventative strategies, as there is currently no standardized test to identify HPV-associated premalignant oropharyngeal lesions.
Another area of HNSCC immunotherapeutics requiring further study is the ability to identify, a priori, which patients are likely to benefit from a particular immunotherapeutic approach. Indeed, for many immunotherapeutics only a subset of patients respond to any given therapy, likely due to variations in HLA subtype or other immunomodulatory proteins. In addition, the specific mutational and gene expression profile of a given tumor may also have significant impacts on the effectiveness of immunotherapy. For some immunotherapeutics, such as the Allovectin-7 vaccine, there are described molecular predictors of immune response, however for a vast majority of therapies the specific molecular and genetic characteristics that predict response remain to be defined. Therefore, integrated genomic characterization of both germline and tumor tissues from HNSCC patients may lead to the identification of molecular profiles that predict responses to a given therapy. However, it should be noted that due to the divergent mechanisms of action among many immunotherapeutics, molecular profiles that predict response to one agent may not be broadly predictive for general responses to immunotherapies.
8. Conclusions
Immunotherapy represents a promising avenue for the treatment of head and neck cancers, with several treatment regimens showing significant promise in clinical trials. When combined with traditional approaches including chemotherapy, radiation therapy, and surgery, these immunotherapies have the potential to reduce the morbidity associated with HNSCC and improve survival. It should be noted, however, that immunotherapies often produce a response profile that is often significantly different from that associated with traditional chemotherapy. Indeed, many immunotherapeutics require a longer period of time to achieve clinical response, and may even induce tumor pseudo-progression, when compared to traditional chemotherapeutic approaches. Therefore, these agents may not be the modality of choice in cases where the tumor is endangering critical structures or there is impending airway compromise. Thus, initial surgical debulking may still play an important role for HNSCC patients treated with immunotherapy. Importantly, one particularly exciting feature of immunotherapy may be longer-term responses compared to other modalities, although further data are required to determine whether immunotherapy, particularly vaccine therapies, can produce durable responses.
Acknowledgments
Darrin V. Bann would like to acknowledge the Pennsylvania Department of Health Tobacco Settlement CURE Fund for providing research funding.
Author Contributions
Neerav Goyal and Darrin V. Bann conceived the manuscript. Darrin V. Bann, Neerav Goyal, and Daniel G. Deschler wrote and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. The treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48. [Google Scholar] [PubMed]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.C.; Kao, J.; Sikora, A.G. Recognizing and reversing the immunosuppressive tumor microenvironment of head and neck cancer. Immunol. Res. 2012, 54, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Cohen, R.; Morrow, M.P.; Bauml, J.; Weinstein, G.; Boyer, J.; Shen, X.; Yan, J.; Goldenberg, J.; Nashit, D.; et al. Immunotherapy with VGX-3100 (HPV16 and HPV18 plasmids) + INO-9012 (DNA encoding IL-12) in human papillomavirus (HPV) associated head and neck squamous cell carcinoma (HNSCCa): Interim safety and immunogenicity results. In Proceedings of the 30th Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer, National Harbor, MD, USA, 4–8 November 2015.
- Yang, Z.; Aggarwal, C.; Cohen, R.; Morrow, M.P.; Bauml, J.; Weinstein, G.; Boyer, J.; Lee, J.; Weiner, D.; Bagarazzi, M.L. Immunotherapy with INO-3112 (HPV16 and HPV18 plasmids + IL-12 DNA) in human papillomavirus (HPV) associated head and neck squamous cell carcinoma (HNSCCa). Ann. Oncol. 2015, 26, viii1. [Google Scholar] [CrossRef]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef]
- Esteban, F.; Concha, A.; Delgado, M.; Perez-Ayala, M.; Ruiz-Cabello, F.; Garrido, F. Lack of MHC class I antigens and tumour aggressiveness of the squamous cell carcinoma of the larynx. Br. J. Cancer 1990, 62, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Mattijssen, V.; De Mulder, P.H.; Schalkwijk, L.; Manni, J.J.; Van’t Hof-Grootenboer, B.; Ruiter, D.J. HLA antigen expression in routinely processed head and neck squamous cell carcinoma primary lesions of different sites. Int. J. Cancer Suppl. 1991, 6, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Gleich, L.L.; Gluckman, J.L.; Nemunaitis, J.; Suen, J.Y.; Hanna, E.; Wolf, G.T.; Coltrera, M.D.; Villaret, D.B.; Wagman, L.; Castro, D.; et al. Clinical experience with HLA-B7 plasmid DNA/lipid complex in advanced squamous cell carcinoma of the head and neck. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 775–779. [Google Scholar] [PubMed]
- Andtbacka, R.H.I.; Gonzalez, R.; Wloch, M.K.; Strause, L.; Stardal, K.; Chu, A.; Rolland, A.; Agarwala, S.S. A phase 3 clinical trial to evaluate the safety and efficacy of treatment with 2 mg intralesional allovectin-7® compared to dacarbazine (DTIC) or temozolomide (TMZ) in subjects with recurrent metastatic melanoma. In Proceedings of the 10th International Meeting of the Society for Melanoma Research 2013, Philadelphia, PA, USA, 17–20 November 2013.
- Vansteenkiste, J.F.; Cho, B.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H.; et al. MAGRIT, a double-blind, randomized, placebo-controlled Phase III study to assess the efficacy of the recMAGE-A3 + AS15 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small cell lung cancer (NSCLC). Ann. Oncol. 2014, 25, iv409–iv416. [Google Scholar]
- Van den Eynde, B.J.; van der Bruggen, P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997, 9, 684–693. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Aqui, N.A.; Stadtmauer, E.A.; Vogl, D.T.; Xu, Y.Y.; Kalos, M.; Cai, L.; Fang, H.B.; Weiss, B.M.; Badros, A.; et al. Combination immunotherapy after ASCT for multiple myeloma using MAGE-A3/poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin. Cancer Res. 2014, 20, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Kruit, W.H.; Suciu, S.; Dreno, B.; Mortier, L.; Robert, C.; Chiarion-Sileni, V.; Maio, M.; Testori, A.; Dorval, T.; Grob, J.J.; et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: Results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 2413–2420. [Google Scholar] [CrossRef] [PubMed]
- Zandberg, D.P.; Rollins, S.; Goloubeva, O.; Morales, R.E.; Tan, M.; Taylor, R.; Wolf, J.S.; Schumaker, L.M.; Cullen, K.J.; Zimrin, A.; et al. A phase I dose escalation trial of MAGE-A3- and HPV16-specific peptide immunomodulatory vaccines in patients with recurrent/metastatic (RM) squamous cell carcinoma of the head and neck (SCCHN). Cancer Immunol. Immunother. CII 2015, 64, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.; Zielinski, M.; Linder, A.; Dahabreh, J.; Gonzalez, E.E.; Malinowski, W.; Lopez-Brea, M.; Vanakesa, T.; Jassem, J.; Kalofonos, H.; et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: Phase II randomized study results. J. Clin. Oncol. 2013, 31, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Pillai, K.; Pourgholami, M.H.; Chua, T.C.; Morris, D.L. MUC1 as a potential target in anticancer therapies. Am. J. Clin. Oncol. 2015, 38, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, V.; Thompson, P.; Wolfert, M.A.; Buskas, T.; Bradley, J.M.; Pathangey, L.B.; Madsen, C.S.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc. Natl. Acad. Sci. USA 2012, 109, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The Prioritization of Cancer Antigens: A National Cancer Institute Pilot Project for the Acceleration of Translational Research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Har-Noy, M.; Slavin, S. The anti-tumor effect of allogeneic bone marrow/stem cell transplant without graft vs. host disease toxicity and without a matched donor requirement? Med. Hypotheses 2008, 70, 1186–1192. [Google Scholar] [CrossRef] [PubMed]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Drijfhout, J.W.; Wafelman, A.R.; Oostendorp, J.; Fleuren, G.J.; et al. Phase I immunotherapeutic trial with long peptides spanning the E6 and E7 sequences of high-risk human papillomavirus 16 in end-stage cervical cancer patients shows low toxicity and robust immunogenicity. Clin. Cancer Res. 2008, 14, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Van Poelgeest, M.I.; Welters, M.J.; Vermeij, R.; Stynenbosch, L.F.; Loof, N.M.; Berends-van der Meer, D.M.; Lowik, M.J.; Hamming, I.L.; van Esch, E.M.; Hellebrekers, B.W.; et al. Vaccination against oncoproteins of HPV16 for noninvasive vulvar/vaginal lesions: Lesion clearance is related to the strength of the T-cell response. Clin. Cancer Res. 2016, 22, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Speetjens, F.; Welters, M.; Gelderblom, H.; Roozen, I.; van der Velden, L.; Melief, C.J.; Zandvliet, M.; van der Burg, S.; Ossendorp, F. A phase I study in patients with a human papillomavirus type 16 positive oropharyngeal tumor treated with second generation synthetic long peptide vaccine conjugated to a defined adjuvant. J. Clin. Oncol. 2016, 34. Abstrct TPS3113. [Google Scholar]
- Wallecha, A.; French, C.; Petit, R.; Singh, R.; Amin, A.; Rothman, J. Lm-LLO-based immunotherapies and HPV-associated disease. J. Oncol. 2012, 2012, 542851. [Google Scholar] [CrossRef] [PubMed]
- Maciag, P.C.; Radulovic, S.; Rothman, J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: A Phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine 2009, 27, 3975–3983. [Google Scholar] [CrossRef] [PubMed]
- Huh, W. ADXS11-001 immunotherapy in the treatment of persistent/recurrent metastatic squamous or non-squamous cell carcinoma of the cervix: Group GOG/NRG-0265 study. In Proceedings of the 2015 AGOS Annual Meeting, Half Moon Bay, CA, USA, 17–19 September 2015.
- Cohen, E.P. Cancer Immunotherapy with Semi-Allogeneic Cells. U.S. Patent 6,187,307 B1, 2001. Filing Date 30 Jananuary 1998. [Google Scholar]
- De Zoeten, E.; Carr-Brendel, V.; Markovic, D.; Taylor-Papadimitriou, J.; Cohen, E.P. Treatment of breast cancer with fibroblasts transfected with DNA from breast cancer cells. J. Immunol. 1999, 162, 6934–6941. [Google Scholar] [PubMed]
- O-Sullivan, I.; Kim, T.S.; Chopra, A.; Cohen, E.P. Therapeutic properties of DNA-based fibroblast and dendritic cell vaccines in mice with squamous carcinoma. Anticancer Res. 2006, 26, 873–884. [Google Scholar] [PubMed]
- Kim, T.S.; Jung, M.Y.; Cho, D.; Cohen, E.P. Prolongation of the survival of breast cancer-bearing mice immunized with GM-CSF-secreting syngeneic/allogeneic fibroblasts transfected with a cDNA expression library from breast cancer cells. Vaccine 2006, 24, 6564–6573. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; Jones, C.M.; Baille, J.P. Characteristics of a human diploid cell designated MRC-5. Nature 1970, 227, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.; Muhsin, M.; Kirkpatrick, P. Cetuximab. Nat. Rev. Drug Discov. 2004, 3, 549–550. [Google Scholar] [PubMed]
- Weidhaas, J.B.; Harris, J.; Axelrod, R.; El-Naggar, A.K.; Singh, A.; Galloway, T.; Raben, D.; Wang, D.; Herman, T.S.; Lee, R.J.; et al. The KRAS-variant and cetuximab response in RTOG 0522. J. Clin. Oncol. 2014, 32. Abstract 6000. [Google Scholar]
- Burtness, B.; Goldwasser, M.A.; Flood, W.; Mattar, B.; Forastiere, A.A.; Eastern Cooperative Oncology Group. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: An Eastern Cooperative Oncology Group study. J. Clin. Oncol. 2005, 23, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Ang, K.K.; Zhang, Q.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Sherman, E.J.; Weber, R.S.; Galvin, J.M.; Bonner, J.A.; Harris, J.; El-Naggar, A.K.; et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J. Clin. Oncol. 2014, 32, 2940–2950. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, C.A.; Nicolini, V.G.; Herter, S.; van Puijenbroek, E.; Lang, S.; Roemmele, M.; Moessner, E.; Freytag, O.; Friess, T.; Ries, C.H.; et al. GA201 (RG7160): A novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin. Cancer Res. 2013, 19, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.G.; Gomez-Roca, C.; Delord, J.P.; Cervantes, A.; Markman, B.; Corral, J.; Soria, J.C.; Berge, Y.; Roda, D.; Russell-Yarde, F.; et al. Phase I pharmacokinetic and pharmacodynamic dose-escalation study of RG7160 (GA201), the first glycoengineered monoclonal antibody against the epidermal growth factor receptor, in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 3783–3790. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Tabernero, J.; Garcia-Carbonero, R.; Cervantes, A.; Gomez-Roca, C.; Berge, Y.; Capdevila, J.; Paz-Ares, L.; Roda, D.; Delmar, P.; et al. Open-label, multicentre expansion cohort to evaluate imgatuzumab in pre-treated patients with KRAS-mutant advanced colorectal carcinoma. Eur. J. Cancer 2014, 50, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Allan, D.G. Nimotuzumab: Evidence of clinical benefit without rash. Oncologist 2005, 10, 760–761. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.S.; Eswaraiah, A.; Crombet, T.; Piedra, P.; Saurez, G.; Iyer, H.; Arvind, A.S. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. mAbs 2009, 1, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.K.M.; Lokesh, V.; Vidyasagar, M.S.; Shenoy, K.; Babu, K.G.; Shenoy, A.; Naveen, T.; Joseph, B.; Bonanthaya, R.; Nanjundappa; et al. Nimotuzumab provides survival benefit to patients with inoperable advanced squamous cell carcinoma of the head and neck: A randomized, open-label, phase IIb, 5-year study in Indian patients. Oral Oncol. 2014, 50, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Nakagawa, T. The current state of molecularly targeted drugs targeting HGF/Met. Jpn. J. Clin. Oncol. 2014, 44, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Sclafani, F.; Cunningham, D. Emerging molecular targets in oncology: Clinical potential of met/hepatocyte growth-factor inhibitors. OncoTargets Ther. 2014, 7, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Meetze, K.A.; Boudrow, A.; Connolly, K.; Huang, R.; Rideout, W., III; Gyuris, J.; Han, M. Anti-tumor activity of SCH 900105 (AV299), an anti-HGF antibody, in non-small cell lung cancer models. Mol. Cancer Ther. 2009, 12, C173. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Park, K.; Geater, S.L.; Agarwal, S.; Han, M.; Komarnitsky, P.; Credi, M.; McKee, K.; Kuriyama, N.; Slichenmyer, W.; et al. A randomized phase 2 study with exploratory biomarker analysis of ficlatuzumab, a humanized hepatocyte growth factor (HGF) inhibitory monoclonal antibody, in combination with gefitinib versus gefitinib alone in Asian patients with lung adenocarcinoma. Ann. Oncol. 2012, 23. Abstract 1198P. [Google Scholar]
- McDermott, D.F.; Atkins, M.B. PD-1 as a potential target in cancer therapy. Cancer Med. 2013, 2, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Haddad, R.I.; Gupta, S.; Mehra, R.; Tahara, M.; Berger, R.; Lee, S.H.; Burtness, B.; Le, D.T.; Heath, K.; et al. Antitumor activity and safety of pembrolizumab in patients (pts) with advanced squamous cell carcinoma of the head and neck (SCCHN): Preliminary results from KEYNOTE-012 expansion cohort. J. Clin. Oncol. 2015, 33, LBA6008. [Google Scholar]
- Starr, P. Encouraging results for pembrolizumab in head and neck cancer. Am. Health Drug Benefits 2015, 8, 16. [Google Scholar] [PubMed]
- Bristol-Myers Squibb Company. CheckMate-141, a Pivotal Phase 3 Opdivo (nivolumab) Head and Neck Cancer Trial, Stopped Early. Available online: http://investor.bms.com/investors/news-and-events/press-releases/press-release-details/2016/CheckMate--141-a-Pivotal-Phase-3-Opdivo-nivolumab-Head-and-Neck-Cancer-Trial-Stopped-Early/default.aspx (accessed on 1 March 2016).
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Friedman, E.R.; Richards, J.; Tsang, K.Y.; Heery, C.R.; Schlom, J.; Hodge, J.W. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Spigel, D.; Kelly, K.; Chandler, J.C.; Rajan, A.; Hassan, R.; Wong, D.J.L.; Leach, J.; Edenfield, W.J.; Wang, D.; et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in advanced NSCLC patients: A phase 1B, open-label expansion trial in patients progressing after platinum-based chemotherapy. J. Clin. Oncol. 2015, 33. Abstract 8034. [Google Scholar]
- Kelly, K.; Patel, M.R.; Infante, J.R.; Iannotti, N.; Nikolinakos, P.; Leach, J.; Wang, D.; Chandler, J.C.; Jerusalem, G.H.M.; Gurtler, J.S.; et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with metastatic or locally advanced solid tumors: Assessment of safety and tolerability in a phase I, open-label expansion study. J. Clin. Oncol. 2015, 33. Abstract 8034. [Google Scholar]
- Blank, C.U.; Enk, A. Therapeutic use of anti-CTLA-4 antibodies. Int. Immunol. 2015, 27, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbe, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Neal, J.; Lu, H.; Cuillerot, J.M.; et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIb/IV non-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase II study. J. Clin. Oncol. 2012, 30, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Lu, H.; Cuillerot, J.M.; Lynch, T.J. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase 2 trial. Ann. Oncol. 2013, 24, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A. Immune-mediated adverse events associated with ipilimumab CTLA-4 blockade therapy: The underlying mechanisms and clinical management. Scientifica 2013, 2013, 857519. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, G.; Ronchetti, S.; Petrillo, M.G.; Riccardi, C. Pharmacological modulation of GITRL/GITR system: Therapeutic perspectives. Br. J. Pharmacol. 2012, 165, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Montagut, T.; Chow, A.; Hirschhorn-Cymerman, D.; Terwey, T.H.; Kochman, A.A.; Lu, S.; Miles, R.C.; Sakaguchi, S.; Houghton, A.N.; van den Brink, M.R. Glucocorticoid-induced TNF receptor family related gene activation overcomes tolerance/ignorance to melanoma differentiation antigens and enhances antitumor immunity. J. Immunol. 2006, 176, 6434–6442. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Schaer, D.A.; Liu, C.; Li, Y.; Hirschhorn-Cymmerman, D.; Kim, S.C.; Diab, A.; Rizzuto, G.; Duan, F.; Perales, M.A.; et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS ONE 2010, 5, e10436. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.H.; Moon, A.; Burke, J.; Ribas, A.; Stephenson, J.; Breitbach, C.J.; Daneshmand, M.; De Silva, N.; Parato, K.; Diallo, J.S.; et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol. Ther. 2011, 19, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Hwang, T.; Park, B.H.; Bell, J.; Kirn, D.H. The Targeted Oncolytic Poxvirus JX-594 Demonstrates Antitumoral, Antivascular, and Anti-HBV Activities in Patients with Hepatocellular Carcinoma. Mol. Ther. 2008, 16, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.S.; Falls, T.; Burns, J.; Garcia, V.; et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.H.; Sommermann, E.M.; Maruri Avidal, L.; et al. Phase 1b trial of biweekly intravenous Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus in colorectal cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Bilusic, M.; Heery, C.; Schlom, J.; Gulley, J.L. Clinical evaluation of TRICOM vector therapeutic cancer vaccines. Semin. Oncol. 2012, 39, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Mohebtash, M.; Tsang, K.Y.; Madan, R.A.; Huen, N.Y.; Poole, D.J.; Jochems, C.; Jones, J.; Ferrara, T.; Heery, C.R.; Arlen, P.M.; et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin. Cancer Res. 2011, 17, 7164–7173. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, K.; Bloy, N.; Buque, A.; Cremer, I.; Eggermont, A.; Fridman, W.H.; Fucikova, J.; Galon, J.; Spisek, R.; Zitvogel, L.; et al. Trial watch: Immunostimulation with Toll-like receptor agonists in cancer therapy. Oncoimmunology 2016, 5, e1088631. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Khan, Z.; Warnakulasuriya, S. Cancer-associated toll-like receptor modulation and insinuation in infection susceptibility: Association or coincidence? Ann. Oncol. 2016, 27, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Dietsch, G.N.; Matthews, M.A.; Yang, Y.; Ghanekar, S.; Inokuma, M.; Suni, M.; Maino, V.C.; Henderson, K.E.; Howbert, J.J.; et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin. Cancer Res. 2012, 18, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, R.M.; Lim, C.M.; Matthews, M.; Dietsch, G.; Hershberg, R.; Ferris, R.L. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol. Immunother. CII 2013, 62, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Northfelt, D.W.; Ramanathan, R.K.; Cohen, P.A.; Von Hoff, D.D.; Weiss, G.J.; Dietsch, G.N.; Manjarrez, K.L.; Randall, T.D.; Hershberg, R.M. A phase I dose-finding study of the novel toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clin. Cancer Res. 2014, 20, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Dietsch, G.N.; Lu, H.; Yang, Y.; Morishima, C.; Chow, L.Q.; Disis, M.L.; Hershberg, R.M. Coordinated activation of Toll-like receptor8 (TLR8) and NLRP3 by the TLR8 agonist, VTX-2337, ignites tumoricidal natural killer cell activity. PLoS ONE 2016, 11, e0148764. [Google Scholar] [CrossRef] [PubMed]
- Van Ojik, H.H.; Bevaart, L.; Dahle, C.E.; Bakker, A.; Jansen, M.J.; van Vugt, M.J.; van de Winkel, J.G.; Weiner, G.J. CpG-A and B oligodeoxynucleotides enhance the efficacy of antibody therapy by activating different effector cell populations. Cancer Res. 2003, 63, 5595–5600. [Google Scholar] [PubMed]
- Wang, D.; Li, Y.; Yu, D.; Song, S.S.; Kandimalla, E.R.; Agrawal, S. Immunopharmacological and antitumor effects of second-generation immunomodulatory oligonucleotides containing synthetic CPR motifs. Int. J. Oncol. 2004, 24, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Kwak, E.L.; Hwang, J.; Heiskala, M.; de La Bourdonnaye, G.; Mita, M. Open-label phase 1b study of FOLFIRI plus cetuximab plus IMO-2055 in patients with colorectal cancer who have progressed following chemotherapy for advanced or metastatic disease. Cancer Chemother. Pharmacol. 2015, 75, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Ruzsa, A.; Sen, M.; Evans, M.; Lee, L.W.; Hideghety, K.; Rottey, S.; Klimak, P.; Holeckova, P.; Fayette, J.; Csoszi, T.; et al. Phase 2, open-label, 1:1 randomized controlled trial exploring the efficacy of EMD 1201081 in combination with cetuximab in second-line cetuximab-naive patients with recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN). Investig. New Drugs 2014, 32, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, V.; Paz-Ares, L.; Boyer, M.; Rosell, R.; Middleton, G.; Eberhardt, W.E.; Szczesna, A.; Reiterer, P.; Saleh, M.; Arrieta, O.; et al. Randomized phase III trial of paclitaxel/carboplatin with or without PF-3512676 (Toll-like receptor 9 agonist) as first-line treatment for advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Manegold, C.; van Zandwijk, N.; Szczesna, A.; Zatloukal, P.; Au, J.S.; Blasinska-Morawiec, M.; Serwatowski, P.; Krzakowski, M.; Jassem, J.; Tan, E.H.; et al. A phase III randomized study of gemcitabine and cisplatin with or without PF-3512676 (TLR9 agonist) as first-line treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2012, 23, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Machiels, J.P.; Kaminsky, M.C.; Keller, U.; Brummendorf, T.H.; Goddemeier, T.; Forssmann, U.; Delord, J.P. Phase Ib trial of the toll-like receptor 9 agonist IMO-2055 in combination with 5-fluorouracil, cisplatin, and cetuximab as first-line palliative treatment in patients with recurrent/metastatic squamous cell carcinoma of the head and neck. Investig. New Drugs 2013, 31, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Ryoma, Y.; Moriya, Y.; Okamoto, M.; Kanaya, I.; Saito, M.; Sato, M. Biological effect of OK-432 (picibanil) and possible application to dendritic cell therapy. Anticancer Res. 2004, 24, 3295–3301. [Google Scholar] [PubMed]
- Brewis, C.; Pracy, J.P.; Albert, D.M. Treatment of lymphangiomas of the head and neck in children by intralesional injection of OK-432 (picibanil). Clin. Otolaryngol. Allied Sci. 2000, 25, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Rebuffini, E.; Zuccarino, L.; Grecchi, E.; Carinci, F.; Merulla, V.E. Picibanil (OK-432) in the treatment of head and neck lymphangiomas in children. Dent. Res. J. 2012, 9, S192–S196. [Google Scholar]
- Tano, T.; Okamoto, M.; Kan, S.; Nakashiro, K.; Shimodaira, S.; Yamashita, N.; Kawakami, Y.; Hamakawa, H. Growth inhibition and apoptosis by an active component of OK-432, a streptococcal agent, via Toll-like receptor 4 in human head and neck cancer cell lines. Oral Oncol. 2012, 48, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Tano, T.; Okamoto, M.; Kan, S.; Bando, T.; Goda, H.; Nakashiro, K.; Shimodaira, S.; Koido, S.; Homma, S.; Fujita, T.; et al. Immunochemoradiotherapy for patients with oral squamous cell carcinoma: Augmentation of OK-432-induced helper T cell 1 response by 5-FU and X-ray irradiation. Neoplasia 2013, 15, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Historical insights into cytokines. Eur. J. Immunol. 2007, 37, S34–S45. [Google Scholar] [CrossRef] [PubMed]
- Lippitz, B.E. Cytokine patterns in patients with cancer: A systematic review. Lancet Oncol. 2013, 14, e218–e228. [Google Scholar] [CrossRef]
- Chuang, T.F.; Lee, S.C.; Liao, K.W.; Hsiao, Y.W.; Lo, C.H.; Chiang, B.L.; Lin, X.Z.; Tao, M.H.; Chu, R.M. Electroporation-mediated IL-12 gene therapy in a transplantable canine cancer model. Int. J. Cancer 2009, 125, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.E.; Quadrini, K.J.; Santiago-Schwarz, F.; Hadden, J.W.; Brandwein, H.J.; Signorelli, K.L. IRX-2, a novel in vivo immunotherapeutic, induces maturation and activation of human dendritic cells in vitro. J. Immunother. 2007, 30, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Naylor, P.H.; Hernandez, K.E.; Nixon, A.E.; Brandwein, H.J.; Haas, G.P.; Wang, C.Y.; Hadden, J.W. IRX-2 increases the T cell-specific immune response to protein/peptide vaccines. Vaccine 2010, 28, 7054–7062. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Halstead, E.S.; Schuler, P.; Harasymczuk, M.; Egan, J.E.; Whiteside, T.L. IRX-2, a novel immunotherapeutic, enhances and protects NK-cell functions in cancer patients. Cancer Immunol. Immunother. CII 2012, 61, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Harasymczuk, M.; Schuler, P.; Egan, J.; Ferrone, S.; Whiteside, T.L. IRX-2, a novel immunotherapeutic, enhances functions of human dendritic cells. PLoS ONE 2013, 8, e47234. [Google Scholar] [CrossRef] [PubMed]
- Hadden, J.; Verastegui, E.; Barrera, J.L.; Kurman, M.; Meneses, A.; Zinser, J.W.; de la Garza, J.; Hadden, E. A trial of IRX-2 in patients with squamous cell carcinomas of the head and neck. Int. Immunopharmacol. 2003, 3, 1073–1081. [Google Scholar] [CrossRef]
- Freeman, S.M.; Franco, J.L.; Kenady, D.E.; Baltzer, L.; Roth, Z.; Brandwein, H.J.; Hadden, J.W. A phase 1 safety study of an IRX-2 regimen in patients with squamous cell carcinoma of the head and neck. Am. J. Clin. Oncol. 2011, 34, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Gertig, D.M.; Brotherton, J.M.; Budd, A.C.; Drennan, K.; Chappell, G.; Saville, A.M. Impact of a population-based HPV vaccination program on cervical abnormalities: A data linkage study. BMC Med. 2013, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M. HPV vaccines and potential prevention of HPV-positive head and neck cancer. In Primary End-Points for Prophylactic HPV Vaccine Trials; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).




{kind=link}