
Cancer Stem Cell Plasticity Drives Therapeutic Resistance

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Connecting the Dots: CSC Properties and Epithelial-Mesenchymal Transition (EMT)
3. Mirroring Metastasis: Trends in Tumor Recurrence
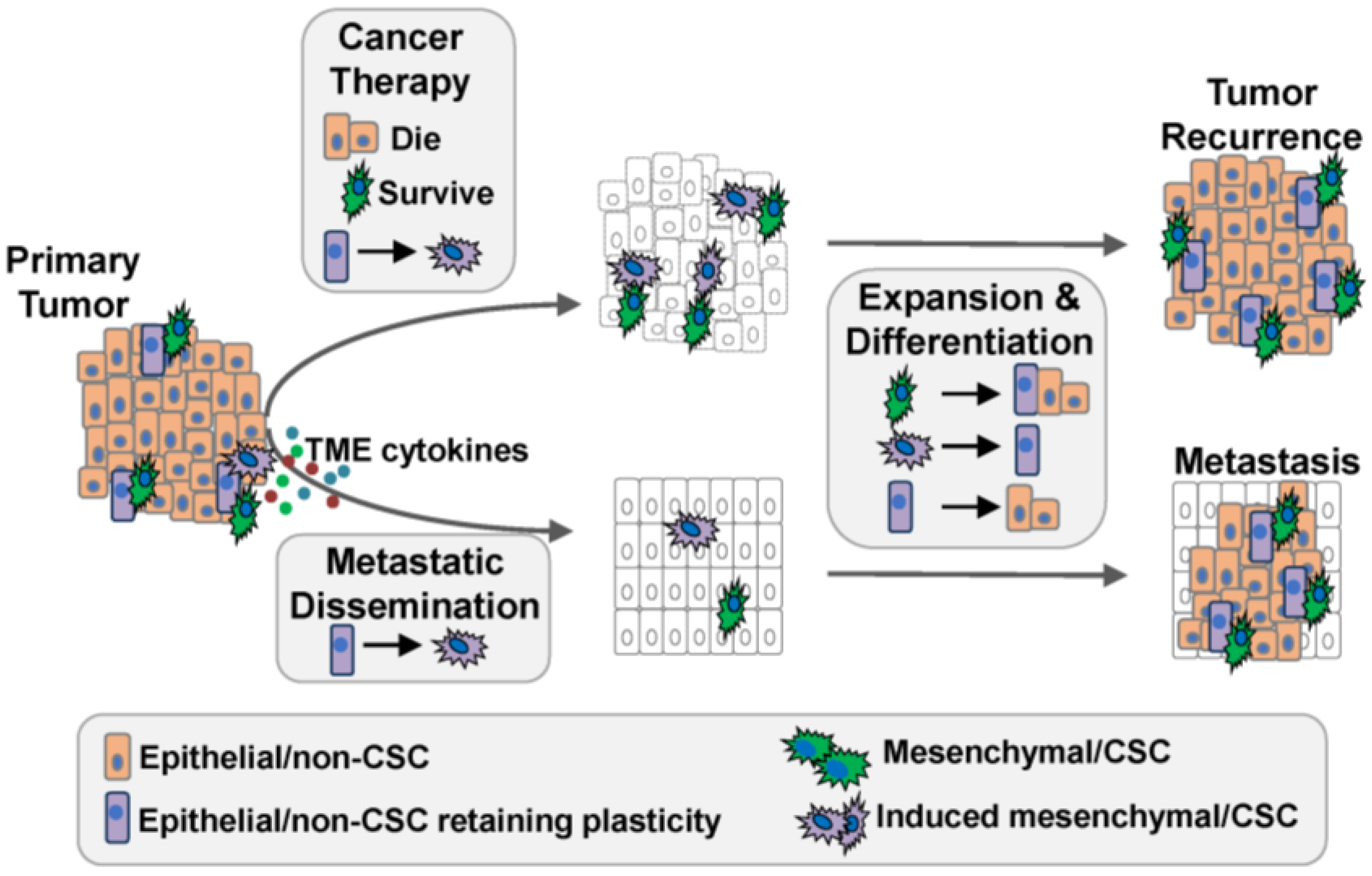
4. De Novo Generation of CSC from non-CSC
5. The TME; Supporting E-M and CSC Plasticity
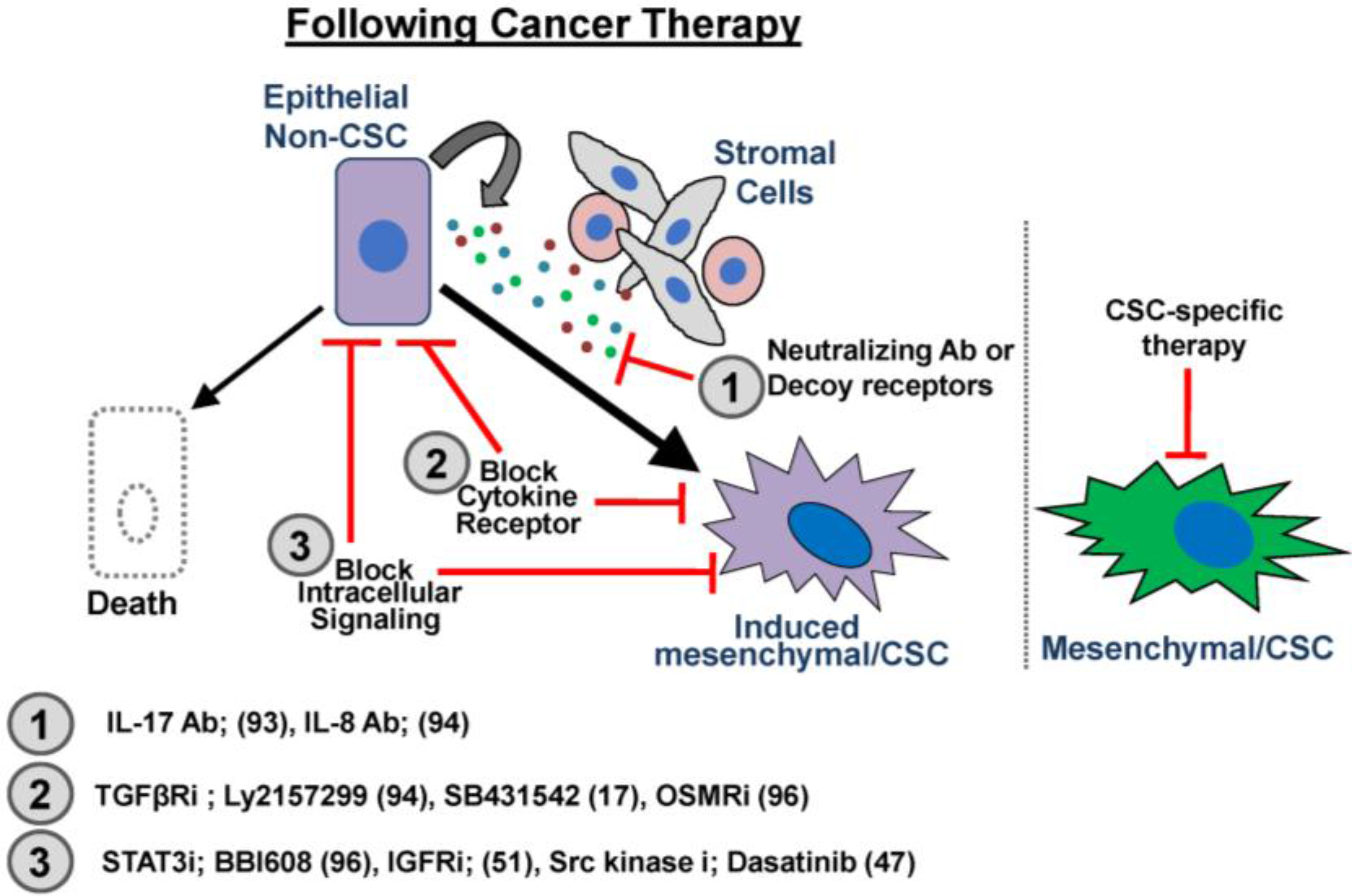
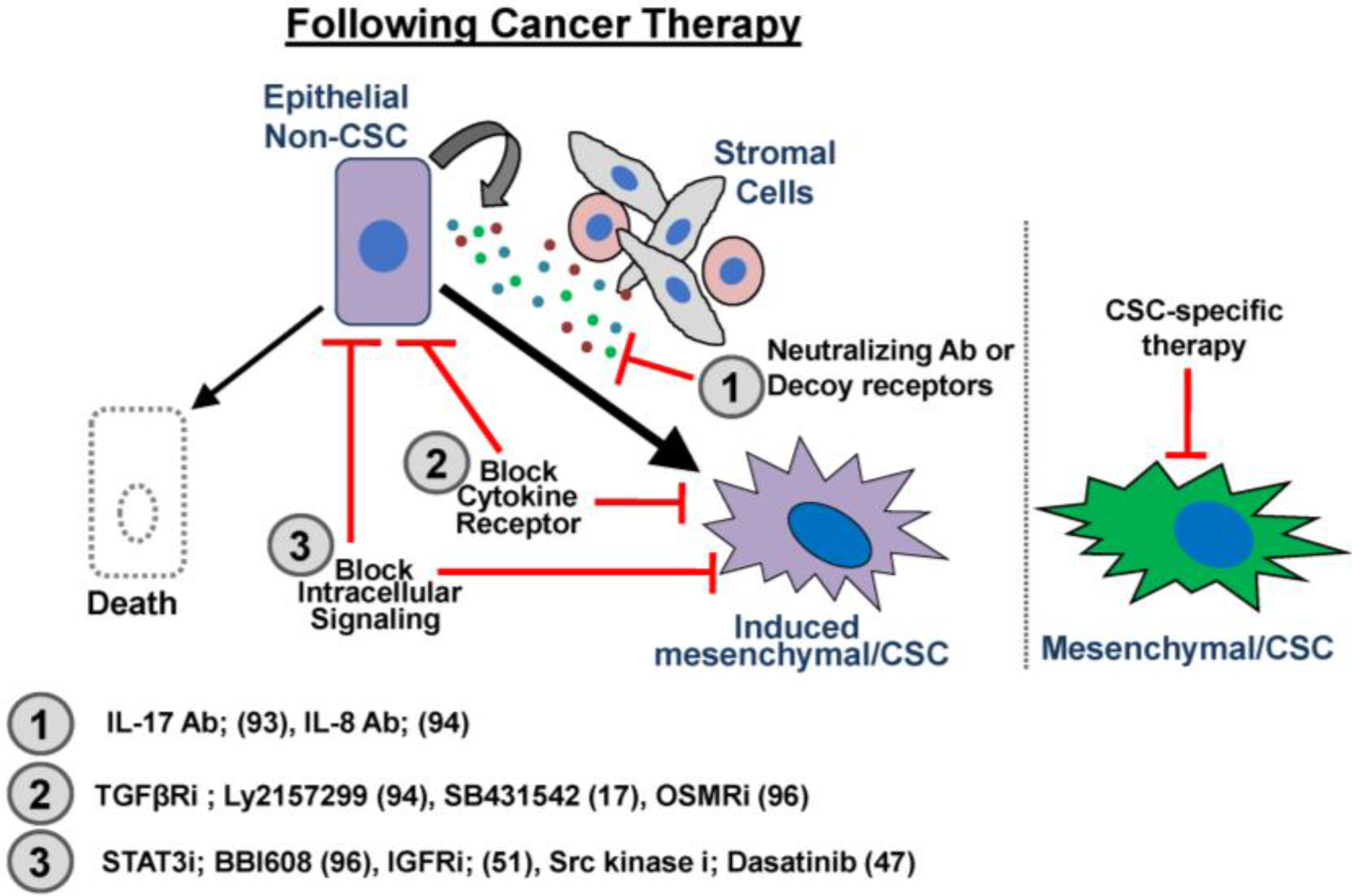
6. Conclusion: Targeting CSC Plasticity to Suppress Therapeutic Resistance
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weigelt, B.; Peterse, J.L.; van’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Guarneri, V.; Broglio, K.; Kau, S.W.; Cristofanilli, M.; Buzdar, A.U.; Valero, V.; Buchholz, T.; Meric, F.; Middleton, L.; Hortobagyi, G.N.; et al. Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J. Clin. Oncol. 2006, 24, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Kuerer, H.M.; Newman, L.A.; Smith, T.L.; Ames, F.C.; Hunt, K.K.; Dhingra, K.; Theriault, R.L.; Singh, G.; Binkley, S.M.; Sneige, N.; et al. Clinical course of breast cancer patients with complete pathologic primary tumor and axillary lymph node response to doxorubicin-based neoadjuvant chemotherapy. J. Clin. Oncol. 1999, 17, 460–469. [Google Scholar] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Adorno-Cruz, V.; Kibria, G.; Liu, X.; Doherty, M.; Junk, D.J.; Guan, D.; Hubert, C.; Venere, M.; Mulkearns-Hubert, E.; Sinyuk, M.; et al. Cancer stem cells: Targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015, 75, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Venere, M.; Rao, M.S.; Rich, J.N. Seeing is believing: Are cancer stem cells the loch ness monster of tumor biology? Stem Cell Rev. 2011, 7, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; ito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Tang, D.G. Cell-of-origin of cancer versus cancer stem cells: Assays and interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of cancer stem cells by cytokine networks: Attacking cancer’s inflammatory roots. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Bertucci, F.; Cabaud, O.; Wicinski, J.; Finetti, P.; Josselin, E.; Adelaide, J.; Nguyen, T.T.; Monville, F.; et al. ALDH1-positive cancer stem cells predict engraftment of primary breast tumors and are governed by a common stem cell program. Cancer Res. 2013, 73, 7290–7300. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Cipriano, R.; Bryson, B.L.; Gilmore, H.L.; Jackson, M.W. Tumor microenvironmental signaling elicits epithelial-mesenchymal plasticity through cooperation with transforming genetic events. Neoplasia 2013, 15, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the cd44hi/cd24lo/− stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSC in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Hollier, B.G.; Tinnirello, A.A.; Werden, S.J.; Evans, K.W.; Taube, J.H.; Sarkar, T.R.; Sphyris, N.; Shariati, M.; Kumar, S.V.; Battula, V.L.; et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013, 73, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Badea, L.; Herlea, V.; Dima, S.O.; Dumitrascu, T.; Popescu, I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato-gastroenterology 2008, 55, 2016–2027. [Google Scholar] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nature Rev. Mol. cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.; Evans, A.J.; Rakha, E.A.; Green, A.R.; Ellis, I.O. Influence of E-cadherin expression on the mammographic appearance of invasive nonlobular breast carcinoma detected at screening. Radiology 2009, 253, 51–55. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rakha, E.A.; Abd El Rehim, D.; Pinder, S.E.; Lewis, S.A.; Ellis, I.O. E-cadherin expression in invasive non-lobular carcinoma of the breast and its prognostic significance. Histopathology 2005, 46, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Patel, A.; Powe, D.G.; Benhasouna, A.; Green, A.R.; Lambros, M.B.; Reis-Filho, J.S.; Ellis, I.O. Clinical and biological significance of E-cadherin protein expression in invasive lobular carcinoma of the breast. Am. J. Surg. Pathol. 2010, 34, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, X.; Shang, M.; Zhang, Y.; Xia, B.; Niu, M.; Liu, Y.; Pang, D. Dysregulated expression of slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J. Surg. Oncol. 2013, 107, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, A.; Welnicka-Jaskiewicz, M.; Seroczynska, B.; Skokowski, J.; Majewska, H.; Szade, J.; Zaczek, A.J. Epithelial-mesenchymal transition markers in lymph node metastases and primary breast tumors-relation to dissemination and proliferation. Am. J. Transl. Res. 2014, 6, 793–808. [Google Scholar] [PubMed]
- Wu, S.; Liu, S.; Liu, Z.; Huang, J.; Pu, X.; Li, J.; Yang, D.; Deng, H.; Yang, N.; Xu, J. Classification of circulating tumor cells by epithelial-mesenchymal transition markers. PLoS NOE 2015, 10, e0123976. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Mitra, A.; Brownlee, Z.; Xia, X.; Bellister, S.; Overman, M.J.; Kopetz, S.; Ellis, L.M.; Meng, Q.H.; Li, S. Epithelial-mesenchymal transitioned circulating tumor cells capture for detecting tumor progression. Clin. Cancer Res. 2015, 21, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Ito, S.; Nakamura, H. Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013, 73, 4112–4122. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.T.; Lippitt, J.M.; Guzman-Ramirez, N.; Hamdy, F.C.; Eaton, C.L.; Thalmann, G.N.; Cecchini, M.G.; et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010, 70, 5163–5173. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Signore, M.; Ricci-Vitiani, L.; De Maria, R. Targeting apoptosis pathways in cancer stem cells. Cancer Lett. 2013, 332, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharmacol. Ther. 2011, 89, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.; Liang, X.; Gammon, L.; Fazil, B.; Harper, L.J.; Emich, H.; Costea, D.E.; Mackenzie, I.C. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011, 71, 5317–5326. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yao, Y.; Liu, B.; Lin, Z.; Lin, L.; Yang, M.; Zhang, W.; Chen, W.; Pan, C.; Liu, Q.; et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene 2012, 31, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Pisco, A.O.; Huang, S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: “What does not kill me strengthens me”. Br. J. Cancer 2015, 112, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ghisolfi, L.; Keates, A.C.; Zhang, J.; Xiang, S.; Lee, D.K.; Li, C.J. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012, 11, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Ghisolfi, L.; Keates, A.C.; Hu, X.; Lee, D.K.; Li, C.J. Ionizing radiation induces stemness in cancer cells. PLoS ONE 2012, 7, e43628. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ginestier, C.; Ou, S.J.; Clouthier, S.G.; Patel, S.H.; Monville, F.; Korkaya, H.; Heath, A.; Dutcher, J.; Kleer, C.G.; et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011, 71, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Celis, J.E.; Gromov, P.; Cabezon, T.; Moreira, J.M.; Ambartsumian, N.; Sandelin, K.; Rank, F.; Gromova, I. Proteomic characterization of the interstitial fluid perfusing the breast tumor microenvironment: A novel resource for biomarker and therapeutic target discovery. Mol. Cell. Proteom. 2004, 3, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Moreno, O.; Lecanda, J.; Green, J.E.; Segura, V.; Catena, R.; Serrano, D.; Calvo, A. Vegf elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (pin)-like cells via an autocrine loop. Exp. Cell Res. 2010, 316, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. Inflammatory cytokines augments TGF-β1-induced epithelial-mesenchymal transition in A549 cells by up-regulating TβR-I. Cell Motil. Cytoskelet. 2008, 65, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Waerner, T.; Alacakaptan, M.; Tamir, I.; Oberauer, R.; Gal, A.; Brabletz, T.; Schreiber, M.; Jechlinger, M.; Beug, H. ILEI: A cytokine essential for emt, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 2006, 10, 227–239. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Murray, J.I.; Watson, P.H. Oncostatin-m promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014, 33, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Techasen, A.; Loilome, W.; Namwat, N.; Dokduang, H.; Jongthawin, J.; Yongvanit, P. Cytokines released from activated human macrophages induce epithelial mesenchymal transition markers of cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 115–118. [Google Scholar] [PubMed]
- Kan, C.E.; Cipriano, R.; Jackson, M.W. C-myc functions as a molecular switch to alter the response of human mammary epithelial cells to oncostatin M. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ng, G.; Winder, D.; Muralidhar, B.; Gooding, E.; Roberts, I.; Pett, M.; Mukherjee, G.; Huang, J.; Coleman, N. Gain and overexpression of the oncostatin M receptor occur frequently in cervical squamous cell carcinoma and are associated with adverse clinical outcome. J. Pathol. 2007, 212, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Kiba, A.; Zong, Y.; Witte, O.N. Interleukin-6 and oncostatin-M synergize with the PI3K/AKT pathway to promote aggressive prostate malignancy in mouse and human tissues. Mol. Cancer Res. 2013, 11, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines oncostatin m and eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef] [PubMed]
- Lauber, S.; Wong, S.; Cutz, J.C.; Tanaka, M.; Barra, N.; Lhotak, S.; Ashkar, A.; Richards, C.D. Novel function of oncostatin m as a potent tumour-promoting agent in lung. Int. J. Cancer 2015, 136, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Botelho, F.M.; Rodrigues, R.M.; Richards, C.D. Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab. Investig. 2014, 94, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; Yang, W.T.; Rosen, D.G.; Landis, M.D.; Wong, H.; Lewis, M.T.; Creighton, C.J.; Sexton, K.R.; Hilsenbeck, S.G.; Sahin, A.A.; et al. Cancer stem cell markers are enriched in normal tissue adjacent to triple negative breast cancer and inversely correlated with DNA repair deficiency. Breast cancer Res. 2013, 15, R77. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, J.; Sun, F.; Liu, L.; Liu, X.; Yue, Y. Oncostatin m promotes proliferation of ovarian cancer cells through signal transducer and activator of transcription 3. Int. J. Mol. Med. 2011, 28, 101–108. [Google Scholar] [PubMed]
- Zhu, M.; Che, Q.; Liao, Y.; Wang, H.; Wang, J.; Chen, Z.; Wang, F.; Dai, C.; Wan, X. Oncostatin m activates STAT3 to promote endometrial cancer invasion and angiogenesis. Oncol. Rep. 2015, 34, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Bolin, C.; Tawara, K.; Sutherland, C.; Redshaw, J.; Aranda, P.; Moselhy, J.; Anderson, R.; Jorcyk, C.L. Oncostatin m promotes mammary tumor metastasis to bone and osteolytic bone degradation. Genes Cancer 2012, 3, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Holzer, R.G.; Ryan, R.E.; Tommack, M.; Schlekeway, E.; Jorcyk, C.L. Oncostatin M stimulates the detachment of a reservoir of invasive mammary carcinoma cells: Role of cyclooxygenase-2. Clin. Exp. Metastasis 2004, 21, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Jorcyk, C.L.; Holzer, R.G.; Ryan, R.E. Oncostatin m induces cell detachment and enhances the metastatic capacity of T-47D human breast carcinoma cells. Cytokine 2006, 33, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast cancer cells stimulate neutrophils to produce oncostatin m: Potential implications for tumor progression. Cancer Res. 2005, 65, 8896–8904. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.E.; Martin, B.; Mellor, L.; Jacob, R.B.; Tawara, K.; McDougal, O.M.; Oxford, J.T.; Jorcyk, C.L. Oncostatin M binds to extracellular matrix in a bioactive conformation: Implications for inflammation and metastasis. Cytokine 2015, 72, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Coleman, N. Oncostatin m receptor is a novel therapeutic target in cervical squamous cell carcinoma. J. Pathol. 2014, 232, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Winder, D.M.; Chattopadhyay, A.; Muralidhar, B.; Bauer, J.; English, W.R.; Zhang, X.; Karagavriilidou, K.; Roberts, I.; Pett, M.R.; Murphy, G.; et al. Overexpression of the oncostatin m receptor in cervical squamous cell carcinoma cells is associated with a pro-angiogenic phenotype and increased cell motility and invasiveness. J. Pathol. 2011, 225, 448–462. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Murphy, L.C.; Watson, P.H. Oncostatin m suppresses oestrogen receptor-alpha expression and is associated with poor outcome in human breast cancer. Endocr.-Relat. Cancer 2012, 19, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Gurluler, E.; Tumay, L.V.; Guner, O.S.; Kucukmetin, N.T.; Hizli, B.; Zorluoglu, A. Oncostatin-M as a novel biomarker in colon cancer patients and its association with clinicopathologic variables. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2042–2047. [Google Scholar] [PubMed]
- Garcia-Tunon, I.; Ricote, M.; Ruiz, A.; Fraile, B.; Paniagua, R.; Royuela, M. Osm, lif, its receptors, and its relationship with the malignance in human breast carcinoma (in situ and in infiltrative). Cancer Investig. 2008, 26, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Royuela, M.; Ricote, M.; Parsons, M.S.; Garcia-Tunon, I.; Paniagua, R.; de Miguel, M.P. Immunohistochemical analysis of the IL-6 family of cytokines and their receptors in benign, hyperplasic, and malignant human prostate. J. Pathol. 2004, 202, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Perales, S.; Alejandre, M.J.; Iglesias, J.; Palomino, R.J.; Martin, M.; Caba, O.; Prados, J.C.; Aranega, A.; Delgado, J.R.; et al. Serum cytokine profile in patients with pancreatic cancer. Pancreas 2014, 43, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Vlaicu, P.; Mertins, P.; Mayr, T.; Widschwendter, P.; Ataseven, B.; Hogel, B.; Eiermann, W.; Knyazev, P.; Ullrich, A. Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific EGFR ligands and a STAT3 activator. BMC Cancer 2013, 13, 197. [Google Scholar] [CrossRef] [PubMed]
- Grenier, A.; Combaux, D.; Chastre, J.; Gougerot-Pocidalo, M.A.; Gibert, C.; Dehoux, M.; Chollet-Martin, S. Oncostatin m production by blood and alveolar neutrophils during acute lung injury. Lab. Investig. 2001, 81, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Grenier, A.; Dehoux, M.; Boutten, A.; Arce-Vicioso, M.; Durand, G.; Gougerot-Pocidalo, M.A.; Chollet-Martin, S. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood 1999, 93, 1413–1421. [Google Scholar] [PubMed]
- Zhu, Q.; Zhang, X.; Zhang, L.; Li, W.; Wu, H.; Yuan, X.; Mao, F.; Wang, M.; Zhu, W.; Qian, H.; et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell. Death Dis. 2014, 5, e1295. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Li, Z.Z.; Gong, J.; Xiang, M.; Zhang, P.; Zhao, G.N.; Li, M.; Zheng, A.; Zhu, X.; Lei, H.; et al. Oncostatin m confers neuroprotection against ischemic stroke. J. Neurosci. 2015, 35, 12047–12062. [Google Scholar] [CrossRef] [PubMed]
- Levano, K.S.; Jung, E.H.; Kenny, P.A. Breast cancer subtypes express distinct receptor repertoires for tumor-associated macrophage derived cytokines. Biochem. Biophys. Res. Commun. 2011, 411, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.A.; Sodhi, A. Cisplatin-treated macrophages produce oncostatin m: Regulation by serine/threonine and protein tyrosine kinases/phosphatases and Ca2+/calmodulin. Immunol. Lett. 1998, 62, 159–164. [Google Scholar] [CrossRef]
- Sodhi, A.; Shishodia, S.; Shrivastava, A. Cisplatin-stimulated murine bone marrow-derived macrophages secrete oncostatin m. Immunol. Cell. Biol. 1997, 75, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.D. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm. 2013, 2013, 512103. [Google Scholar] [CrossRef] [PubMed]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-H.; Chen, M.-T.; Wang, M.-L.; Lee, Y.-Y.; Chiou, G.-Y.; Chien, C.-S.; Huang, P.-I.; Chen, Y.-W.; Huang, M.-C.; Chiou, S.-H.; et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget 2015, 6, 1750–1768. [Google Scholar] [CrossRef] [PubMed]
- Repovic, P.; Fears, C.Y.; Gladson, C.L.; Benveniste, E.N. Oncostatin-M induction of vascular endothelial expression in astroglioma cells. Oncogene 2003, 22, 8117–8124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Lee, H.J.; Lulla, A.; Hernandez, L.; Gokare, P.; Lim, B. Current treatment of early breast cancer: Adjuvant and neoadjuvant therapy. F1000Research 2014, 3, 198. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Li, Q.; Liu, S.; Ning, N.; Zhang, X.; Xu, Y.; Chang, A.E.; Wicha, M.S. Concise review: Targeting cancer stem cells using immunologic approaches. Stem Cells 2015, 33, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, S.; Nandy, A.; Hor, P.; Mukhopadhyay, A. Breast cancer stem cells: A novel therapeutic target. Clin. Breast Cancer 2013, 13, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Kolev, V.N.; Wright, Q.G.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 2015, 75, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Prasetyanti, P.R.; de Lau, W.; Rodermond, H.; Clevers, H.; Medema, J.P. Monoclonal antibodies against LGR5 identify human colorectal cancer stem cells. Stem Cells 2012, 30, 2378–2386. [Google Scholar] [CrossRef] [PubMed]
- Sachlos, E.; Risueno, R.M.; Laronde, S.; Shapovalova, Z.; Lee, J.H.; Russell, J.; Malig, M.; McNicol, J.D.; Fiebig-Comyn, A.; Graham, M.; et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012, 149, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting wnt, notch, and hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8, 8. https://doi.org/10.3390/cancers8010008
Doherty MR, Smigiel JM, Junk DJ, Jackson MW. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers. 2016; 8(1):8. https://doi.org/10.3390/cancers8010008
Chicago/Turabian StyleDoherty, Mary R., Jacob M. Smigiel, Damian J. Junk, and Mark W. Jackson. 2016. "Cancer Stem Cell Plasticity Drives Therapeutic Resistance" Cancers 8, no. 1: 8. https://doi.org/10.3390/cancers8010008
APA StyleDoherty, M. R., Smigiel, J. M., Junk, D. J., & Jackson, M. W. (2016). Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers, 8(1), 8. https://doi.org/10.3390/cancers8010008