
Next-Generation Sequencing in Clinical Molecular Diagnostics of Cancer: Advantages and Challenges


Abstract
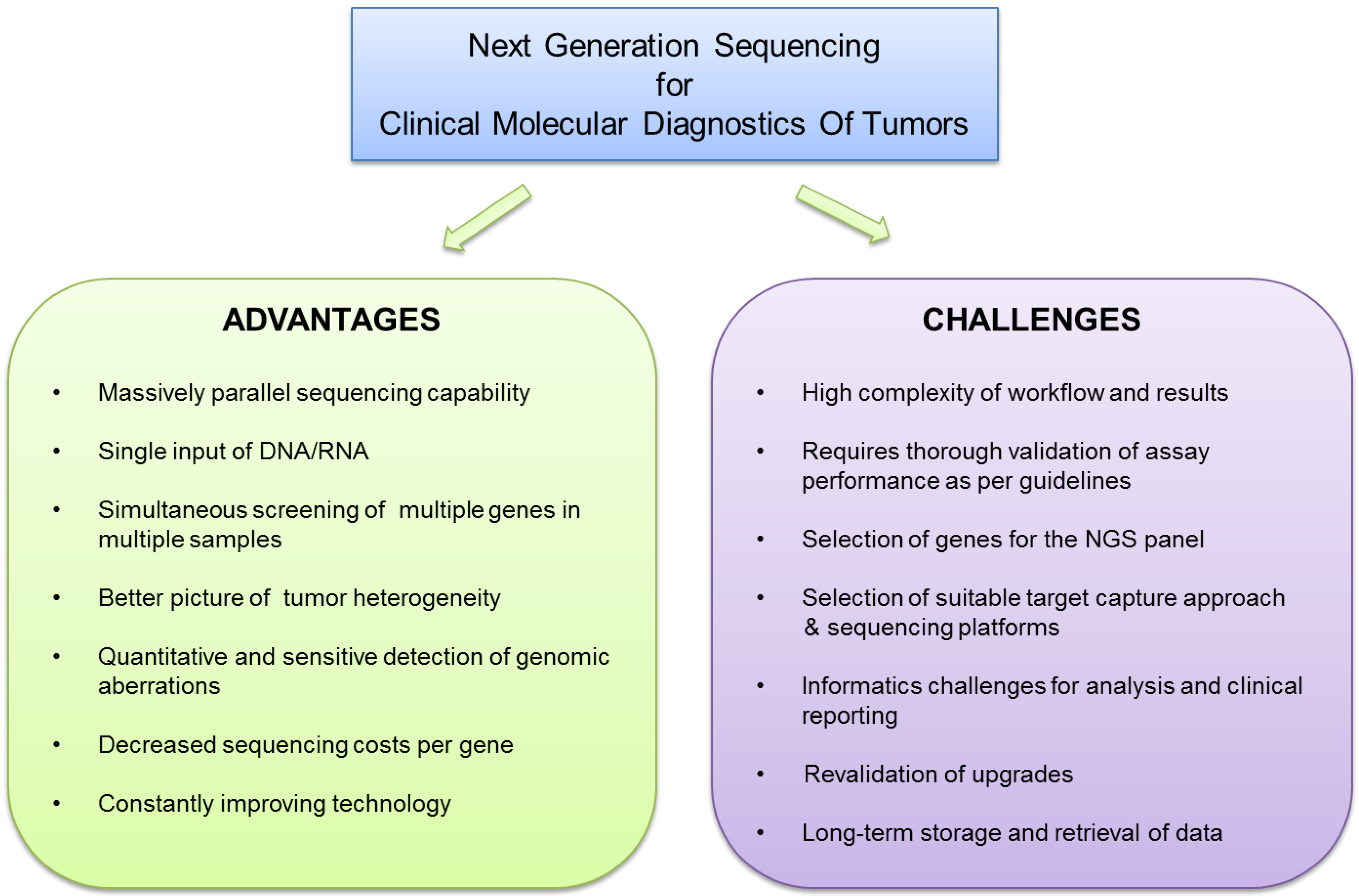
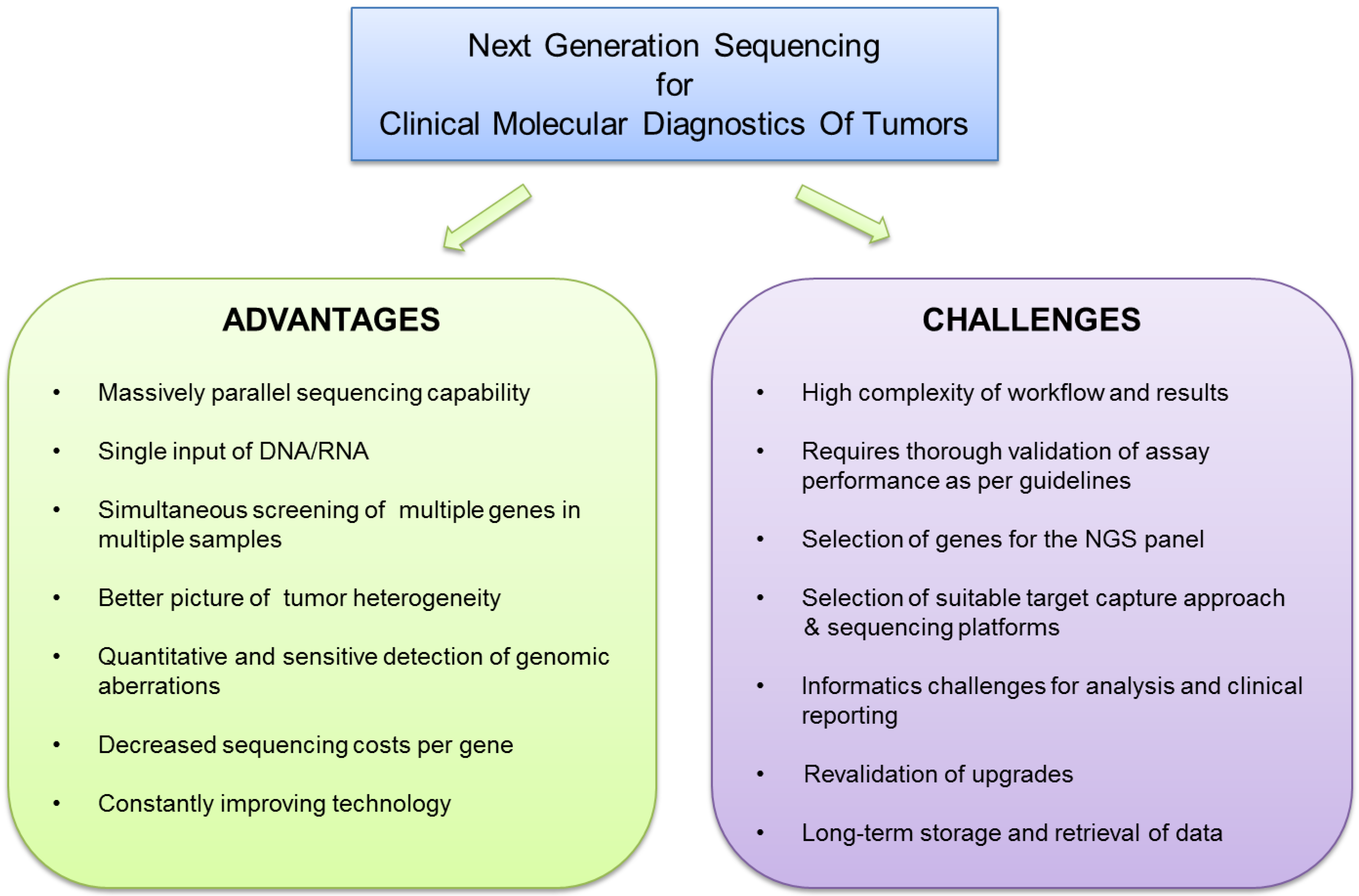
:1. Introduction
1.1. Performance Validation

{kind=link}
| Performance Parameters | Explanation | Purpose |
|---|---|---|
| Analytical Sensitivity* | Portion of samples in the validation set that are positive for mutations, as detected by a validated platform, and are correctly identified as positive | Ability of the assay to detect true sequence variants (false-negative rate) |
| Analytical Specificity | Portion of samples in the validation set that are negative for mutations, as established by a validated platform, and are accurately classified as negative | Probability of the assay to not detect mutations where none are present (false-positive rate) |
| Accuracy | Concordance between the genomic sequences obtained by the NGS assay and the reference sequence | Measure of sequencing accuracy and error rates |
| Precision | The tendency of achieving accurate results regarding detection of mutations across users and sequencing runs | Measure of reproducibility of mutation detection by the assay and inter-user reproducibility |
| Limit-of-detection * | The lower limit of mutation detection | To establish the detection limit for different variants such SNVs, MNVs, insertions, deletions, CNVs, and gene fusions |
| Sequencing depth and allelic frequency cutoffs | Define the minimum sequencing coverage necessary for confident detection and calling of variants | Needs to be established for different variants such as SNVs, MNVs, insertions, deletions, CNVs, and gene fusions |
1.2. Choices and Challenges of NGS Technology
| Company | Sequencer | Sequencing Technology | Comments |
|---|---|---|---|
| Illumina Inc | MiSeq HiSeq NextSeq | Sequencing-by-Synthesis (Reversible terminator-Based) |
|
| Life Technologies | Ion Torrent Personal Genome Machine (PGM) Ion Proton | Sequencing-by-Synthesis (Semiconductor-based) |
|
| Pacific Biosciences | PACBIO RSII | Single molecule realtime (SMRT) sequencing |
|
2. Choices for NGS Panel Content and Target Capture Technology
| Company | Enrichment Technology | Enrichment Approach | Options, recommended DNA input and comments |
|---|---|---|---|
| Illumina Inc | TruSeq | DNA probe-based capture | TruSeq Amlicon Kit—(50 ng–250 ng high quality DNA, 250 ng FFPE DNA) |
| TruSeq DNA PCR free (Low Throughput)—(1 µg) | |||
| TruSeq DNA PCR free (High Throughput)—(2 µg) | |||
| TruSeq NANO Low and High throughput kit (LT and HT)—(100–200 ng) | |||
| NeoPrep System—Automated enrichment and library preparation system | |||
| Life Technologies | AmpliSeq | PCR-based amplification | 10 ng DNA per primer pool ( up to 6000 primers) |
| Well-suited for low quantity and quality DNA samples like FFPE samples | |||
| Agilent Technologies | SureSelect | Hybridization and capture using cRNA-baits | 200 ng–3 µg DNA input |
| Haloplex | Restriction enzyme digested DNA used as template | 200 ng DNA input | |
| Circularization probe-based target enrichment | |||
| Qiagen | GeneRead | PCR-based amplification | 40 ng DNA input |
| Integrated DNA Technologies | Xgen Lockdown probes | DNA probe-based capture | 500 ng DNA input |
| RainDance Technologies | ThunderStorm and ThunderBolt systems | Droplet PCR-based amplification | ThunderBolt system 10–50 ng for limited gene panel size. |
| ThunderStorm system 500 ng–1 µg depending on the gene panel size |
3. NGS Data Analysis and Clinical Reporting
| Steps in NGS workflow | QC Metric | Method/Indicator | Comments |
|---|---|---|---|
| Nucleic Acid Quantification | Nucleic acid quality and quantity | Fluorimetric dye-based or qPCR based quantification |
|
| Genomic library Preparation | Genomic library quality and quantity | Gel-based systems or qPCR based quantification |
|
| Sequencing | Run and sample level sequencing output and quality |
|
|
| Variant detection and clinical reporting | Variant detection and reporting confidence |
|
|
4. Potential for the Future
Supplementary Files
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Roychowdhury, S.; Chinnaiyan, A.M. Translating genomics for precision cancer medicine. Annu. Rev. Genomics Hum. Genet. 2014, 15, 395–415. [Google Scholar] [PubMed]
- Previati, M.; Manfrini, M.; Galasso, M.; Zerbinati, C.; Palatini, J.; Gasparini, P.; Volinia, S. Next generation analysis of breast cancer genomes for precision medicine. Cancer Lett. 2013, 339, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Byron, S.A.; Tran, N.L.; Phillips, J.J.; Molinaro, A.M.; Ligon, K.L.; Wen, P.Y.; Kuhn, J.G.; Mellinghoff, I.K.; de Groot, J.F.; et al. Toward precision medicine in glioblastoma: The promise and the challenges. Neuro Oncol. 2015, 17, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Richer, A.L.; Friel, J.M.; Carson, V.M.; Inge, L.J.; Whitsett, T.G. Genomic profiling toward precision medicine in non-small cell lung cancer: Getting beyond EGFR. Pharmgenomics Pers. Med. 2015, 8, 63–79. [Google Scholar] [PubMed]
- Intlekofer, A.M.; Younes, A. Precision therapy for lymphoma—Current state and future directions. Nat. Rev. Clin. Oncol. 2014, 11, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Roper, N.; Stensland, K.D.; Hendricks, R.; Galsky, M.D. The landscape of precision cancer medicine clinical trials in the United States. Cancer Treat. Rev. 2015, 41, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Wilson, R.K. Cancer genome sequencing: A review. Hum. Mol. Genet. 2009, 18, R163–R168. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.; van Deerlin, V.M.; Gulley, M.L. Recommended principles and practices for validating clinical molecular pathology tests. Arch. Pathol. Lab. Med. 2009, 133, 743–755. [Google Scholar] [PubMed]
- Aziz, N.; Zhao, Q.; Bry, L.; Driscoll, D.K.; Funke, B.; Gibson, J.S.; Grody, W.W.; Hegde, M.R.; Hoeltge, G.A.; Leonard, D.G.; et al. College of American Pathologists’ Laboratory Standards for Next-Generation Sequencing Clinical Tests. Arch. Pathol. Lab. Med. 2015, 139, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E.; et al. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, I.; Aziz, N.; Farkas, D.H.; Furtado, M.; Gonzalez, A.F.; Greiner, T.C.; Grody, W.W.; Hambuch, T.; Kalman, L.; Kant, J.A.; et al. Opportunities and challenges associated with clinical diagnostic genome sequencing: A report of the Association for Molecular Pathology. J. Mol. Diagn. 2012, 14, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Beadling, C.; Neff, T.L.; Heinrich, M.C.; Rhodes, K.; Thornton, M.; Leamon, J.; Andersen, M.; Corless, C.L. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J. Mol. Diagn. 2013, 15, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, I.S.; Devarakonda, S.; Lockwood, C.M.; Spencer, D.H.; Guebert, K.; Bredemeyer, A.J.; Al-Kateb, H.; Nguyen, T.T.; Duncavage, E.J.; Cottrell, C.E.; et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer 2014, 121, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Luthra, R.; Patel, K.P.; Reddy, N.G.; Haghshenas, V.; Routbort, M.J.; Harmon, M.A.; Barkoh, B.A.; Kanagal-Shamanna, R.; Ravandi, F.; Cortes, J.E.; et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: Applicability for diagnostics and disease monitoring. Haematologica 2014, 99, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Aldape, K.; Lu, X.; Manekia, J.; Abraham, R.; Reddy, N.G.; Barkoh, B.A.; Veliyathu, J.; et al. Clinical massively parallel next-generation sequencing analysis of 409 cancer-related genes for mutations and copy number variations in solid tumours. Br. J. Cancer 2014, 111, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D.; et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn. 2013, 15, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Kanagal-Shamanna, R.; Portier, B.P.; Singh, R.R.; Routbort, M.J.; Aldape, K.D.; Handal, B.A.; Rahimi, H.; Reddy, N.G.; Barkoh, B.A.; Mishra, B.M.; et al. Next-generation sequencing-based multi-gene mutation profiling of solid tumors using fine needle aspiration samples: Promises and challenges for routine clinical diagnostics. Mod. Pathol. 2014, 27, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, P.L.; Reinhold, W.C.; Varma, S.; Hutchinson, A.A.; Pommier, Y.; Chanock, S.J.; Weinstein, J.N. DNA fingerprinting of the NCI-60 cell line panel. Mol. Cancer Ther. 2009, 8, 713–724. [Google Scholar] [CrossRef] [PubMed]
- International HapMap Consortium. The International HapMap Project 2003, 426, 789–796.
- Han, S.J.; Rutledge, W.C.; Molinaro, A.M.; Chang, S.M.; Clarke, J.L.; Prados, M.D.; Taylor, J.W.; Berger, M.S.; Butowski, N.A. The Effect of Timing of Concurrent Chemoradiation in Patients with Newly Diagnosed Glioblastoma. Neurosurgery 2015, 77, 248–253. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Merriman, B.; Rothberg, J.M. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis 2012, 33, 3397–3417. [Google Scholar] [CrossRef] [PubMed]
- Kilianski, A.; Haas, J.L.; Corriveau, E.J.; Liem, A.T.; Willis, K.L.; Kadavy, D.R.; Rosenzweig, C.N.; Minot, S.S. Bacterial and viral identification and differentiation by amplicon sequencing on the MinION nanopore sequencer. Gigascience 2015, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Nivon, L.; Brandin, E.; Golovchenko, J.; Branton, D. Rapid nanopore discrimination between single polynucleotide molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Vercoutere, W.; Winters-Hilt, S.; Olsen, H.; Deamer, D.; Haussler, D.; Akeson, M. Rapid discrimination among individual DNA hairpin molecules at single-nucleotide resolution using an ion channel. Nat. Biotechnol. 2001, 19, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Bodi, K.; Perera, A.G.; Adams, P.S.; Bintzler, D.; Dewar, K.; Grove, D.S.; Kieleczawa, J.; Lyons, R.H.; Neubert, T.A.; Noll, A.C.; et al. Comparison of commercially available target enrichment methods for next-generation sequencing. J. Biomol. Tech. 2013, 24, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Mertes, F.; Elsharawy, A.; Sauer, S.; van Helvoort, J.M.; van der Zaag, P.J.; Franke, A.; Nilsson, M.; Lehrach, H.; Brookes, A.J. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief. Funct. Genomics 2011, 10, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wallis, J.W.; Kandoth, C.; Kalicki-Veizer, J.M.; Mungall, K.L.; Mungall, A.J.; Jones, S.J.; Marra, M.A.; Ley, T.J.; Mardis, E.R.; et al. BreakFusion: Targeted assembly-based identification of gene fusions in whole transcriptome paired-end sequencing data. Bioinformatics 2012, 28, 1923–1924. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Hormozdiari, F.; Zayed, A.; Giuliany, R.; Ha, G.; Sun, M.G.; Griffith, M.; Heravi Moussavi, A.; Senz, J.; Melnyk, N.; et al. deFuse: An algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 2011, 7, e1001138. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Wain, H.M.; Bruford, E.A.; Lovering, R.C.; Lush, M.J.; Wright, M.W.; Povey, S. Guidelines for human gene nomenclature. Genomics 2002, 79, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- Forbes, S.A.; Tang, G.; Bindal, N.; Bamford, S.; Dawson, E.; Cole, C.; Kok, C.Y.; Jia, M.; Ewing, R.; Menzies, A.; et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010, 38, D652–D657. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.; Chen, H.; Andrews, J.; Naser, R.; Pao, W.; Horn, L. DNA-Mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): A catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapy. Clin. Cancer Res. 2013, 19, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Bale, S.; Bayrak-Toydemir, P.; Gibson, J.; Jeng, L.J.; Joseph, L.; Laser, J.; Lubin, I.M.; Miller, C.E.; Ross, L.F.; et al. Reporting incidental findings in genomic scale clinical sequencing—A clinical laboratory perspective: A report of the Association for Molecular Pathology. J. Mol. Diagn. 2015, 17, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Deverka, P.A.; Dreyfus, J.C. Clinical integration of next generation sequencing: Coverage and reimbursement challenges. J. Law Med. Ethics 2014, 42, S22–S41. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luthra, R.; Chen, H.; Roy-Chowdhuri, S.; Singh, R.R. Next-Generation Sequencing in Clinical Molecular Diagnostics of Cancer: Advantages and Challenges. Cancers 2015, 7, 2023-2036. https://doi.org/10.3390/cancers7040874
Luthra R, Chen H, Roy-Chowdhuri S, Singh RR. Next-Generation Sequencing in Clinical Molecular Diagnostics of Cancer: Advantages and Challenges. Cancers. 2015; 7(4):2023-2036. https://doi.org/10.3390/cancers7040874
Chicago/Turabian StyleLuthra, Rajyalakshmi, Hui Chen, Sinchita Roy-Chowdhuri, and R. Rajesh Singh. 2015. "Next-Generation Sequencing in Clinical Molecular Diagnostics of Cancer: Advantages and Challenges" Cancers 7, no. 4: 2023-2036. https://doi.org/10.3390/cancers7040874
APA StyleLuthra, R., Chen, H., Roy-Chowdhuri, S., & Singh, R. R. (2015). Next-Generation Sequencing in Clinical Molecular Diagnostics of Cancer: Advantages and Challenges. Cancers, 7(4), 2023-2036. https://doi.org/10.3390/cancers7040874