Abstract
Discussions of therapeutic suppression of hedgehog (Hh) signaling almost exclusively focus on receptor antagonism; however, hedgehog’s biosynthesis represents a unique and potentially targetable aspect of this oncogenic signaling pathway. Here, we review a key biosynthetic step called cholesterolysis from the perspectives of structure/function and small molecule inhibition. Cholesterolysis, also called cholesteroylation, generates cholesterol-modified Hh ligand via autoprocessing of a hedgehog precursor protein. Post-translational modification by cholesterol appears to be restricted to proteins in the hedgehog family. The transformation is essential for Hh biological activity and upstream of signaling events. Despite its decisive role in generating ligand, cholesterolysis remains conspicuously unexplored as a therapeutic target.
Keywords:
hedgehog; autoprocessing; cholesterol; cancer; cholesterolysis; cholesteroylation; signaling 1. Introduction
In general, cell/cell signaling by hedgehog (Hh) proteins is beneficial, particularly during early development [1,2,3,4,5,6,7,8]. Severe congenital malformations are linked to mutations in the three human Hh genes—Sonic, Indian, and Desert [9,10,11]. However, in the adult persistent Hh signaling has pathogenic effects in multiple cancers. Recent studies connect Hh overexpression, predominantly sonic Hh, to sporadic tumorigenesis and metastasis in prostate, breast, and lung tissue, among others [12,13,14,15,16,17,18,19]. Validation of Hh signaling as a target for anti-cancer therapy reached a milestone in 2012 with the approval of Erivedge (Vismodegib) for the treatment of advanced basal cell carcinoma [20,21,22,23]. The path of Hh research from developmental biology to approved cancer drug is a case study in basic scientific inquiry with translational purpose [24,25,26,27,28].
Erivedge is representative of a general class of Hh therapeutics, where the mechanism of action involves inhibiting Hh signal transduction (Figure 1). The drug interferes with a seven-pass cell surface receptor called Smoothened (SMO), the quintessential target for Hh pathway manipulation [25]. Although Hhs are not bound by SMO, SMO is required for transducing the Hh signal. The intermediary Patched (PTCH1), a 12-pass receptor, antagonizes SMO action in the absence of Hh [29,30,31,32]. When PTCH1 is bound by Hh ligand, the antagonistic interactions cease. Signal transduction events downstream of SMO1 have also ignited drug discovery efforts. The Gli1 and Gli2 transcription factors, which relay Hh signaling at the level of gene expression [33,34], have been blocked by small molecules, GANT58 and GANT61 [35]. The anticancer agent, arsenic trioxide, also seems to compromise GLI action [36], although effects vary with cell type [37]. The accompanying reviews in this issue provide timely updates in this area.
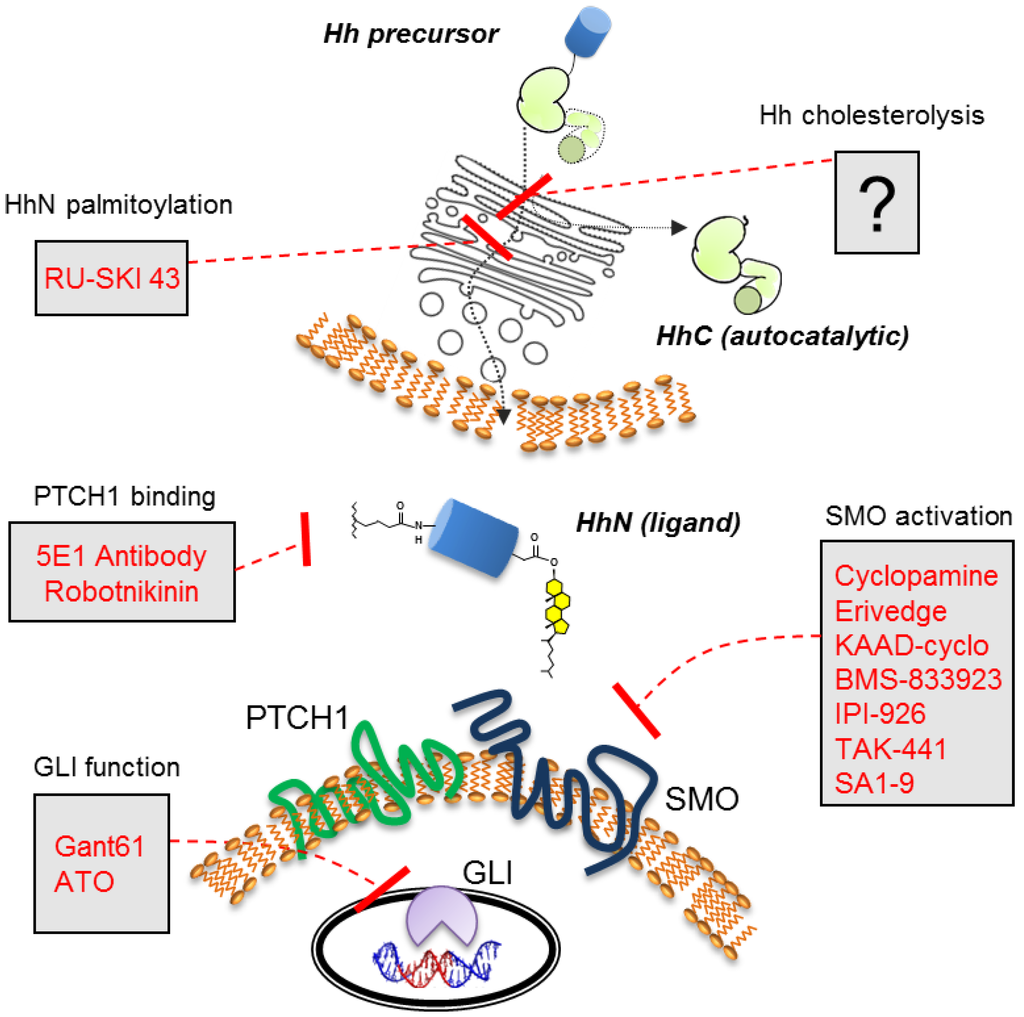
Figure 1.
Components and selected antagonists of the Hh cell-cell signaling pathway. Hh precursor cholesterolysis occurs in the endoplasmic reticulum of the Hh expressing cell (top).
Here the spotlight shines on a less explored opportunity for pathway manipulation, Hh cholesterolysis. This unusual biosynthetic event, involving a precursor form of Hh [38], occurs upstream of Hh ligand secretion, and hence upstream of signal transduction [39,40,41,42]. Cholesterolysis represents one of two lipidation events that the Hh ligand undergoes. The other is N-linked palmitoylation [43,44,45,46]. Unlike the fatty acid modification, the catalyst for cholesterol modification is the Hh protein itself—no cofactors or accessory proteins are required. Self-catalyzed reactions like this one are classified as autoprocessing [47,48]. Mutagenesis studies indicate that diminished cholesterolysis prevents Hh ligand release, leads to premature Hh ligand degradation, and thereby stifles downstream signaling [39,41,42,49].
Small molecules that inhibit the Hh pathway in a similar manner could prove useful therapeutically, particularly in cancers driven by Hh ligand overproduction. Despite the promise, compounds that selectively inhibit cholesterolysis have not been reported. In this work, we review mechanistic details, structure/function data, along with preliminary efforts to discover cholesterolysis inhibitors. Many details are murky, and autoprocessing in general is unexplored as a therapeutic target. As a result, basic and applied research in this area remain at a formative stage since cholesterolysis was described 20 years ago.
2. Cholesterolysis—Liberating the Ligand
Hh proteins are expressed as bifunctional precursors, consisting of the signaling ligand (HhN, 20 kDa) fused to a C-terminal autocatalytic element (HhC, 26 kDa) (Figure 2A). Early studies demonstrated that Hh precursor is short lived in eukaryotic cells, undergoing site-specific cleavage to separate HhN from HhC [39,40]. Beachy and his group revealed two surprising features of this reaction: first, the transformation can be reconstituted in vitro in the absence of accessory proteins; second, the terminal nucleophile that cleaves at the HhN/HhC junction is a sterol [38,39,50]. Cholesterol proved to be most active in the reconstitution experiments, and presumably represents the native substrate in mammals. Thus, a new post-translational modification was discovered that proceeds by peptide bond cholesterolysis [38,51]. Remarkably, two decades from those seminal publications, members of the Hh family still seem to retain “exclusive rights” to this chemistry [53].
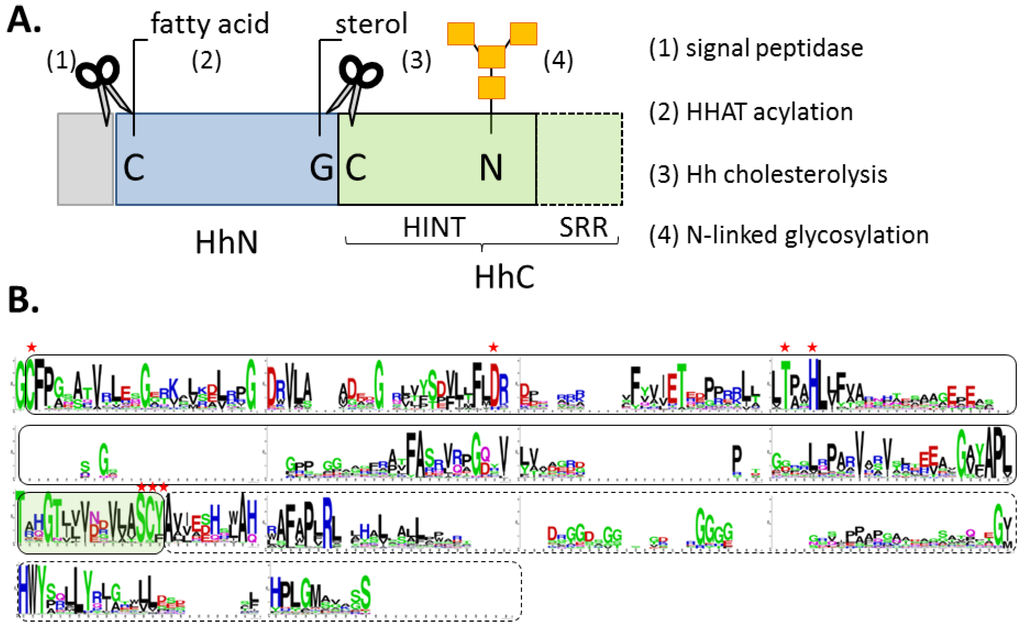
Figure 2.
(A) Block diagram of Hh precursor protein, with signal peptide (grey), signaling ligand (blue), and autocatalytic segment (green). (B) Conserved residues in the autocatalytic segment displayed by Logoplot (http://weblogo.berkeley.edu/logo.cgi). Solid line delineates the HINT domain; hashed line, the SRR region. Residues marked with red asterisks are required for cholesterolysis.
Results of truncation experiments indicate that all catalytic activity essential for cholesterolysis resides in HhC; the N-terminal signaling domain is a bystander [38,53]. HhN can be replaced by arbitrary proteins and peptides without appreciable loss of activity [42,49,54]. HhC exhibits a bipartite organization with a hedgehog/intein domain “HINT” followed by a hydrophobic sterol recognition region, SRR (Figure 2A,B). High-resolution structural data of these segments could be invaluable; however, studies on HhC are incomplete (more below). We lack structural data for a Hh precursor, an intact HhC segment, and a HINT domain from a vertebrate. An atomic structure of the SRR region would be particularly advantageous for understanding the cholesterolysis mechanism and guiding inhibitor development.
The standard mechanism of Hh cholesterolysis involves two steps and requires both the HINT and SRR segments [38,53,55]. As depicted in Figure 3 (Step 1), the sequence begins with the generation of an internal thioester via rearrangement of the backbone peptide bond linking HhN to HhC. The first residue of HhC, invariably cysteine, serves as the nucleophile. It seems conceivable that the scissile amide is strained to facilitate this endergonic rearrangement [56], as has been suggested in related autoprocessing reactions [57,58,59]. Mutagenesis studies have revealed several conserved residues in the HINT domain that are required for the N-S acyl shift [39,42,53]. Mechanistic roles are considered below. In Step 2 of cholesterolysis (Figure 3, Step 2), the thioester linking HhN to HhC is resolved by transesterification to cholesterol. This step liberates HhN from HhC and covalently links the newly formed C-terminus of HhN to substrate cholesterol. Deletion mapping indicate that Step 2 requires the SRR segment, comprising the last ~70 residues of HhC [53]. The source of cholesterol, its binding interactions, and the means by which its C3 hydroxyl group (pKa > 15) is activated remain obscure.
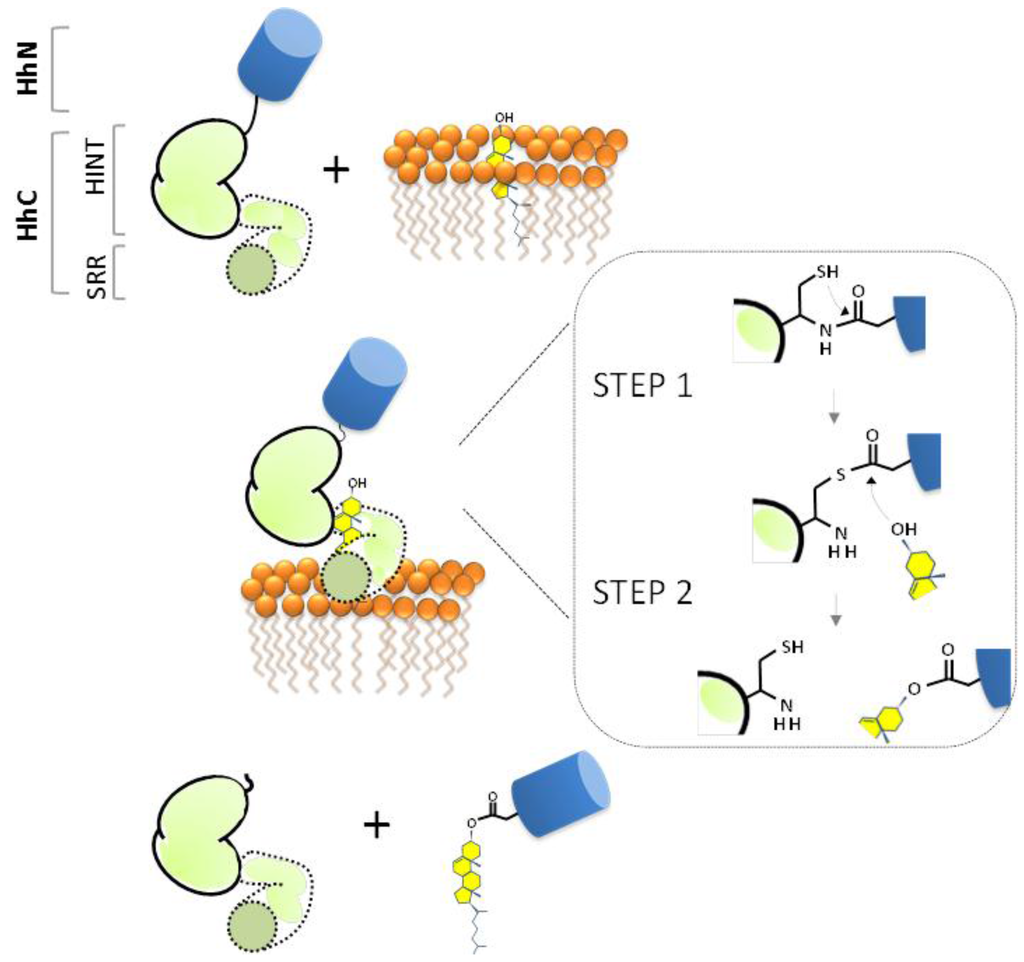
Figure 3.
Proposed mechanism of Hh precursor cholesterolysis as a self-catalyzed event. Inset depicts the two chemical steps: an N-S acyl shift (Step 1) followed by transesterification (Step 2). Signaling ligand, HhN (blue); autocatalytic segment, HhC (green).
2.1. The HINT Domain from Drosophila Melanogaster Hh Protein
The first, and so far only, structure relevant to HhC is that of a HINT domain reported by Hall et al. in 1997 [53]. The domain belongs to the Drosophila melanogaster (Dme) Hh precursor. It is competent to self-catalyze the first step of cholesterolysis, N-S acyl shift, but not the second. Thus, the domain can generate an internal thioester, as apparent from cleavage at the N-terminal HINT junction by water (hydrolysis) and added hydroxylamine (hydroxyaminolysis); however, activity with cholesterol is absent. The HINT domain is predominately β-strand, folded into two symmetrical lobes resembling a baseball catcher’s glove (Figure 4A). Active site residues are arranged in the glove’s pocket (Figure 4B). Striking homology exists between the HINT structure and the self-splicing domain of inteins, pointing to a common ancestor (Figure 4C) [47,53,60]. Catalytic residues in common between Hh HINT and inteins comprise the N-terminal cysteine, a conserved TXXH motif, and second cysteine at the C-terminal end of the domain. Evolutionary divergence is apparent in, for example, an active-site aspartate (Asp303 in full length Dme Hh numbering).
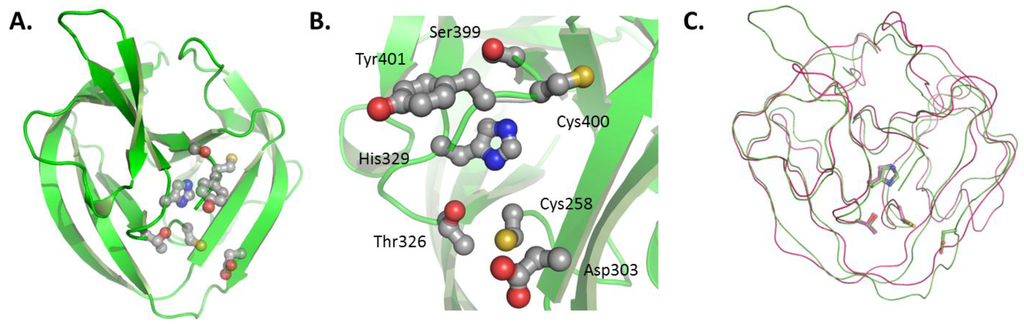
Figure 4.
(A) HINT domain of Drosophila melanogaster Hh precursor (PDB#, 1AT0); (B) conserved catalytic residues of the HINT domain; an (C) alignment of Hh HINT domain with self-splicing intein (PDB#, 2IN0). Figures rendered using PyMol (DeLano Scientific LLC, Palo Alto, CA, USA).
Residual activity of point mutants has helped define mechanistic roles for conserved HINT residues. Mutation of the threonine or histidine residues in the TXXH motif of the HINT compromises N-S acyl shift activity. The threonine is arranged similarly to the corresponding residue in inteins, hydrogen bonding to the nucleophilic cysteine residue (Cys258 in Dme Hh) [61,62]. This Thr residue may have a role in positioning Cys258 for attack and the leaving group, glycine, for departure. The histidine (His329) of the conserved TXXH motif may act as a general base to activate the cysteine for N-S acyl shift [55,63], or promote N-S acyl shift through ground-state destabilization [58,64]. In contrast, mutating the Hh-specific aspartate (Asp303) to alanine inhibits transesterification (Step 2) but not N-S acyl shift, suggesting a role for this residue in coupling the two steps in Hh autoprocessing [53]. Interestingly, an aspartate residue of inteins plays a similar coordinating role for self-splicing [63,65]. The downstream cysteine of the HINT (Cys400) is also intriguing, as it seems to form an intramolecular disulfide bond with Cys258 [42]; recalling the redox regulation of some inteins [59,66,67]. Mutations of Cys400 of HINT domain inhibit Step 1 and Step 2 of cholesterolysis [42].
2.2. Biological Role of Hh Ligand Cholesteroylation
Cholesterol serves as more than just a means for freeing up Hh ligand (HhN) [68,69,70]. Intuitively, covalent modification of soluble protein with cholesterol (LogP ~7) is expected to confer affinity for cellular membranes (Figure 5A). This expectation is borne out in multiple experiments, not only with Hh [71,72,73,74] but also with engineered, cholesterol-modified proteins and peptides [75,76,77,78,79]. Biophysical experiments suggest that membrane partitioning of cholesterol-modified peptides is quasi-irreversible, with half-times for dissociation approaching 3 h [75]. Avid membrane binding may explain the >10 fold greater potency in cell signaling assays of cholesterol-modified HhN compared with unmodified HhN [80,81]. It also helps rationalize why a specialized secretion mechanism is needed to release Hh ligand from the producing cell. Two proteins, Scube and Dispatched, collaborate in this role [49,82]. Cholesterol provides the handle for interactions, which effectively solubilize the Hh ligand [49] (Figure 5B). Other studies suggest that cholesterol leads to association of HhN with caveolin [83] and with lipoproteins [84], influencing intracellular trafficking and extracellular dispersal, respectively.
When the hydrophobic anchor of HhN is not bound by membrane or by protein, it may bind itself to form micelle-like structures. The proposed structure of these soluble aggregates places HhN as the “head group” with attached lipids forming a hydrophobic core (Figure 5C) [85,86]. Supramolecular structures were suggested by the anomalously large apparent molecular weight of Hh ligand interpolated by size exclusion chromatography (exp, 20 KDa; obs, >200 kDa) [52,71,87,88]. An alternative interpretation of these high molecular forms of Hh ligand has also been proposed [89].
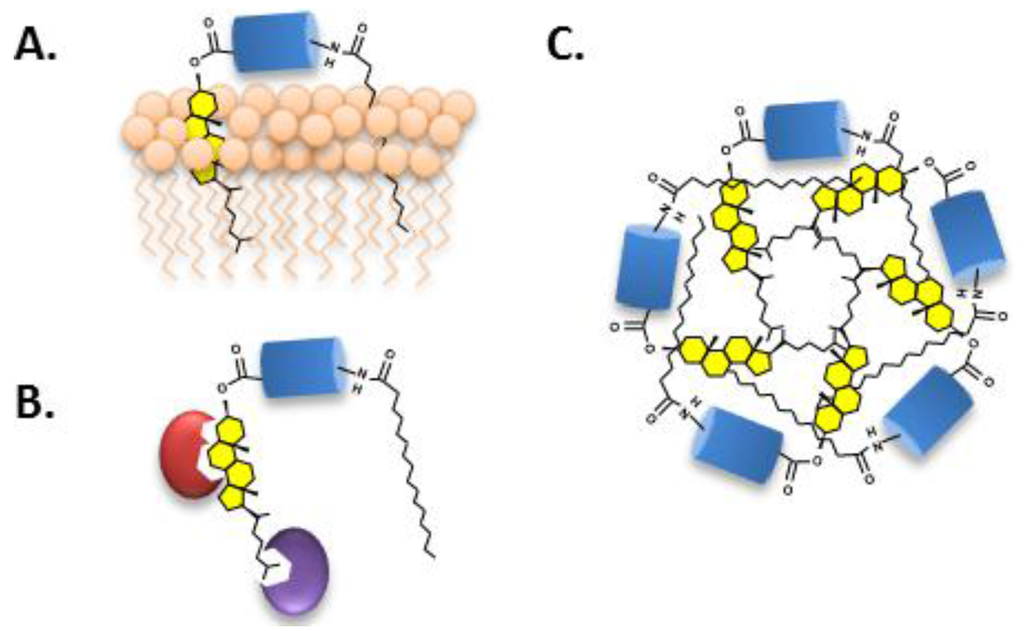
Figure 5.
(A) Cholesterol targets soluble Hh ligand to cell membranes; (B) Proteins associate with Hh ligand via cholesterol; and (C) Micellization of the Hh ligand stabilized by hydrophobic interactions of the attached lipids.
In summary, cholesterol furnishes a membrane anchor, a point of interaction for noncovalent protein association, in addition to promoting ligand self-assembly through a hydrophobic effect. These varied effects of cholesterol on Hh ligand function likely underpin acute developmental anomalies exhibited by humans expressing cholesterolysis-defective Hh proteins [9,90], and transgenic animals expressing engineered, cholesterol-free versions of the ligand [72,91].
3. Targeting Cholesterolysis—A Ligand Deprivation Approach for Hh Ligand Driven Cancers
Cholesterolysis affords an attractive target in the pathway to mitigate oncogenic ligand-driven Hh signaling. Aberrant pathway activity can involve paracrine communication from tumor-to-tumor or from tumor-to-stroma; as well as tumor self-signaling through an autocrine pathway [19,92]. In these scenarios, depleting Hh ligand is expected to restrain and in some cases reverse tumor growth. Prostate cancer provides an instructive example (for reviews, see [12,93,94]). Here, two-way communication develops between transformed epithelial cells, serving as the Hh ligand producers, and benign stromal cells, the Hh responders [95]. Paracrine signaling may explain why xenografts of prostate tumors grow at accelerated rates when overexpressing Sonic Hh [96]. In addition, antibodies against Hh ligand suppress growth of primary prostate tumors and prostate cell lines [97]. The oncogenic signaling can also be diminished to some extent with SMO antagonists in animals bearing prostate tumor xenografts [98,99].
Recent evidence suggests that stromal cells secrete growth factors in response to Hh stimulation [100,101]; a paracrine effect replicated in controlled co-culture experiments [102]. While the reciprocal signal(s) sent from the stroma remains to be identified, several candidates exist [103]. In addition, Hh ligand released from transformed prostate epithelial cells can activate steroidogenesis in neighboring stromal cells, creating a microenvironment hospitable for tumor growth [104,105]. Crosstalk between Hh signaling and steroidogenesis is a running theme [106], as pituitary cancer and ovarian cancer exhibit similar behavior [107,108,109]. In prostate cancer, given the oncogenic activity of androgens [110], linking Hh signaling to steroidogenesis seems especially pertinent to understanding disease progression.
3.1. Target Attributes that Mitigate Risk
There are several attributes of cholesterolysis that garner attention for therapeutic targeting: (1) its decisive role in generating Hh ligand; (2) its position in the pathway upstream of signaling; and (3) the apparent specificity of cholesterolysis to the Hh family. These three aspects of the reaction are considered below.
3.1.1. Decisive Role Played by Cholesterolysis in Hh Signaling
The importance of cholesterolysis for Hh function emerged from genotypic studies that linked a severe congenital malformation of the brain to mutations in human sonic hedgehog exon 3, encoding HhC [9]. Analogous brain malformations had also been observed in patients carrying mutations in exon 1 and 2, encoding HhN [10]. That correspondence indicated that mutations in HhN can be phenocopied by mutations in HhC. Mechanistic data to connect genotype with phenotype followed from cellular studies of cholesterolysis-defective Hh precursor proteins. Guy examined the fate of Hh precursor protein that failed to react with cholesterol due to site-directed mutation or acute sterol depletion [41]. In both instances, Hh precursor remained associated with producing cells, with consequent loss of paracrine signaling [41].
More recent work by Salic and his coworkers underscored and expanded on the imperative of cholesterolysis for Hh function [42]. Among their results, the cellular site of cholesterolysis was localized to the endoplasmic reticulum (ER). Further, a retention mechanism was proposed that prevents HhC as well as cholesterolysis-defective Hh precursor from advancing from the ER to the Golgi. Sequestered in the ER, Hh is poly-ubiqutinated by ERAD, the organelle’s protein surveillance system [111]. HhC and cholesterolysis-defective precursor are shuttled by ERAD to the cytosol for proteasomal degradation. This transformation removes signaling ligand irreversibly. Surveillance by ERAD is sufficiently robust, the authors speculated that HhC “never leaves the ER and is not secreted.” Some evidence does exist to support limited cell-contact mediated signaling by cholesterolysis-defective precursor [112,113]. Nonetheless, the studies outlined above, and others, support the notion that blocking cholesterolysis stops paracrine Hh cell signaling, the pathway most closely linked to cancer.
3.1.2. Upstream Target in the Pathway
Canonical signal transduction begins when secreted Hh ligand is bound by its cell surface receptor, PTCH1. With few exceptions, antagonists target events downstream of this event. In the absence of activating mutations in SMO or GLI, ligand depletion should stifle pathway activity before signaling starts and then amplifies. The monoclonal antibody 5E1, which complexes with Hh ligand in a manner that prevents interaction with PTCH1 [114], provides proof of concept [97,115]. A more upstream target, the Hh palmitoyl transferase HHAT [46], has also been targeted with small molecules by Resh and her colleagues [116]. RU-SKI-43 inhibits autocrine and paracrine signaling, and has shown exciting activity against cancers of the breast and pancreas [117,118].
Cholesterolysis appears to be the most upstream transformation targeted to date. The reaction occurs very early in the secretory pathway [41,42]. Cellular studies with brefeldin A, a fungal compound that inhibits ER to Golgi protein transport, support this notion [42]. At concentrations of brefeldin A sufficient to cause disappearance of the Golgi, Hh cholesterolysis proceeds unabated. Additional evidence for ER localization includes the involvement of ERAD in Hh degradation and association of Hh with PDI, an oxidoreductase residing in the ER [42,119,120]. Thus, cholesterolysis occurs early in the secretory pathway, soon after Hh precursor translation, and well before the first signaling event.
3.1.3. A Transformation Unique to Hedgehog
Since the original reporting of Hh modification by cholesterol, attempts have been made to uncover examples of this lipidation outside the Hh family; none have been identified [121]. Proteomic screens have found numerous proteins that interact with sterols [122,123]; however, all seem to interact with this abundant lipid noncovalently. Sequence searches using human Sonic HhC turn up few hits outside the paralogues, Indian Hh and Desert Hh. Cholesterol binding proteins (enzymes, transporters, toxins, sensors) are absent from the results of our database searches, suggesting that HhC harbors a unique scaffold for sterol interaction. This aspect bodes well for the development of selective Hh inhibitors. Finally, we note that the homology of the three human HhC segments raises the possibility of designing a single cholesterolysis inhibitor active against all three human Hh precursors. A broad-spectrum antagonist could prove crucial to combat functional compensation, which has been seen in transgenic knockout mice [124].
3.2. Assays for Cholesterolysis Amenable to High Throughput Screening
To facilitate screening of chemical libraries for potential Hh cholesterolysis inhibitors, we as well as others have introduced optical activity assays suitable for microplate readers. These assays replace the conventional means of assaying Hh cholesterolysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Because throughput is low and kinetic analyses are discontinuous, SDS-PAGE is not considered a viable platform for chemical screening campaigns. The newer assay systems employ fluorescent labels to monitor cholesterolysis continuously in multi-well plates, improving accuracy and throughput.
The first microplate assay makes use of changes in fluorescent polarization (FP) that accompany cholesterolysis of an engineered Hh precursor protein [125]. The construct consists of HhC from Drosophila melanogaster that is equipped with an N-terminal fluorescent FlAsh-tag (Figure 6A). The FlAsh-tag replaces the Hh ligand sequence (HhN). As mentioned above, HhN is not essential for HhC activity. Cholesterolysis of the engineered precursor liberates the FLAsh tag peptide, resulting in a decrease in fluorescence anisotropy that can be measured by FP. This assay was used to screen 446 compounds in the NIH Clinical Collection, resulting in the identification of two potential inhibitors, Zafirlukast (PubChem CID, 5717) and Honokiol (PubChem CID, 72303). Larger screening efforts have not been reported. Although attractive in many respects, the FP assay does require refolding of the precursor construct, labeling with arsenic-based reagents, and suffers from a small dynamic range.
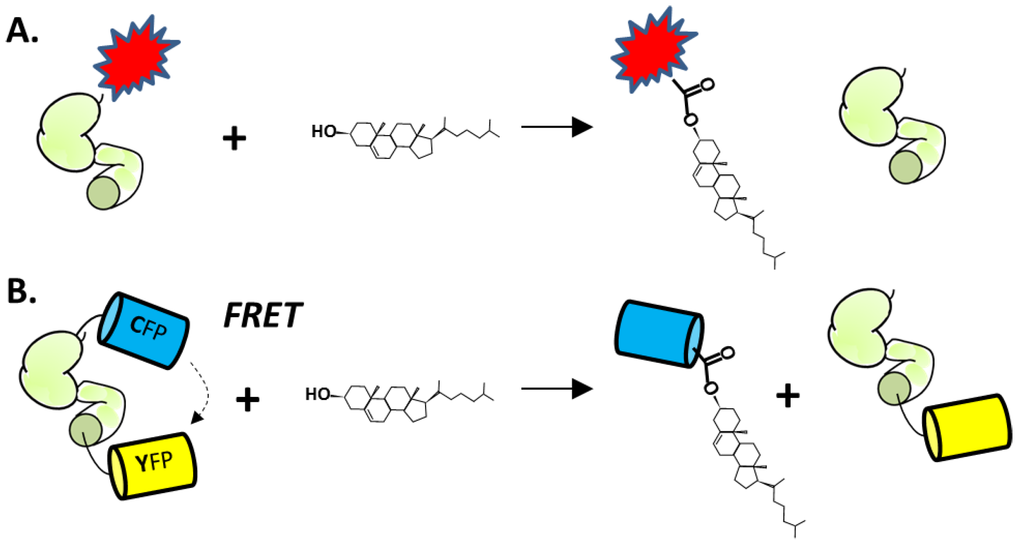
Figure 6.
(A) Cholesterolysis assay using HhC (green) fused to the FLaSH, peptide-dye complex (red). (B) Cholesterolysis assay using HhC fused to cyan and yellow fluorescent proteins.
More recently, Owen et al. described an optical cholesterolysis assay that employs Förster resonance energy transfer (FRET) [126]. An engineered fluorescent Hh precursor is again utilized, except that the labels are soluble recombinant proteins. The key construct, termed C-H-Y, is composed of cyan (C) and yellow (Y) fluorescent proteins (CFP and YFP, respectively) joined to the amino and carboxyl termini of HhC (Figure 6B). Adding cholesterol to buffered solutions of purified C-H-Y, leads to a fourfold attenuation of the FRET ratio, consistent with liberation of CFP from the precursor by substrate cholesterol. Kinetics of C-H-Y cholesterolysis at pH 7.1, the prevailing pH of the endoplasmic reticulum (ER) [127], are remarkably similar to processing measured by pulse-chase experiments in cultured mammalian cells (0.0006 s−1) [41]. The observed rate is also within range of autocatalytic “protein splicing” reactions brought about by inteins [128,129,130,131], as discussed in Section 2.1.
The FRET precursor C-H-Y serves double duty, providing both HTS-compatible activity assay, while also reporting on small-molecule binding to HhC. The latter feature was uncovered in studies with the Hh cholesterolysis inhibitor, phenyl arsine oxide (PhAsIII) (PubChem CID, 4778) [132]. When added to solutions containing C-H-Y, PhAsIII induced quenching of the construct’s FRET, suggesting a change in protein conformation. NMR analysis identified multiple conserved residues at the active site of HhC that are bound by PhAsIII, including two cysteine residues (Cys303 and Cys400). Accordingly, no FRET quenching was observed in control experiments with a C303A C-H-Y mutant, where the nucleophilic cysteine of HhC is replaced by alanine. In a pilot small molecule screen using C-H-Y, we observed similar quenching by a second compound, butyl 3,5-dinitro-4-(1-phenyltetrazol-5-yl)sulfanylbenzoate (PubChem CID 4358899), that inhibits cholesterolysis irreversibly. The binding site of this latter compound was mapped to Cys400 in HhC, a conserved cysteine residue whose mechanistic role in cholesterolysis remains enigmatic. The dual function of the FRET system as activity assay and binding reporter make it an attractive platform for high throughput screening.
3.3. Endogenous Regulators of Hh Cholesterolysis
Along with searches for inhibitors in libraries of synthetic compounds, it seems worthwhile to consider the possibility of endogenous regulators of Hh cholesterolysis. Recently, Xie et al. demonstrated that zinc, the ubiquitous divalent cation, could inhibit Hh autoprocessing in vitro and in cells [133]. Zinc is known to inhibit protein splicing mediated by inteins [134,135,136]. Because inteins and Hh HINT domains are homologous, it seemed plausible that zinc could also inhibit Hh autoprocessing. Indeed, Hh autoprocessing assays indicate inhibition by zinc with a Ki of 2 μM. Zinc also inhibits Hh autoprocessing in a cellular environment with primary rat astrocyte culture. Solution NMR revealed that zinc inhibits autoprocessing by binding to active site residues of the Hh HINT domain.
Zinc deficiency is found in many types of cancer, including prostate, lung and ovarian cancer [137,138]. In these three cancers, Hh ligand is overproduced resulting in abnormal activation of Hh signaling pathway [97,139,140,141,142,143,144]. The data by Xie et al. suggests that there is a mechanistic link between zinc deficiency and Hh-ligand dependent activation of the Hh signaling pathway [133]. Thus, low zinc concentration in tissues can deregulate Hh autoprocessing, boosting Hh ligand production. The zinc/Hh axis (Figure 7) is now being investigated in these cancers.
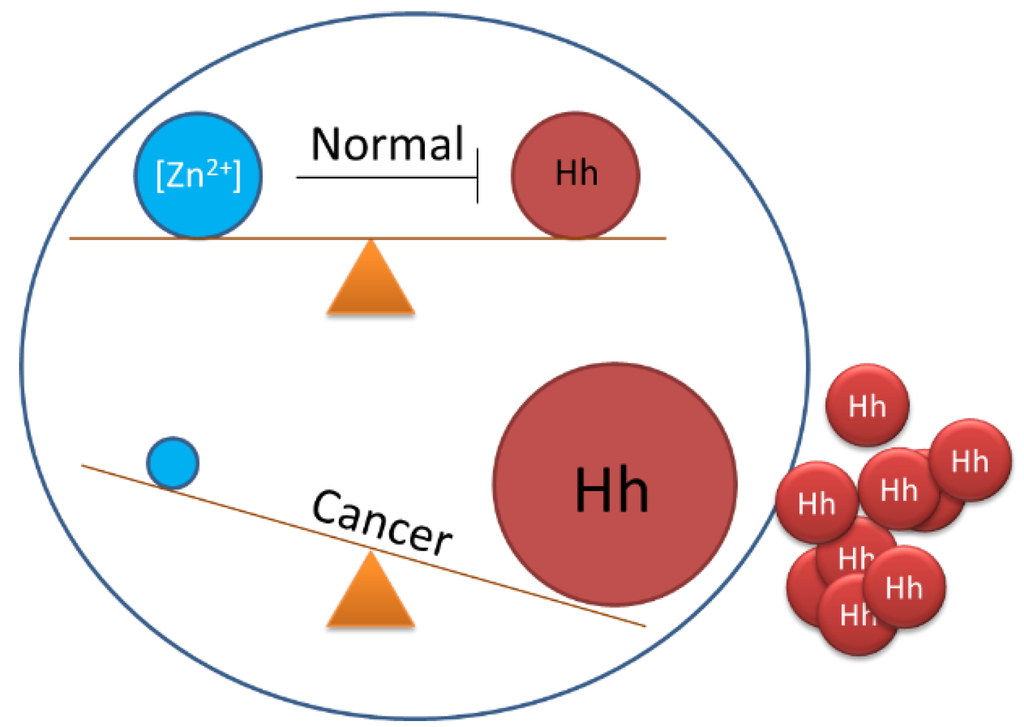
Figure 7.
Proposed balancing of Hedgehog biosynthesis by cellular zinc concentrations and disruption in cancer.
4. Conclusions and Outlook
It seems certain that no single target will suffice for blocking every type of Hh-driven malignancy. Antagonists of the receptor Smoothened (SMO), for example, can generate dramatic remissions, yet efficacy can also be compromised by SMO mutation [145], and against tumors driven by the Hh ligand, responses have been disappointing [146]. Cholesterolysis offers an alternative target for intervention. It supplies a transformation that is early in the pathway, essential, and uniquely hedgehog. Devising selective cholesterolysis inhibitors represents one of several outstanding challenges for this ligand deprivation approach (Table 1). Here again though, cholesterolysis inhibitors are expected to show therapeutic limits, particularly for ligand-independent Hh cancers. The successes and the shortcomings of Hh inhibitors, as a whole, should serve to highlight cancer’s heterogeneity and encourage bi- and tri-target treatment strategies.

Table 1.
Areas for future work.
|
Acknowledgments
We thank Tim Owen for help preparing Figure 2. BPC is supported by start-up funds from Binghamton University and by the Office of the Assistant Secretary of Defense for Health Affairs, through the Prostate Cancer Research Program, grant W81XWH-14-1-0155.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Briscoe, J.; Therond, P.P. The mechanisms of hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Scott, M.P. Hedgehog and patched in neural development and disease. Neuron 1998, 21, 1243–1257. [Google Scholar] [CrossRef]
- Harmon, E.B.; Ko, A.H.; Kim, S.K. Hedgehog signaling in gastrointestinal development and disease. Curr. Mol. Med. 2002, 2, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- VanHook, A.M. Focus issue: Fine-tuning hedgehog signaling in development and disease. Sci. Signal. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Dakubo, G.; Howley, P.; Campsall, K.D.; Mazarolle, C.J.; Shiga, S.A.; Lewis, P.M.; McMahon, A.P.; Wallace, V.A. Development of normal retinal organization depends on sonic hedgehog signaling from ganglion cells. Nat. Neurosci. 2002, 5, 831–832. [Google Scholar] [PubMed]
- Parkin, C.A.; Ingham, P.W. The adventures of sonic hedgehog in development and repair. I. Hedgehog signaling in gastrointestinal development and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G363–G367. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhou, Y.; Beachy, P.A.; Moses, K. The segment polarity gene hedgehog is required for progression of the morphogenetic furrow in the developing drosophila eye. Cell 1993, 75, 927–938. [Google Scholar] [PubMed]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Vargas, F.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the C-terminal domain of sonic hedgehog cause holoprosencephaly. Hum. Mol. Genet. 1997, 6, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the human sonic hedgehog gene cause holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, E.; Hui, C.C. Hedgehog signaling and congenital malformations. Clin. Genet. 2005, 67, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Hedgehog signaling in prostate cancer and its therapeutic implication. Int. J. Mol. Sci. 2013, 14, 13979–14007. [Google Scholar] [CrossRef] [PubMed]
- Sims-Mourtada, J.; Yang, D.; Tworowska, I.; Larson, R.; Smith, D.; Tsao, N.; Opdenaker, L.; Mourtada, F.; Woodward, W. Detection of canonical hedgehog signaling in breast cancer by 131-iodine-labeled derivatives of the sonic hedgehog protein. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Sanchez-Mejias, A.; Wang, Z.; Flaveny, C.; Long, J.; Singh, S.; Rodriguez-Blanco, J.; Tokhunts, R.; Giambelli, C.; Briegel, K.J.; et al. Hedgehog signaling regulates bladder cancer growth and tumorigenicity. Cancer Res. 2012, 72, 4449–4458. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Feuerstein, M.A.; Levina, E.; Baghel, P.S.; Carkner, R.D.; Tanner, M.J.; Shtutman, M.; Vacherot, F.; Terry, S.; de la Taille, A.; et al. Hedgehog/Gli supports androgen signaling in androgen deprived and androgen independent prostate cancer cells. Mol. Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Zunich, S.M.; Douglas, T.; Valdovinos, M.; Chang, T.; Bushman, W.; Walterhouse, D.; Iannaccone, P.; Lamm, M.L. Paracrine sonic hedgehog signalling by prostate cancer cells induces osteoblast differentiation. Mol. Cancer 2009. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Di Magliano, M.P.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, J.W.; de Sauvage, F.J. Paracrine hedgehog signaling in cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef] [PubMed]
- Low, J.A.; de Sauvage, F.J. Clinical experience with hedgehog pathway inhibitors. J. Clin. Oncol. 2010, 28, 5321–5326. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; de Sauvage, F.J. Targeting the hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Guha, M. Hedgehog inhibitor gets landmark skin cancer approval, but questions remain for wider potential. Nat. Rev. Drug Discov. 2012, 11, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.E.; Low, J.A.; Marsters, J.C., Jr.; Robarge, K.; Rubin, L.L.; de Sauvage, F.J.; Sutherlin, D.P.; Wong, H.; Yauch, R.L. Discovery and preclinical development of vismodegib. Expert Opin. Drug Discov. 2014, 9, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [PubMed]
- Gould, A.; Missailidis, S. Targeting the hedgehog pathway: The development of cyclopamine and the development of anti-cancer drugs targeting the hedgehog pathway. Mini Rev. Med. Chem. 2011, 11, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Cyclopamine and hedgehog signaling: Chemistry, biology, medical perspectives. Angew. Chem. Int. Ed. Engl. 2010, 49, 3418–3427. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Struhl, G. Dual roles for patched in sequestering and transducing hedgehog. Cell 1996, 87, 553–563. [Google Scholar] [CrossRef]
- Hahn, H.; Christiansen, J.; Wicking, C.; Zaphiropoulos, P.G.; Chidambaram, A.; Gerrard, B.; Vorechovsky, I.; Bale, A.E.; Toftgard, R.; Dean, M.; et al. A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J. Biol. Chem. 1996, 271, 12125–12128. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Davey, R.A.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that patched is the hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Struhl, G. In vivo evidence that patched and smoothened constitute distinct binding and transducing components of a hedgehog receptor complex. Development 1998, 125, 4943–4948. [Google Scholar] [PubMed]
- Zhu, H.; Carpenter, R.L.; Han, W.; Lo, H.W. The GLI1 splice variant TGLI1 promotes glioblastoma angiogenesis and growth. Cancer Lett. 2014, 343, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Li, H.; Kozul, C.D.; Black, K.E.; Singh, S.; Gosse, J.A.; DiRenzo, J.; Martin, K.A.; Wang, B.; Hamilton, J.W.; et al. Activation of hedgehog signaling by the environmental toxicant arsenic may contribute to the etiology of arsenic-induced tumors. Cancer Res. 2010, 70, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Ekker, S.C.; von Kessler, D.P.; Porter, J.A.; Sun, B.I.; Beachy, P.A. Autoproteolysis in hedgehog protein biogenesis. Science 1994, 266, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Bumcrot, D.A.; Takada, R.; McMahon, A.P. Proteolytic processing yields two secreted forms of sonic hedgehog. Mol. Cell Biol. 1995, 15, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.K. Inhibition of sonic hedgehog autoprocessing in cultured mammalian cells by sterol deprivation. Proc. Natl. Acad. Sci. USA 2000, 97, 7307–7312. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tukachinsky, H.; Huang, C.H.; Jao, C.; Chu, Y.R.; Tang, H.Y.; Mueller, B.; Schulman, S.; Rapoport, T.A.; Salic, A. Processing and turnover of the hedgehog protein in the endoplasmic reticulum. J. Cell Biol. 2011, 192, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Zeng, C.; Wen, D.; Rayhorn, P.; Baker, D.P.; Williams, K.P.; Bixler, S.A.; Ambrose, C.M.; Garber, E.A.; Miatkowski, K.; et al. Identification of a palmitic acid-modified form of human sonic Hedgehog. J. Biol. Chem. 1998, 273, 14037–14045. [Google Scholar] [CrossRef] [PubMed]
- Buglino, J.A.; Resh, M.D. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J. Biol. Chem. 2008, 283, 22076–22088. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, Z.; Mann, R.K.; Nellen, D.; von Kessler, D.P.; Bellotto, M.; Beachy, P.A.; Basler, K. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science 2001, 293, 2080–2084. [Google Scholar] [CrossRef] [PubMed]
- Buglino, J.A.; Resh, M.D. Palmitoylation of hedgehog proteins. Vitam. Horm. 2012, 88, 229–252. [Google Scholar] [PubMed]
- Perler, F.B. Protein splicing of inteins and hedgehog autoproteolysis: Structure, function, and evolution. Cell 1998, 92, 1–4. [Google Scholar] [CrossRef]
- Paulus, H. Protein splicing and related forms of protein autoprocessing. Annu. Rev. Biochem. 2000, 69, 447–496. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Kuzmickas, R.P.; Jao, C.Y.; Liu, J.; Salic, A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2012, 2, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Von Kessler, D.P.; Ekker, S.C.; Young, K.E.; Lee, J.J.; Moses, K.; Beachy, P.A. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature 1995, 374, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.K.; Beachy, P.A. Cholesterol modification of proteins. Biochim. Biophys. Acta 2000, 1529, 188–202. [Google Scholar] [CrossRef]
- Ciepla, P.; Konitsiotis, A.D.; Serwa, R.A.; Masumoto, N.; Leong, W.P.; Dallman, M.J.; Magee, A.I.; Tate, E.W. New chemical probes targeting cholesterylation of sonic hedgehog in human cells and zebrafish. Chem. Sci. 2014, 5, 4249–4259. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.M.; Porter, J.A.; Young, K.E.; Koonin, E.V.; Beachy, P.A.; Leahy, D.J. Crystal structure of a hedgehog autoprocessing domain: Homology between hedgehog and self-splicing proteins. Cell 1997, 91, 85–97. [Google Scholar] [CrossRef]
- Vincent, S.; Thomas, A.; Brasher, B.; Benson, J.D. Targeting of proteins to membranes through hedgehog auto-processing. Nat. Biotechnol. 2003, 21, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Ekker, S.C.; Park, W.J.; von Kessler, D.P.; Young, K.E.; Chen, C.H.; Ma, Y.; Woods, A.S.; Cotter, R.J.; Koonin, E.V.; et al. Hedgehog patterning activity: Role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell 1996, 86, 21–34. [Google Scholar] [CrossRef]
- Lewis, C.A., Jr.; Wolfenden, R. Amide bonds to the nitrogen atoms of cysteine and serine as "weak points" in the backbones of proteins. Biochemistry 2011, 50, 7259–7264. [Google Scholar] [CrossRef] [PubMed]
- Johansson, D.G.; Wallin, G.; Sandberg, A.; Macao, B.; Aqvist, J.; Hard, T. Protein autoproteolysis: Conformational strain linked to the rate of peptide cleavage by the pH dependence of the N –> O acyl shift reaction. J. Am. Chem. Soc. 2009, 131, 9475–9477. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, A.; Shekhtman, A.; Cowburn, D.; Muir, T.W. Semisynthesis of a segmental isotopically labeled protein splicing precursor: NMR evidence for an unusual peptide bond at the N-extein-intein junction. Proc. Natl. Acad. Sci. USA 2004, 101, 6397–6402. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.P.; Topilina, N.I.; Stanger, M.J.; Van Roey, P.; Belfort, M. Structure of catalytically competent intein caught in a redox trap with functional and evolutionary implications. Nat. Struct. Mol. Biol. 2011, 18, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Perler, F.B.; Xu, M.-Q.; Paulus, H. Protein splicing and autoproteolysis mechanisms. Curr. Opin. Chem. Biol. 1997, 1, 292–299. [Google Scholar] [CrossRef]
- Dearden, A.K.; Callahan, B.; Roey, P.V.; Li, Z.; Kumar, U.; Belfort, M.; Nayak, S.K. A conserved threonine spring-loads precursor for intein splicing. Protein Sci. 2013, 22, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Liu, J.; Albracht, C.D.; Hsu, A.; Chen, W.; Marieni, M.D.; Colelli, K.M.; Williams, J.E.; Reitter, J.N.; Mills, K.V.; et al. Structural and mutational studies of a hyperthermophilic intein from DNA polymerase ii of pyrococcus abyssi. J. Biol. Chem. 2011, 286, 38638–38648. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Liu, Y.; Ban, D.; Lopez, M.M.; Belfort, M.; Wang, C. Backbone dynamics and global effects of an activating mutation in minimized Mtu RecA inteins. J. Mol. Biol. 2010, 400, 755–767. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Binschik, J.; Mootz, H.D. Chemical bypass of intein-catalyzed N-S acyl shift in protein splicing. Angew. Chem. Int. Ed. Engl. 2013, 52, 4260–4264. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, P.; Pereira, B.; Li, Z.; Hiraga, K.; Belfort, M.; Derbyshire, V. Crystallographic and mutational studies of mycobacterium tuberculosis recA mini-inteins suggest a pivotal role for a highly conserved aspartate residue. J. Mol. Biol. 2007, 367, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Nicastri, M.C.; Xega, K.; Li, L.; Xie, J.; Wang, C.; Linhardt, R.J.; Reitter, J.N.; Mills, K.V. Internal disulfide bond acts as a switch for intein activity. Biochemistry 2013, 52, 5920–5927. [Google Scholar] [CrossRef] [PubMed]
- Topilina, N.I.; Green, C.M.; Jayachandran, P.; Kelley, D.S.; Stanger, M.J.; Piazza, C.L.; Nayak, S.; Belfort, M. SufB intein of Mycobacterium tuberculosis as a sensor for oxidative and nitrosative stresses. Proc. Natl. Acad. Sci. USA 2015, 112, 10348–10353. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A. Cholesterol and its derivatives in sonic hedgehog signaling and cancer. Curr. Opin. Pharmacol. 2012, 12, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; McMahon, A.P. Cholesterol modification of hedgehog family proteins. J. Clin. Investig. 2002, 110, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Gallet, A. Hedgehog morphogen: From secretion to reception. Trends Cell Biol. 2011, 21, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Gallet, A.; Ruel, L.; Staccini-Lavenant, L.; Therond, P.P. Cholesterol modification is necessary for controlled planar long-range activity of hedgehog in Drosophila epithelia. Development 2006, 133, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, I.; Chiang, C. A conserved mechanism of hedgehog gradient formation by lipid modifications. Trends Cell Biol. 2007, 17, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S. Multiple roles for lipids in the hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 2008, 9, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Grover, V.K.; Valadez, J.G.; Bowman, A.B.; Cooper, M.K. Lipid modifications of sonic hedgehog ligand dictate cellular reception and signal response. PLoS ONE 2011, 6, e21353. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Wolf, A.; Wagner, M.; Kuhlmann, J.; Waldmann, H. The cholesterol membrane anchor of the hedgehog protein confers stable membrane association to lipid-modified proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 8531–8536. [Google Scholar] [CrossRef] [PubMed]
- Teruya, K.; Nishizawa, K.; Doh-ura, K. Semisynthesis of a protein with cholesterol at the C-terminal, targeted to the cell membrane of live cells. Protein J. 2010, 29, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Schneider, A.; Schlechtingen, G.; Weidlich, S.; Ries, J.; Braxmeier, T.; Schwille, P.; Schulz, J.B.; Schroeder, C.; Simons, M.; et al. Efficient inhibition of the Alzheimer's disease beta-secretase by membrane targeting. Science 2008, 320, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Miller, G.M.; Grotenbreg, G.M.; Ploegh, H.L. Lipid modification of proteins through sortase-catalyzed transpeptidation. J. Am. Chem. Soc. 2008, 130, 16338–16343. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.P.; Taylor, F.R.; Pepinsky, R.B. Purifying the hedgehog protein and its variants. Methods Mol. Biol. 2007, 397, 1–22. [Google Scholar] [PubMed]
- Taylor, F.R.; Wen, D.; Garber, E.A.; Carmillo, A.N.; Baker, D.P.; Arduini, R.M.; Williams, K.P.; Weinreb, P.H.; Rayhorn, P.; Hronowski, X.; et al. Enhanced potency of human sonic hedgehog by hydrophobic modification. Biochemistry 2001, 40, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Creanga, A.; Glenn, T.D.; Mann, R.K.; Saunders, A.M.; Talbot, W.S.; Beachy, P.A. Scube/you activity mediates release of dually lipid-modified hedgehog signal in soluble form. Genes Dev. 2012, 26, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Diehl, A.M.; Li, Y.X. Sonic hedgehog ligand partners with caveolin-1 for intracellular transport. Lab. Investig. 2009, 89, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Panakova, D.; Sprong, H.; Marois, E.; Thiele, C.; Eaton, S. Lipoprotein particles are required for hedgehog and wingless signalling. Nature 2005, 435, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Goetz, J.A.; Suber, L.M.; Scott, W.J., Jr.; Schreiner, C.M.; Robbins, D.J. A freely diffusible form of sonic hedgehog mediates long-range signalling. Nature 2001, 411, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Farzan, S.F.; Singh, S.; Schilling, N.S.; Robbins, D.J. The adventures of sonic hedgehog in development and repair. III. Hedgehog processing and biological activity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G844–G849. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.; Goswami, D.; Manonmani, A.; Sharma, P.; Ranganath, H.A.; VijayRaghavan, K.; Shashidhara, L.S.; Sowdhamini, R.; Mayor, S. Nanoscale organization of hedgehog is essential for long-range signaling. Cell 2008, 133, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; White, B.; Tyurina, O.V.; Guner, B.; Larson, T.; Lee, H.Y.; Karlstrom, R.O.; Kohtz, J.D. Synergistic and antagonistic roles of the Sonic hedgehog N- and C-terminal lipids. Development 2004, 131, 4357–4370. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Swierczynska, M.M.; Kumari, V.; Ehrhart-Bornstein, M.; Bornstein, S.R.; Eaton, S. Secretion and signaling activities of lipoprotein-associated hedgehog and non-sterol-modified hedgehog in flies and mammals. PLoS Biol. 2013, 11, e1001505. [Google Scholar] [CrossRef] [PubMed]
- Traiffort, E.; Dubourg, C.; Faure, H.; Rognan, D.; Odent, S.; Durou, M.R.; David, V.; Ruat, M. Functional characterization of sonic hedgehog mutations associated with holoprosencephaly. J. Biol. Chem. 2004, 279, 42889–42897. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.M.; Dunn, M.P.; McMahon, J.A.; Logan, M.; Martin, J.F.; St-Jacques, B.; McMahon, A.P. Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell 2001, 105, 599–612. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Ruiz i Altaba, A. Interference with HH-GLI signaling inhibits prostate cancer. Trends Mol. Med. 2005, 11, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Carkner, R.; Buttyan, R. The hedgehog/Gli signaling paradigm in prostate cancer. Expert Rev. Endocrinol. Metab 2011, 6, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.C.; Joyner, A.L. Hedgehog signaling in prostate epithelial-mesenchymal growth regulation. Dev. Biol. 2015, 400, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Hernandez, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONICHEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed]
- Ibuki, N.; Ghaffari, M.; Pandey, M.; Iu, I.; Fazli, L.; Kashiwagi, M.; Tojo, H.; Nakanishi, O.; Gleave, M.E.; Cox, M.E. Tak-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int. J. Cancer 2013, 133, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- McKee, C.M.; Xu, D.; Cao, Y.; Kabraji, S.; Allen, D.; Kersemans, V.; Beech, J.; Smart, S.; Hamdy, F.; Ishkanian, A.; et al. Protease nexin 1 inhibits hedgehog signaling in prostate adenocarcinoma. J. Clin. Investig. 2012, 122, 4025–4036. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Gipp, J.; Bushman, W. The sonic hedgehog pathway stimulates prostate tumor growth by paracrine signaling and recapitulates embryonic gene expression in tumor myofibroblasts. Oncogene 2009, 28, 4480–4490. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, notch, and hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Domenech, M.; Bjerregaard, R.; Bushman, W.; Beebe, D.J. Hedgehog signaling in myofibroblasts directly promotes prostate tumor cell growth. Integr. Biol. 2012, 4, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, T.; Eastham-Anderson, J.; Dunlap, D.; Alicke, B.; Nannini, M.; Gould, S.; Yauch, R.; Modrusan, Z.; DuPree, K.J.; et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9589–9594. [Google Scholar] [CrossRef] [PubMed]
- Levina, E.; Chen, M.; Carkner, R.; Shtutman, M.; Buttyan, R. Paracrine hedgehog increases the steroidogenic potential of prostate stromal cells in a Gli-dependent manner. Prostate 2012, 72, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tanner, M.; Levine, A.C.; Levina, E.; Ohouo, P.; Buttyan, R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle 2009, 8, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Finco, I.; LaPensee, C.R.; Krill, K.T.; Hammer, G.D. Hedgehog signaling and steroidogenesis. Annu. Rev. Physiol. 2015, 77, 105–129. [Google Scholar] [CrossRef] [PubMed]
- Spicer, L.J.; Sudo, S.; Aad, P.Y.; Wang, L.S.; Chun, S.Y.; Ben-Shlomo, I.; Klein, C.; Hsueh, A.J. The hedgehog-patched signaling pathway and function in the mammalian ovary: A novel role for hedgehog proteins in stimulating proliferation and steroidogenesis of theca cells. Reproduction 2009, 138, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Vila, G.; Theodoropoulou, M.; Stalla, J.; Tonn, J.C.; Losa, M.; Renner, U.; Stalla, G.K.; Paez-Pereda, M. Expression and function of Sonic Hedgehog pathway components in pituitary adenomas: Evidence for a direct role in hormone secretion and cell proliferation. J. Clin. Endocrinol. MeTable 2005, 90, 6687–6694. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Pan, Y.; Luo, H.; Xiong, W.; Zhu, H.; Ruan, H.; Wang, J.; Zou, C.; Tang, L.; Iguchi, T.; et al. Hedgehog signaling stimulates the conversion of cholesterol to steroids. Cell Signal 2015, 27, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O. Combination therapy: Abiraterone prolongs survival in metastatic prostate cancer. Nat. Rev. Clin. Oncol. 2011, 8, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. Er-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Tokhunts, R.; Singh, S.; Chu, T.; D'Angelo, G.; Baubet, V.; Goetz, J.A.; Huang, Z.; Yuan, Z.; Ascano, M.; Zavros, Y.; et al. The full-length unprocessed hedgehog protein is an active signaling molecule. J. Biol. Chem. 2010, 285, 2562–2568. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, C.A.; Asp, E.; Emerson, C.P., Jr. A new role for hedgehogs in juxtacrine signaling. Mech. Dev. 2014, 131, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Maun, H.R.; Wen, X.; Lingel, A.; de Sauvage, F.J.; Lazarus, R.A.; Scales, S.J.; Hymowitz, S.G. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J. Biol. Chem. 2010, 285, 26570–26580. [Google Scholar] [CrossRef] [PubMed]
- O'Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [PubMed]
- Petrova, E.; Rios-Esteves, J.; Ouerfelli, O.; Glickman, J.F.; Resh, M.D. Inhibitors of hedgehog acyltransferase block sonic hedgehog signaling. Nat. Chem. Biol. 2013, 9, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Petrova, E.; Matevossian, A.; Resh, M.D. Hedgehog acyltransferase as a target in pancreatic ductal adenocarcinoma. Oncogene 2015, 34, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Matevossian, A.; Resh, M.D. Hedgehog acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Mol. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Chu, Y.R.; Ye, Y.; Chen, X. Role of herp and a herp-related protein in HRD1-dependent protein degradation at the endoplasmic reticulum. J. Biol. Chem. 2014, 289, 4444–4454. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Hsiao, H.T.; Chu, Y.R.; Ye, Y.; Chen, X. Derlin2 protein facilitates HRD1-mediated retro-translocation of sonic hedgehog at the endoplasmic reticulum. J. Biol. Chem. 2013, 288, 25330–25339. [Google Scholar] [CrossRef] [PubMed]
- Heal, W.P.; Jovanovic, B.; Bessin, S.; Wright, M.H.; Magee, A.I.; Tate, E.W. Bioorthogonal chemical tagging of protein cholesterylation in living cells. Chem. Commun. 2011, 47, 4081–4083. [Google Scholar] [CrossRef] [PubMed]
- Hulce, J.J.; Cognetta, A.B.; Niphakis, M.J.; Tully, S.E.; Cravatt, B.F. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 2013, 10, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gianoulis, T.A.; Yip, K.Y.; Gerstein, M.; Snyder, M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell 2010, 143, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Doles, J.; Cook, C.; Shi, X.; Valosky, J.; Lipinski, R.; Bushman, W. Functional compensation in hedgehog signaling during mouse prostate development. Dev. Biol. 2006, 295, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Q.; Paulus, H. A high-throughput, homogeneous, fluorescence polarization assay for inhibitors of hedgehog protein autoprocessing. J. Biomol. Screen. 2010, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Owen, T.S.; Ngoje, G.; Lageman, T.J.; Bordeau, B.M.; Belfort, M.; Callahan, B.P. Forster resonance energy transfer-based cholesterolysis assay identifies a novel hedgehog inhibitor. Anal. Biochem. 2015, 488, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Johannes, L.; Goud, B.; Antony, C.; Lingwood, C.A.; Daneman, R.; Grinstein, S. Noninvasive measurement of the pH of the endoplasmic reticulum at rest and during calcium release. Proc. Natl. Acad. Sci. USA 1998, 95, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.H.; Eryilmaz, E.; Cowburn, D.; Muir, T.W. Extein residues play an intimate role in the rate-limiting step of protein trans-splicing. J. Am. Chem. Soc. 2013, 135, 5839–5847. [Google Scholar] [CrossRef] [PubMed]
- Amitai, G.; Callahan, B.P.; Stanger, M.J.; Belfort, G.; Belfort, M. Modulation of intein activity by its neighboring extein substrates. Proc. Natl. Acad. Sci. USA 2009, 106, 11005–11010. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Shemella, P.T.; Amitai, G.; Belfort, G.; Nayak, S.K.; Belfort, M. Spontaneous proton transfer to a conserved intein residue determines on-pathway protein splicing. J. Mol. Biol. 2011, 406, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.W.; Camarero, J.A. Intein applications: From protein purification and labeling to metabolic control methods. J. Biol. Chem. 2014, 289, 14512–14519. [Google Scholar] [CrossRef] [PubMed]
- Owen, T.S.; Xie, X.J.; Laraway, B.; Ngoje, G.; Wang, C.; Callahan, B.P. Active site targeting of hedgehog precursor protein with phenylarsine oxide. Chembiochem 2015, 16, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Owen, T.; Xia, K.; Singh, A.V.; Tou, E.; Li, L.; Arduini, B.; Li, H.; Wan, L.Q.; Callahan, B.; et al. Zinc inhibits hedgehog autoprocessing: Linking zinc deficiency with hedgehog activation. J. Biol. Chem. 2015, 290, 11591–11600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zheng, Y.; Xi, Z.; Luo, Z.; Xu, X.; Wang, C.; Liu, Y. Metal ions binding to reca inteins from mycobacterium tuberculosis. Mol. Biosyst. 2009, 5, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Ye, S.; Ferrandon, S.; Evans, T.C.; Xu, M.Q.; Rao, Z. Crystal structures of an intein from the split dnae gene of synechocystis sp. Pcc6803 reveal the catalytic model without the penultimate histidine and the mechanism of zinc ion inhibition of protein splicing. J. Mol. Biol. 2005, 353, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Poland, B.W.; Xu, M.Q.; Quiocho, F.A. Structural insights into the protein splicing mechanism of PI-Scei. J. Biol. Chem. 2000, 275, 16408–16413. [Google Scholar] [CrossRef] [PubMed]
- Gumulec, J.; Masarik, M.; Adam, V.; Eckschlager, T.; Provaznik, I.; Kizek, R. Serum and tissue zinc in epithelial malignancies: A meta-analysis. PLoS ONE 2014, 9, e99790. [Google Scholar] [CrossRef] [PubMed]
- Memon, A.U.; Kazi, T.G.; Afridi, H.I.; Jamali, M.K.; Arain, M.B.; Jalbani, N.; Syed, N. Evaluation of zinc status in whole blood and scalp hair of female cancer patients. Clin. Chim. Acta 2007, 379, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Li, C.; Zhang, X.; Chi, S.; He, N.; Chen, K.; McCormick, F.; Gatalica, Z.; Xie, J. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 2004. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Baylin, S.B. Hedgehog signaling: Progenitor phenotype in small-cell lung cancer. Cell Cycle 2003, 2, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Velcheti, V.; Govindan, R. Hedgehog signaling pathway and lung cancer. J. Thorac. Oncol. 2007, 2, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Horiuchi, A.; Kikuchi, N.; Osada, R.; Yoshida, J.; Shiozawa, T.; Konishi, I. Hedgehog signal pathway is activated in ovarian carcinomas, correlating with cell proliferation: It's inhibition leads to growth suppression and apoptosis. Cancer Sci. 2007, 98, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).