
The Role of nAChR and Calcium Signaling in Pancreatic Cancer Initiation and Progression

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nicotine and Pancreatic Cancer Development
2.1. Nicotine Mediated Alterations in Gene Expression Profiles
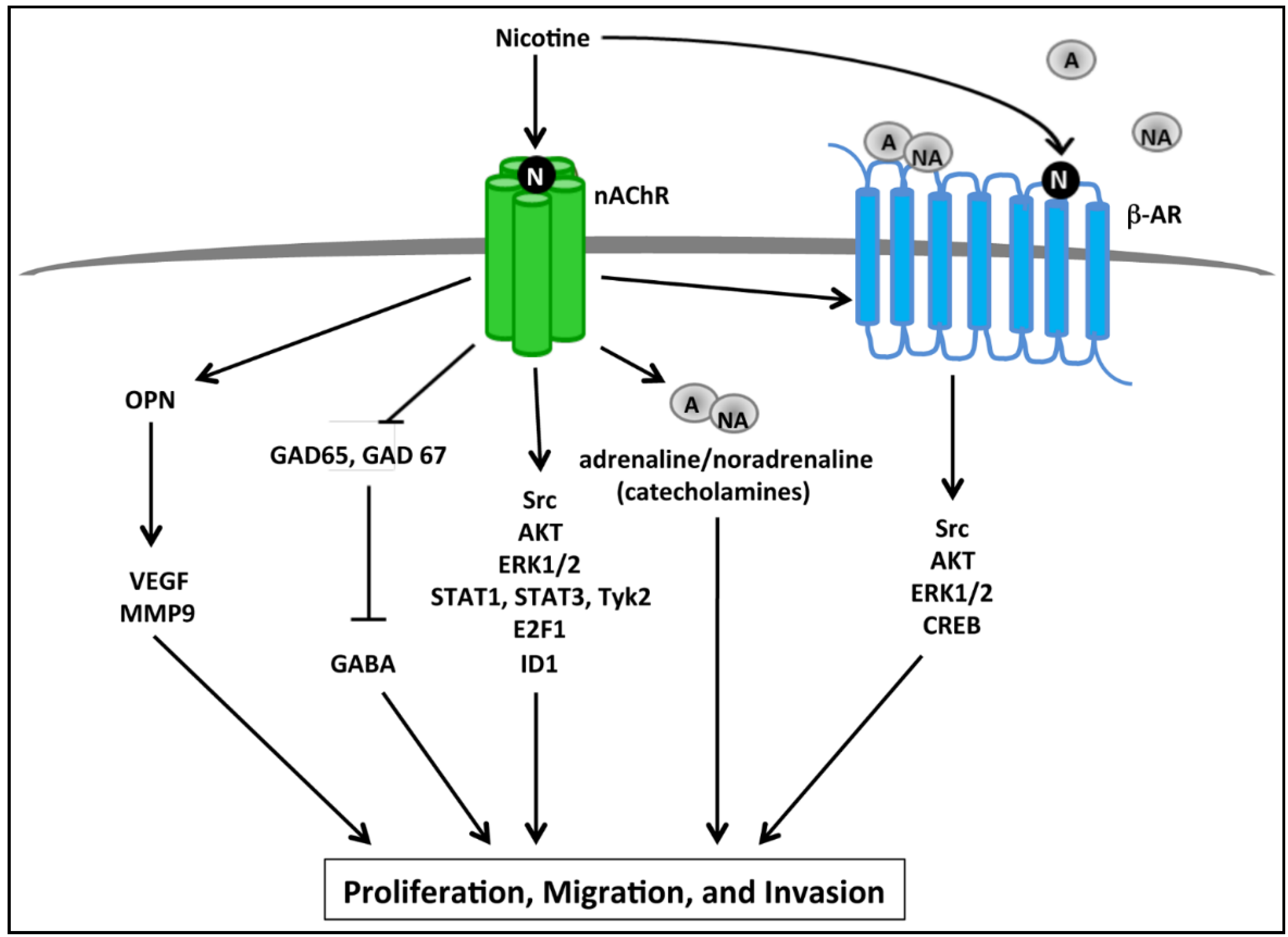
2.2. nAChR Mediated Proliferative and Invasive Signaling Events
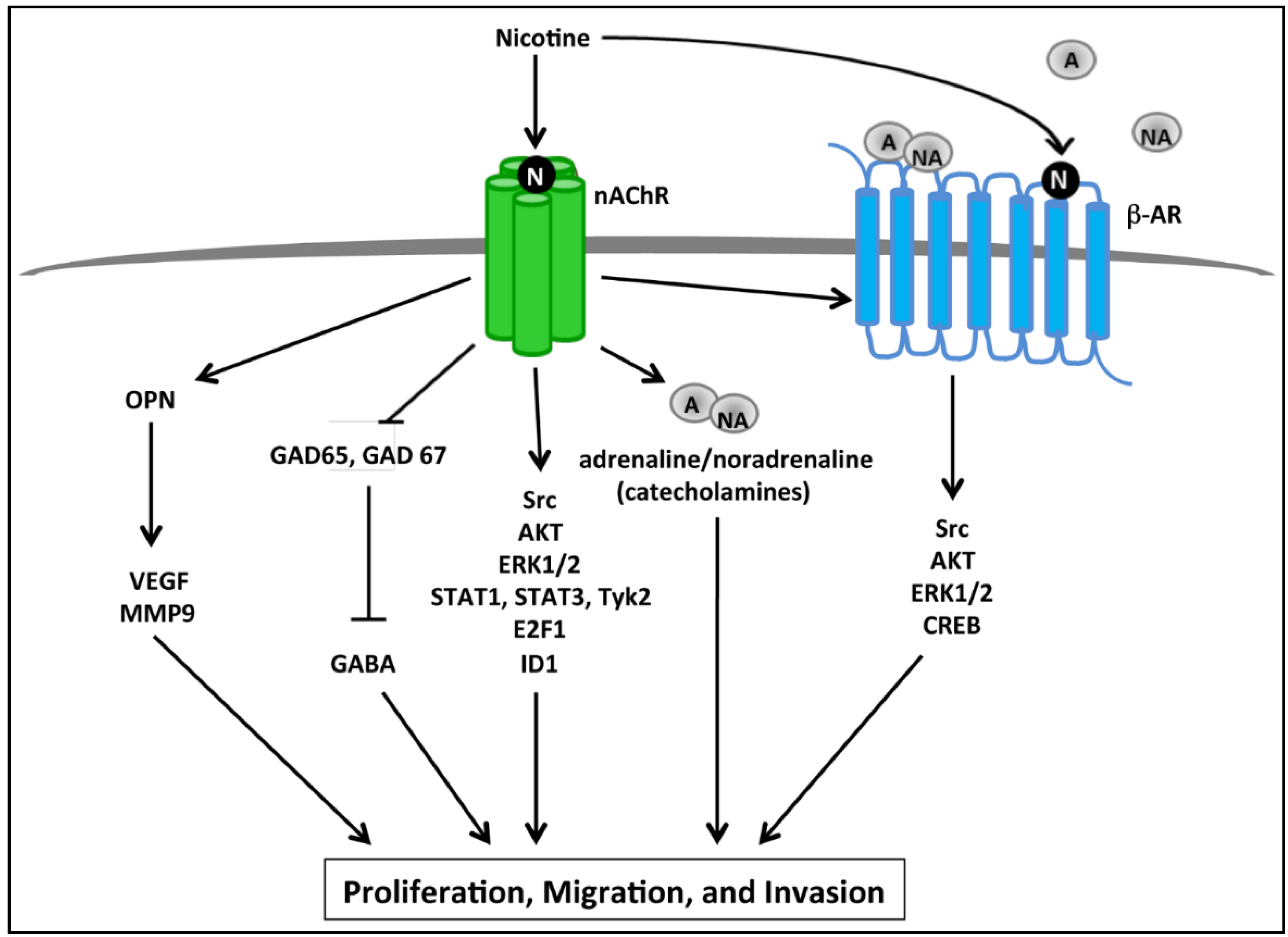
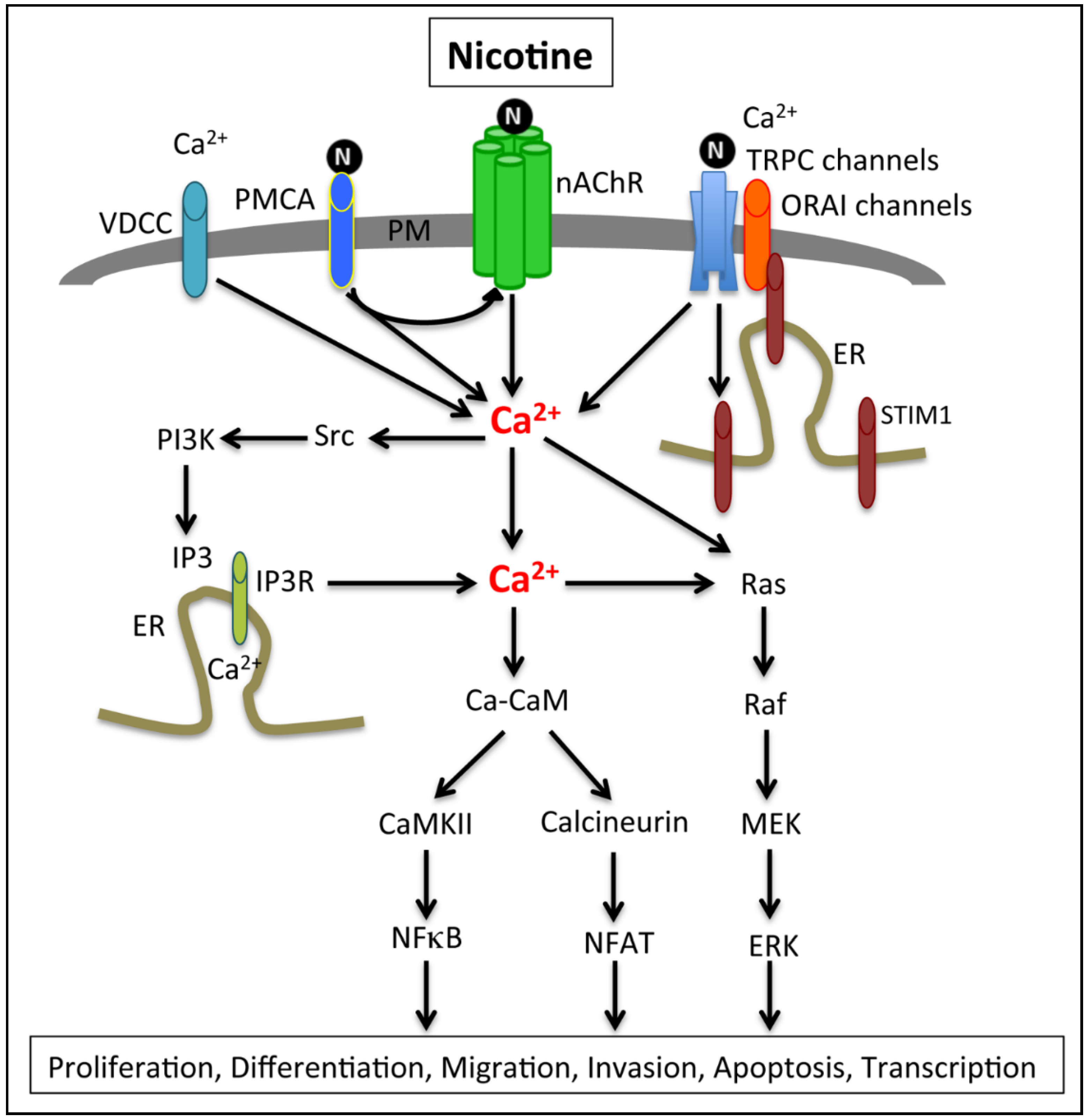
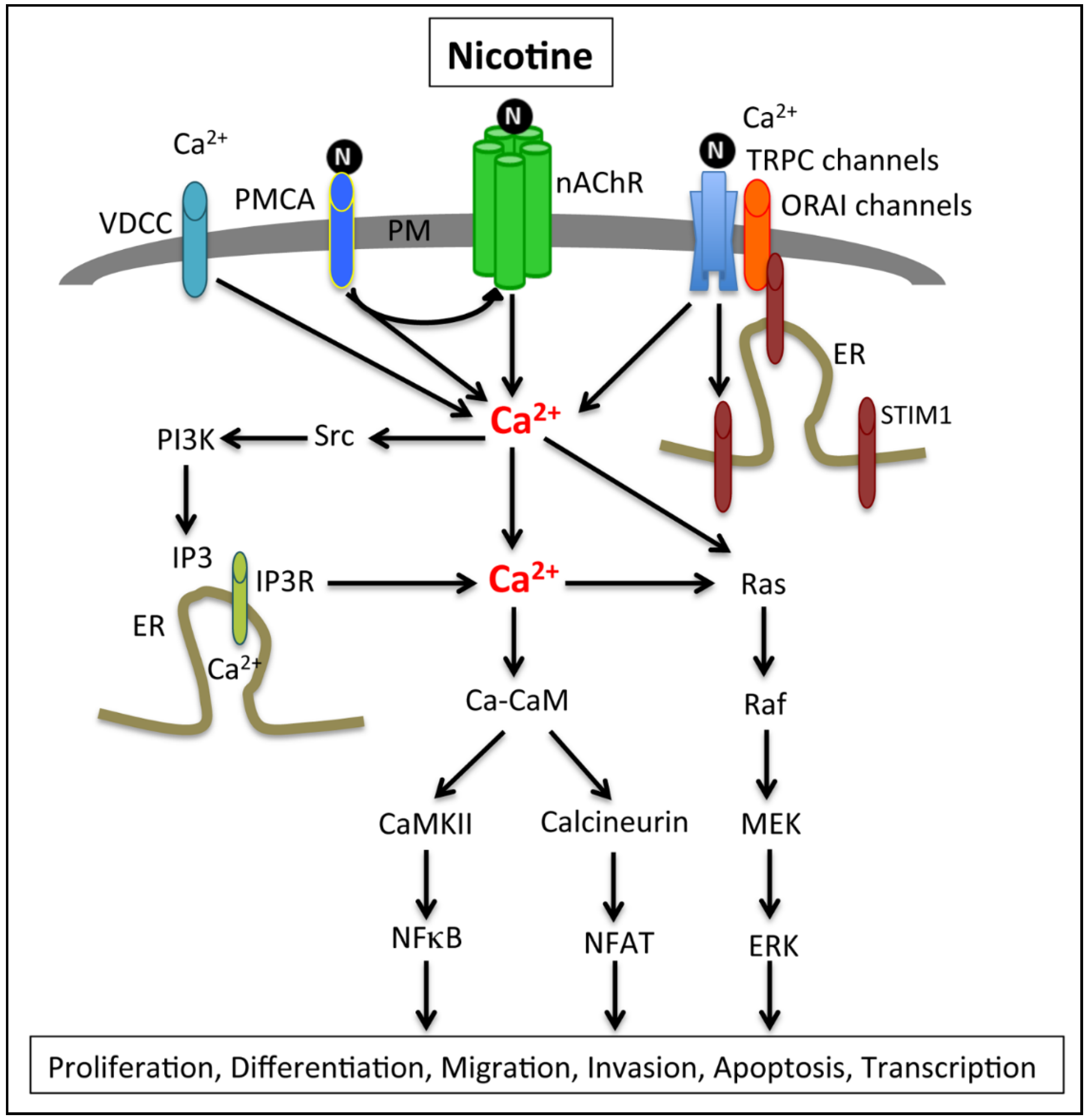
2.3. Calcium Signaling, Smoking and Pancreatic Cancer
2.4. Calcium Channels and Pumps Associated with PDAC Cell Proliferation and EMT
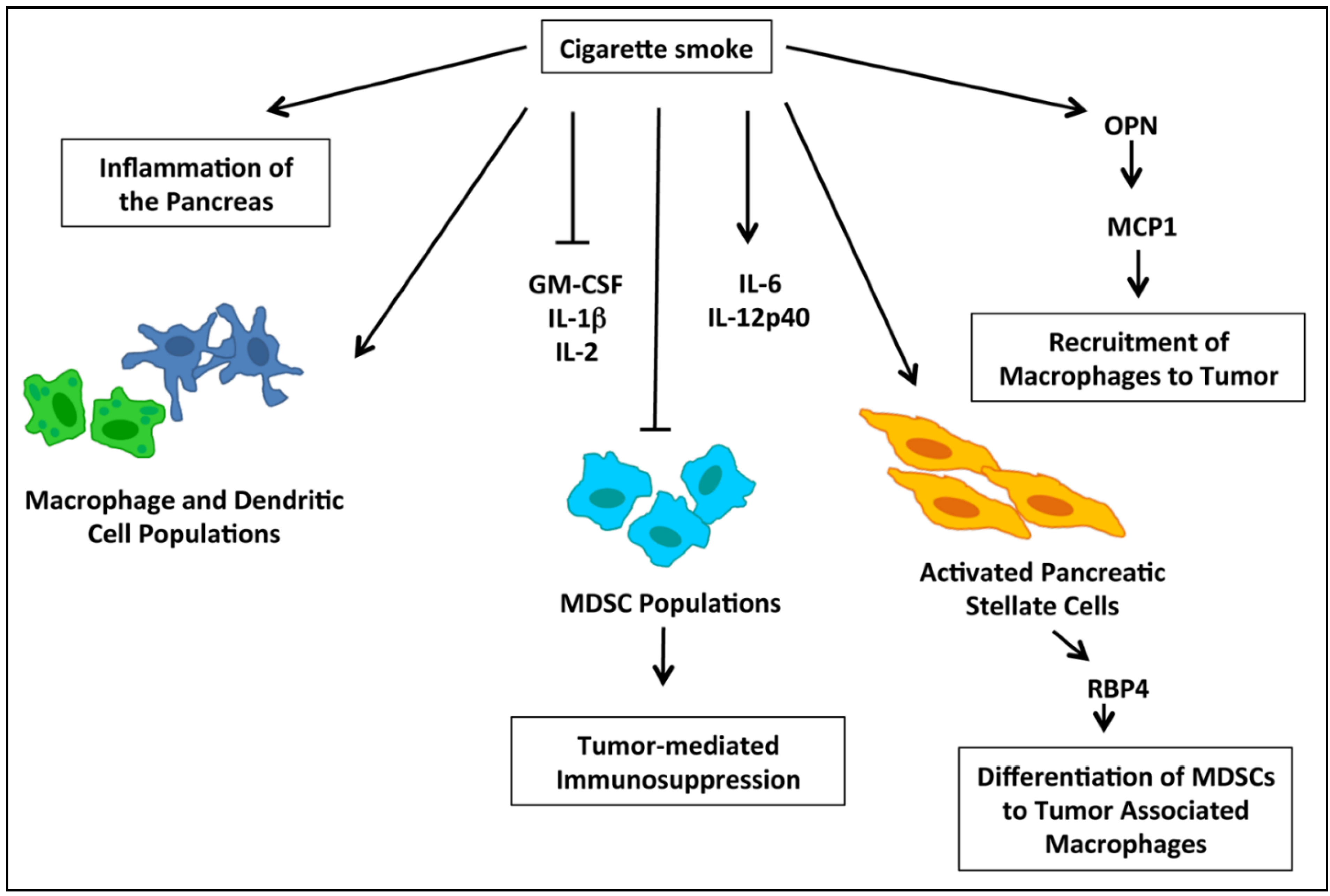
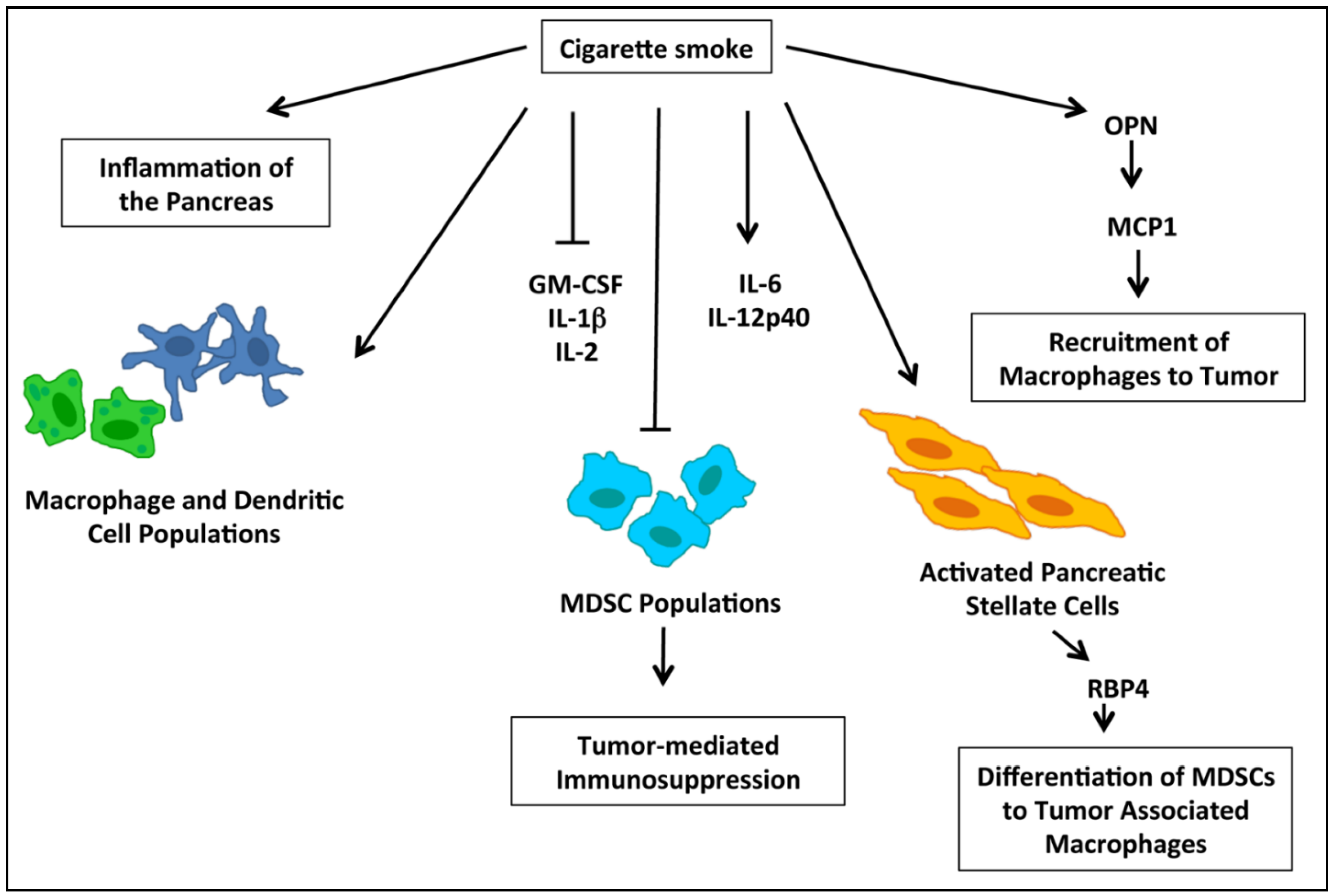
2.5. nAChRs and Immune Modulation of PDACs
2.6. Role of nAChR and Calcium Signaling in Therapeutic Resistance of PDACs
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Crawford, H.C.; Scoggins, C.R.; Washington, M.K.; Matrisian, L.M.; Leach, S.D. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J. Clin. Investig. 2002, 109, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, D.; Saied, A.; Kocher, H.M. Analysis of mortality rates for pancreatic cancer across the world. HPB 2008, 10, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.R.; Jain, M.; Burch, J.D.; Miller, A.B. Cigarette smoking and cancer of the pancreas: Evidence from a population-based case-control study in toronto, canada. Int. J. Cancer 1991, 47, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Mulder, I.; van Genugten, M.L.; Hoogenveen, R.T.; de Hollander, A.E.; Bueno-de-Mesquita, H.B. The impact of smoking on future pancreatic cancer: A computer simulation. Ann. Oncol. 1999, 10, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Doll, R.; Peto, R.; Wheatley, K.; Gray, R.; Sutherland, I. Mortality in relation to smoking: 40 years’ observations on male british doctors. BMJ 1994, 309, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Nakao, M.; Hosono, S.; Ito, H.; Oze, I.; Watanabe, M.; Mizuno, N.; Yatabe, Y.; Yamao, K.; Niimi, A.; Tajima, K.; et al. Cigarette smoking and pancreatic cancer risk: A revisit with an assessment of the nicotine dependence phenotype. Asian Pac. J. Cancer Prev. 2013, 14, 4409–4413. [Google Scholar] [CrossRef] [PubMed]
- Iodice, S.; Gandini, S.; Maisonneuve, P.; Lowenfels, A.B. Tobacco and the risk of pancreatic cancer: A review and meta-analysis. Langenbeck’s Arch. Surg. 2008, 393, 535–545. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, S.L.; Chowdhury, P. The genetics of nicotine dependence: Relationship to pancreatic cancer. World J. Gastroenterol. 2006, 12, 7433–7439. [Google Scholar] [PubMed]
- Phillips, D.H.; Venitt, S. DNA and protein adducts in human tissues resulting from exposure to tobacco smoke. Int. J. Cancer 2012, 131, 2733–2753. [Google Scholar] [CrossRef] [PubMed]
- Talhout, R.; Schulz, T.; Florek, E.; van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galban, S.; Galban, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Galitovskiy, V.; Chernyavsky, A.I.; Edwards, R.A.; Grando, S.A. Muscle sarcomas and alopecia in a/j mice chronically treated with nicotine. Life Sci. 2012, 91, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Connections of nicotine to cancer. Nat. Rev. Cancer 2014, 14, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Chellappan, S.P. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 2014, 12, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Momi, N.; Ponnusamy, M.P.; Kaur, S.; Rachagani, S.; Kunigal, S.S.; Chellappan, S.; Ouellette, M.M.; Batra, S.K. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through alpha7nAChR-mediated MUC4 upregulation. Oncogene 2013, 32, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Trevino, J.G.; Pillai, S.; Kunigal, S.; Singh, S.; Fulp, W.J.; Centeno, B.A.; Chellappan, S.P. Nicotine induces inhibitor of differentiation-1 in a src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia 2012, 14, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pillai, S.; Chellappan, S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J. Oncol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Trevino, J.; Rawal, B.; Singh, S.; Kovacs, M.; Li, X.; Schell, M.; Haura, E.; Bepler, G.; Chellappan, S. Beta-arrestin-1 mediates nicotine-induced metastasis through e2f1 target genes that modulate epithelial-mesenchymal transition. Cancer Res. 2015, 75, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Chellappan, S. Alpha7 nicotinic acetylcholine receptor subunit in angiogenesis and epithelial to mesenchymal transition. Curr. Drug Targets 2012, 13, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M. Beta-adrenergic signaling, a novel target for cancer therapy? Oncotarget 2010, 1, 466–469. [Google Scholar] [PubMed]
- Hecht, S.S.; Abbaspour, A.; Hoffman, D. A study of tobacco carcinogenesis. XLII. Bioassay in A/J mice of some structural analogues of tobacco-specific nitrosamines. Cancer Lett. 1988, 42, 141–145. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Itier, V.; Bertrand, D. Neuronal nicotinic receptors: From protein structure to function. FEBS Lett. 2001, 504, 118–125. [Google Scholar] [CrossRef]
- Lindstrom, J.; Anand, R.; Gerzanich, V.; Peng, X.; Wang, F.; Wells, G. Structure and function of neuronal nicotinic acetylcholine receptors. Prog. Brain Res. 1996, 109, 125–137. [Google Scholar] [PubMed]
- Wallukat, G. The beta-adrenergic receptors. Herz 2002, 27, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Chellappan, S.P. Nicotine-mediated cell proliferation and angiogenesis: New twists to an old story. Cell Cycle 2006, 5, 2324–2328. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wu, C.H.; Ho, Y.S. From smoking to cancers: Novel targets to neuronal nicotinic acetylcholine receptors. J. Oncol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Weddle, D.L.; Tithoff, P.; Williams, M.; Schuller, H.M. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 2001, 22, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, B.J.; Williams, M.; Plummer, H.K.; Schuller, H.M. Activation of voltage-operated Ca2+-channels in human small cell lung carcinoma by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Int. J. Oncol. 2000, 16, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Ushiyama, N.; Fujii, T.; Kawashima, K. Nicotine-induced Ca2+ signaling and down-regulation of nicotinic acetylcholine receptor subunit expression in the cem human leukemic t-cell line. Life Sci. 2003, 72, 2155–2158. [Google Scholar] [CrossRef]
- Schuller, H.M. Nitrosamine-induced lung carcinogenesis and Ca2+/calmodulin antagonists. Cancer Res. 1992, 52, 2723s–2726s. [Google Scholar] [PubMed]
- Del Barrio, L.; Egea, J.; Leon, R.; Romero, A.; Ruiz, A.; Montero, M.; Alvarez, J.; Lopez, M.G. Calcium signalling mediated through alpha7 and non-alpha7 nachr stimulation is differentially regulated in bovine chromaffin cells to induce catecholamine release. Br. J. Pharmacol. 2011, 162, 94–110. [Google Scholar] [CrossRef] [PubMed]
- Del Barrio, L.; Martin-de-Saavedra, M.D.; Romero, A.; Parada, E.; Egea, J.; Avila, J.; McIntosh, J.M.; Wonnacott, S.; Lopez, M.G. Neurotoxicity induced by okadaic acid in the human neuroblastoma sh-sy5y line can be differentially prevented by alpha7 and beta2* nicotinic stimulation. Toxicol. Sci. 2011, 123, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.; Lecchi, M.; Arnaudeau, S.; Bertrand, D.; Demaurex, N. Local and global calcium signals associated with the opening of neuronal alpha7 nicotinic acetylcholine receptors. Cell Calcium 2009, 45, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Fucile, S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 2004, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.X.; Yakel, J.L. Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 2009, 30, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Rooman, I.; Real, F.X. Pancreatic ductal adenocarcinoma and acinar cells: A matter of differentiation and development? Gut 2012, 61, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Bersch, V.P.; Osvaldt, A.B.; Edelweiss, M.I.; Schumacher Rde, C.; Wendt, L.R.; Abreu, L.P.; Blom, C.B.; Abreu, G.P.; Costa, L.; Piccinini, P.; et al. Effect of nicotine and cigarette smoke on an experimental model of intraepithelial lesions and pancreatic adenocarcinoma induced by 7,12-dimethylbenzanthracene in mice. Pancreas 2009, 38, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by k-ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Howes, N.; Lerch, M.M.; Greenhalf, W.; Stocken, D.D.; Ellis, I.; Simon, P.; Truninger, K.; Ammann, R.; Cavallini, G.; Charnley, R.M.; et al. Clinical and genetic characteristics of hereditary pancreatitis in europe. Clin. Gastroenterol. Hepatol. 2004, 2, 252–261. [Google Scholar] [CrossRef]
- Hermann, P.C.; Sancho, P.; Canamero, M.; Martinelli, P.; Madriles, F.; Michl, P.; Gress, T.; de Pascual, R.; Gandia, L.; Guerra, C.; et al. Nicotine promotes initiation and progression of kras-induced pancreatic cancer via gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology 2014, 147, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Torres, M.P.; Kaur, S.; Rachagani, S.; Joshi, S.; Johansson, S.L.; Momi, N.; Baine, M.J.; Gilling, C.E.; Smith, L.M.; et al. Smoking accelerates pancreatic cancer progression by promoting differentiation of mdscs and inducing hb-egf expression in macrophages. Oncogene 2015, 34, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Bavarva, J.H.; Tae, H.; Settlage, R.E.; Garner, H.R. Characterizing the genetic basis for nicotine induced cancer development: A transcriptome sequencing study. PLoS ONE 2013, 8, e67252. [Google Scholar] [CrossRef] [PubMed]
- Laytragoon-Lewin, N.; Bahram, F.; Rutqvist, L.E.; Turesson, I.; Lewin, F. Direct effects of pure nicotine, cigarette smoke extract, swedish-type smokeless tobacco (snus) extract and ethanol on human normal endothelial cells and fibroblasts. Anticancer Res. 2011, 31, 1527–1534. [Google Scholar] [PubMed]
- Bosetti, C.; Lucenteforte, E.; Silverman, D.T.; Petersen, G.; Bracci, P.M.; Ji, B.T.; Negri, E.; Li, D.; Risch, H.A.; Olson, S.H.; et al. Cigarette smoking and pancreatic cancer: An analysis from the international pancreatic cancer case-control consortium (panc4). Ann. Oncol. 2012, 23, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wei, P.; Duell, E.J.; Risch, H.A.; Olson, S.H.; Bueno-de-Mesquita, H.B.; Gallinger, S.; Holly, E.A.; Petersen, G.; Bracci, P.M.; et al. Axonal guidance signaling pathway interacting with smoking in modifying the risk of pancreatic cancer: A gene- and pathway-based interaction analysis of gwas data. Carcinogenesis 2014, 35, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A. Nicotine alters the proteome of two human pancreatic duct cell lines. JOP 2014, 15, 465–474. [Google Scholar] [PubMed]
- Al-Wadei, M.H.; Al-Wadei, H.A.; Schuller, H.M. Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors alpha3, alpha5, and alpha7. Mol. Cancer Res. 2012, 10, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, M.H.; Al-Wadei, H.A.; Schuller, H.M. Effects of chronic nicotine on the autocrine regulation of pancreatic cancer cells and pancreatic duct epithelial cells by stimulatory and inhibitory neurotransmitters. Carcinogenesis 2012, 33, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, H.A.; Plummer, H.K., 3rd; Schuller, H.M. Nicotine stimulates pancreatic cancer xenografts by systemic increase in stress neurotransmitters and suppression of the inhibitory neurotransmitter gamma-aminobutyric acid. Carcinogenesis 2009, 30, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Bose, C.; Zhang, H.; Udupa, K.B.; Chowdhury, P. Activation of p-ERK1/2 by nicotine in pancreatic tumor cell line AR42J: Effects on proliferation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G926–G934. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chowdhury, P.; Walker, A. A cell-based approach to study changes in the pancreas following nicotine exposure in an animal model of injury. Langenbeck’s Arch. Surg. 2008, 393, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Kiehne, K.; Herzig, K.H.; Folsch, U.R. Differential activation of P42ERK2 and P125FAK by cholecystokinin and bombesin in the secretion and proliferation of the pancreatic amphicrine cell line AR42J. Pancreatology 2002, 2, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Al-Wadei, H.A.; Majidi, M. Gaba b receptor is a novel drug target for pancreatic cancer. Cancer 2008, 112, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Al-Salam, S.; Hameed, R.; Parvez, H.S.; Adeghate, E. Diabetes mellitus decreases the expression of calcitonin-gene related peptide, gamma-amino butyric acid and glutamic acid decarboxylase in human pancreatic islet cells. Neuro Endocrinol. Lett. 2009, 30, 506–510. [Google Scholar] [PubMed]
- Bafna, S.; Kaur, S.; Batra, S.K. Membrane-bound mucins: The mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 2010, 29, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Singh, A.P.; Batra, S.K. Structure, evolution, and biology of the MUC4 mucin. FASEB J. 2008, 22, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Finnie, I.A.; Campbell, B.J.; Taylor, B.A.; Milton, J.D.; Sadek, S.K.; Yu, L.G.; Rhodes, J.M. Stimulation of colonic mucin synthesis by corticosteroids and nicotine. Clin. Sci. 1996, 91, 359–364. [Google Scholar] [PubMed]
- Andrianifahanana, M.; Moniaux, N.; Schmied, B.M.; Ringel, J.; Friess, H.; Hollingsworth, M.A.; Buchler, M.W.; Aubert, J.P.; Batra, S.K. Mucin (MUC) gene expression in human pancreatic adenocarcinoma and chronic pancreatitis: A potential role of MUC4 as a tumor marker of diagnostic significance. Clin. Cancer Res. 2001, 7, 4033–4040. [Google Scholar] [PubMed]
- Swartz, M.J.; Batra, S.K.; Varshney, G.C.; Hollingsworth, M.A.; Yeo, C.J.; Cameron, J.L.; Wilentz, R.E.; Hruban, R.H.; Argani, P. MUC4 expression increases progressively in pancreatic intraepithelial neoplasia. Am. J. Clin. Pathol. 2002, 117, 791–796. [Google Scholar] [PubMed]
- Kunigal, S.; Ponnusamy, M.P.; Momi, N.; Batra, S.K.; Chellappan, S.P. Nicotine, ifn-gamma and retinoic acid mediated induction of muc4 in pancreatic cancer requires e2f1 and stat-1 transcription factors and utilize different signaling cascades. Mol. Cancer 2012, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Andrianifahanana, M.; Agrawal, A.; Singh, A.P.; Moniaux, N.; van Seuningen, I.; Aubert, J.P.; Meza, J.; Batra, S.K. Synergistic induction of the MUC4 mucin gene by interferon-gamma and retinoic acid in human pancreatic tumour cells involves a reprogramming of signalling pathways. Oncogene 2005, 24, 6143–6154. [Google Scholar] [CrossRef] [PubMed]
- Chipitsyna, G.; Gong, Q.; Anandanadesan, R.; Alnajar, A.; Batra, S.K.; Wittel, U.A.; Cullen, D.M.; Akhter, M.P.; Denhardt, D.T.; Yeo, C.J.; et al. Induction of osteopontin expression by nicotine and cigarette smoke in the pancreas and pancreatic ductal adenocarcinoma cells. Int. J. Cancer 2009, 125, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chakraborty, G.; Bulbule, A.; Kaur, R.; Kundu, G.C. Osteopontin: An emerging therapeutic target for anticancer therapy. Expert Opin. Ther. Targets 2007, 11, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.A.; Berse, B.; Senger, D.R. Specific reduction in osteopontin synthesis by antisense rna inhibits the tumorigenicity of transformed rat1 fibroblasts. Oncogene 1994, 9, 2321–2326. [Google Scholar] [PubMed]
- Lazar, M.; Sullivan, J.; Chipitsyna, G.; Gong, Q.; Ng, C.Y.; Salem, A.F.; Aziz, T.; Witkiewicz, A.; Denhardt, D.T.; Yeo, C.J.; et al. Involvement of osteopontin in the matrix-degrading and proangiogenic changes mediated by nicotine in pancreatic cancer cells. J. Gastrointest. Surg. 2010, 14, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix metalloproteinases and metastasis. Cancer Chemother. Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Wang, X.; Ling, M.T. Id-1 expression and cell survival. Apoptosis 2004, 9, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Shuno, Y.; Tsuno, N.H.; Okaji, Y.; Tsuchiya, T.; Sakurai, D.; Nishikawa, T.; Yoshikawa, N.; Sasaki, K.; Hongo, K.; Tsurita, G.; et al. Id1/Id3 knockdown inhibits metastatic potential of pancreatic cancer. J. Surg. Res. 2010, 161, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Jager, H.; Dreker, T.; Buck, A.; Giehl, K.; Gress, T.; Grissmer, S. Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol. Pharmacol. 2004, 65, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoeven, D.; Cho, K.J.; Ma, X.; Chigurupati, S.; Parton, R.G.; Hancock, J.F. Fendiline inhibits k-Ras plasma membrane localization and blocks k-Ras signal transmission. Mol. Cell. Biol. 2013, 33, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Bayer, R.; Schwarzmaier, J.; Pernice, R. Basic mechanisms underlying prenylamine-induced “torsade de pointes”: Differences between prenylamine and fendiline due to basic actions of the isomers. Curr. Med. Res. Opin. 1988, 11, 254–272. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Lubnau, E.; Grossmann, M.; Ruck, W. Double-blind, randomized study of the anti-anginal and anti-ischaemic efficacy of fendiline and diltiazem in patients with coronary heart disease. Curr. Med. Res. Opin. 1991, 12, 521–539. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2014. [Google Scholar] [CrossRef] [PubMed]
- Parkash, J.; Asotra, K. Calcium wave signaling in cancer cells. Life Sci. 2010, 87, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Capello, M.; Fredolini, C.; Piemonti, L.; Liotta, L.A.; Novelli, F.; Petricoin, E.F. Proteomic analysis of pancreatic ductal adenocarcinoma cells reveals metabolic alterations. J. Proteome Res. 2011, 10, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Capello, M.; Fredolini, C.; Racanicchi, L.; Piemonti, L.; Liotta, L.A.; Novelli, F.; Petricoin, E.F. Proteomic analysis reveals warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J. Proteome Res. 2012, 11, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Epstein, T.; Xu, L.; Gillies, R.J.; Gatenby, R.A. Separation of metabolic supply and demand: Aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Glycolysis in cancer: A potential target for therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- James, A.D.; Chan, A.; Erice, O.; Siriwardena, A.K.; Bruce, J.I. Glycolytic atp fuels the plasma membrane calcium pump critical for pancreatic cancer cell survival. J. Biol. Chem. 2013, 288, 36007–36019. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. Calcium pump of the plasma membrane. Physiol. Rev. 1991, 71, 129–153. [Google Scholar] [PubMed]
- Carafoli, E. The calcium pumping atpase of the plasma membrane. Annu. Rev. Physiol. 1991, 53, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Kumosani, T.A.; Elshal, M.F.; Al-Jonaid, A.A.; Abduljabar, H.S. The influence of smoking on semen quality, seminal microelements and Ca2+-atpase activity among infertile and fertile men. Clin. Biochem. 2008, 41, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Varela, D.; Schmidt, M.; Schoellerman, J.; Peters, E.C.; Berg, D.K. PMCA2 via PSD-95 controls calcium signaling by alpha7-containing nicotinic acetylcholine receptors on aspiny interneurons. J. Neurosci. 2012, 32, 6894–6905. [Google Scholar] [CrossRef] [PubMed]
- Tojyo, Y.; Morita, T.; Nezu, A.; Tanimura, A. Key components of store-operated Ca2+ entry in non-excitable cells. J. Pharmacol. Sci. 2014, 125, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates crac channels via direct binding of a cytosolic domain to orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Hyzinski-Garcia, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate crac channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: New roles for known actors. Nat. Rev. Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Prevarskaya, N. Study of trp channels in cancer cells. In TRP Channels; Zhu, M.X., Ed.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Lehen’kyi, V.; Prevarskaya, N. Oncogenic TRP channels. Adv. Exp. Med. Biol. 2011, 704, 929–945. [Google Scholar] [PubMed]
- Bidaux, G.; Beck, B.; Zholos, A.; Gordienko, D.; Lemonnier, L.; Flourakis, M.; Roudbaraki, M.; Borowiec, A.S.; Fernandez, J.; Delcourt, P.; et al. Regulation of activity of transient receptor potential melastatin 8 (TRPM8) channel by its short isoforms. J. Biol. Chem. 2012, 287, 2948–2962. [Google Scholar] [CrossRef] [PubMed]
- Wissenbach, U.; Niemeyer, B.A.; Flockerzi, V. TRP channels as potential drug targets. Biol. Cell 2004, 96, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Duncan, L.M.; Deeds, J.; Hunter, J.; Shao, J.; Holmgren, L.M.; Woolf, E.A.; Tepper, R.I.; Shyjan, A.W. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 1998, 58, 1515–1520. [Google Scholar] [PubMed]
- Jiang, J.; Li, M.H.; Inoue, K.; Chu, X.P.; Seeds, J.; Xiong, Z.G. Transient receptor potential melastatin 7-like current in human head and neck carcinoma cells: Role in cell proliferation. Cancer Res. 2007, 67, 10929–10938. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Park, E.J.; Lee, J.H.; Jeon, J.H.; Kim, S.J.; So, I. Suppression of transient receptor potential melastatin 7 channel induces cell death in gastric cancer. Cancer Sci. 2008, 99, 2502–2509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Barritt, G.J. Evidence that TRPM8 is an androgen-dependent Ca2+ channel required for the survival of prostate cancer cells. Cancer Res. 2004, 64, 8365–8373. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S.; Chan, A.S.; Yee, J.D.; Yee, R.K. TRPM7 and TRPM8 ion channels in pancreatic adenocarcinoma: Potential roles as cancer biomarkers and targets. Scientifica 2012. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S.; Zhou, W.; Lee, M. Transient receptor potential channel TRPM8 is over-expressed and required for cellular proliferation in pancreatic adenocarcinoma. Cancer Lett. 2010, 297, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S.; Zhou, W.; Liang, I.C. Transient receptor potential ion channel TRPM7 regulates exocrine pancreatic epithelial proliferation by Mg2+-sensitive Socs3a signaling in development and cancer. Dis. Model. Mech. 2011, 4, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S.; Zhou, W.; Lee, M.; Yee, R.K. Targeted silencing of TRPM7 ion channel induces replicative senescence and produces enhanced cytotoxicity with gemcitabine in pancreatic adenocarcinoma. Cancer Lett. 2012, 318, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.R.; Novak, I.; Pedersen, S.F.; Larsen, E.H.; Hoffmann, E.K. ANO1 (TMEM16A) in pancreatic ductal adenocarcinoma (PDAC). Pflugers Arch. 2015, 467, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Y.; Lin, C.; Jia, J.; Tian, L.; Yang, K.; Zhao, L.; Lai, N.; Jiang, Q.; Sun, Y.; et al. Effects of chronic exposure to cigarette smoke on canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am. J. Physiol. Cell Physiol. 2014, 306, C364–C373. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, W.; Ward, A.; Piggott, B.J.; Larkspur, E.R.; Sternberg, P.W.; Xu, X.Z. A C. elegans model of nicotine-dependent behavior: Regulation by trp-family channels. Cell 2006, 127, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Xu, X.Z. Function and regulation of trp family channels in C. elegans. Pflugers Arch. 2009, 458, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Kelley, A.E. Worms clear the smoke surrounding nicotine addiction. Cell 2006, 127, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Obermann, S.; von Wasielewski, R.; Haile, L.; Manns, M.P.; Korangy, F.; Greten, T.F. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology 2009, 128, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.; Sullivan, J.; Chipitsyna, G.; Aziz, T.; Salem, A.F.; Gong, Q.; Witkiewicz, A.; Denhardt, D.T.; Yeo, C.J.; Arafat, H.A. Induction of monocyte chemoattractant protein-1 by nicotine in pancreatic ductal adenocarcinoma cells: Role of osteopontin. Surgery 2010, 148, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Al-Wadei, H.A.; Al-Wadei, M.H.; Dagnon, K.; Schuller, H.M. Differential modulation of nicotine-induced gemcitabine resistance by gaba receptor agonists in pancreatic cancer cell xenografts and in vitro. BMC Cancer 2014, 14, 725. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Chattopadhyay, K.; Chhabra, J.K.; Chattopadhyay, B. Protein dependent fate of hepatic cells under nicotine induced stress and curcumin ameliorated condition. Eur. J. Pharmacol. 2012, 684, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, J.; Al-Wadei, H.A.; Schuller, H.M. Chronic nicotine inhibits the therapeutic effects of gemcitabine on pancreatic cancer in vitro and in mouse xenografts. Eur. J. Cancer 2013, 49, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaal, C.; Padmanabhan, J.; Chellappan, S. The Role of nAChR and Calcium Signaling in Pancreatic Cancer Initiation and Progression. Cancers 2015, 7, 1447-1471. https://doi.org/10.3390/cancers7030845
Schaal C, Padmanabhan J, Chellappan S. The Role of nAChR and Calcium Signaling in Pancreatic Cancer Initiation and Progression. Cancers. 2015; 7(3):1447-1471. https://doi.org/10.3390/cancers7030845
Chicago/Turabian StyleSchaal, Courtney, Jaya Padmanabhan, and Srikumar Chellappan. 2015. "The Role of nAChR and Calcium Signaling in Pancreatic Cancer Initiation and Progression" Cancers 7, no. 3: 1447-1471. https://doi.org/10.3390/cancers7030845
APA StyleSchaal, C., Padmanabhan, J., & Chellappan, S. (2015). The Role of nAChR and Calcium Signaling in Pancreatic Cancer Initiation and Progression. Cancers, 7(3), 1447-1471. https://doi.org/10.3390/cancers7030845