
Targeting NK-1 Receptors to Prevent and Treat Pancreatic Cancer: a New Therapeutic Approach


{kind=link}
{kind=link}
Abstract
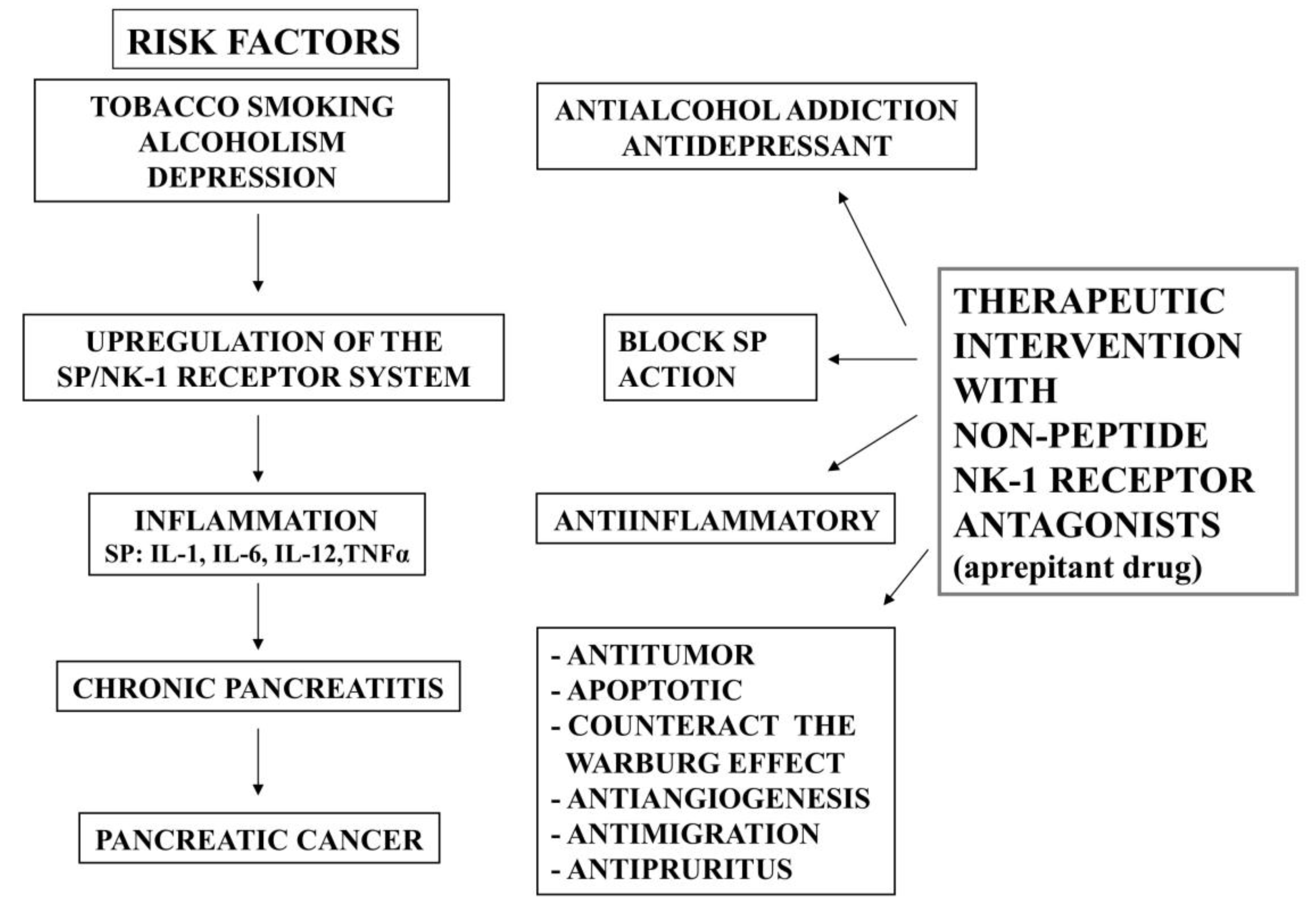
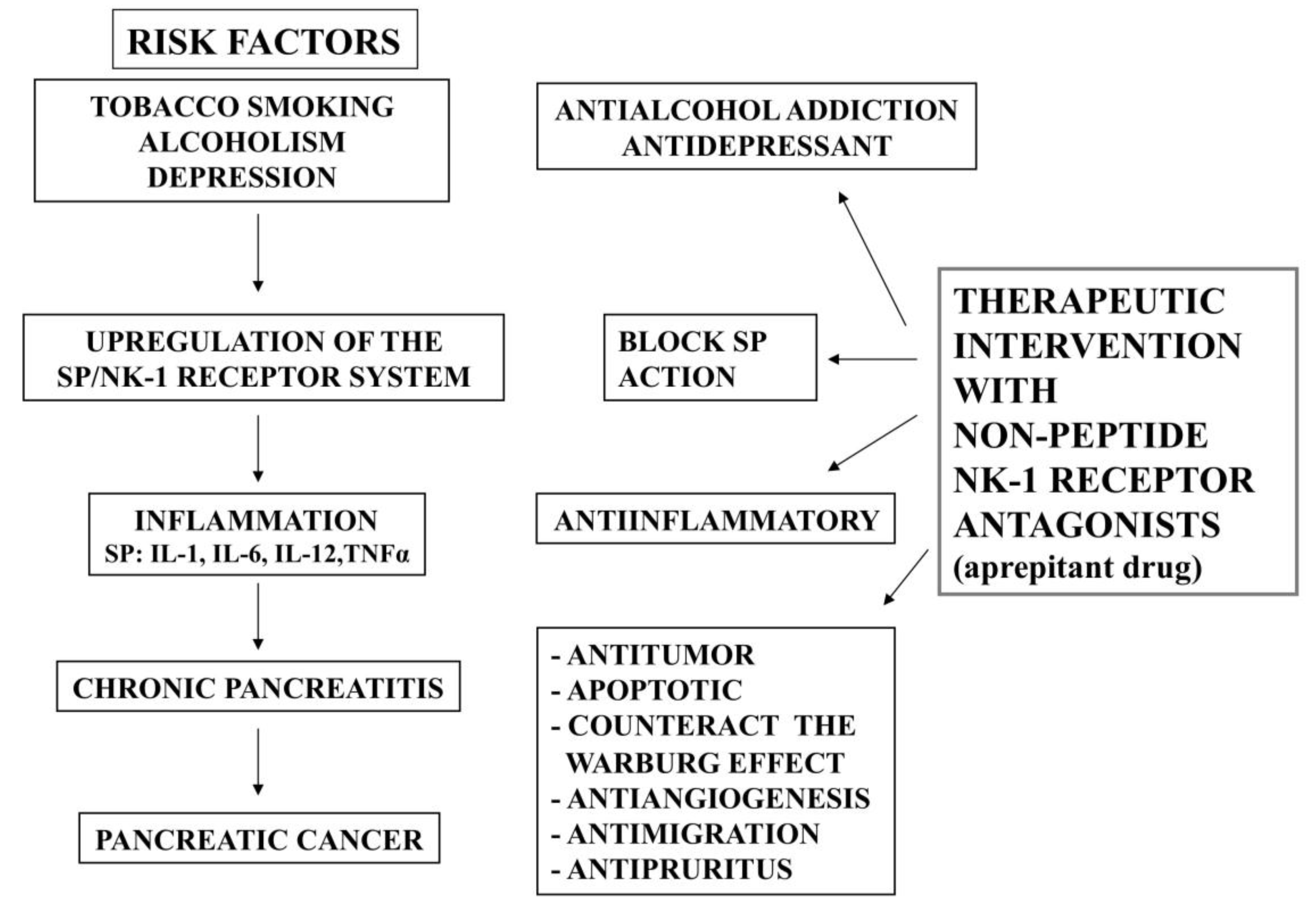
:1. Introduction
2. Substance P and the NK-1 Receptor
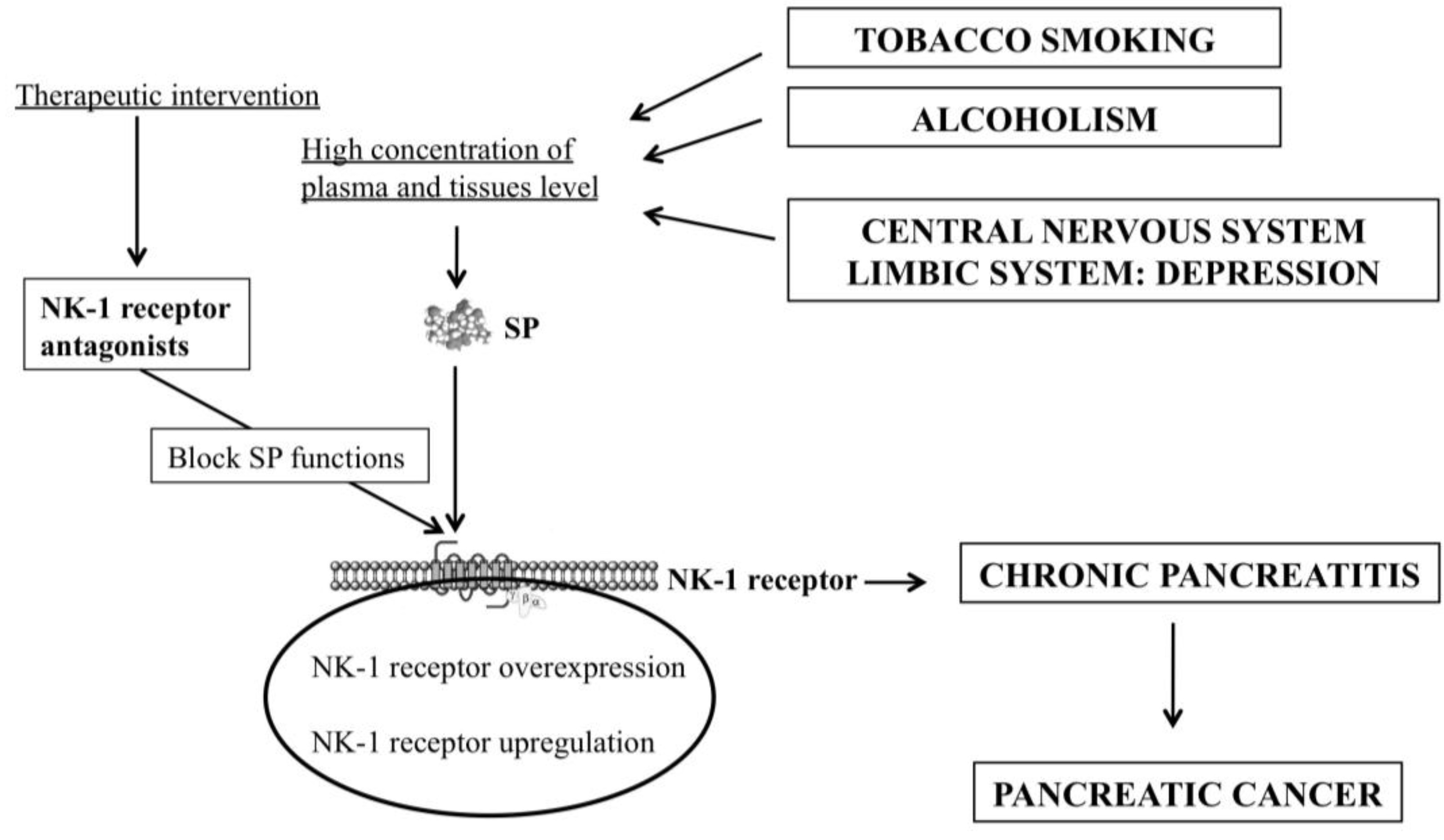
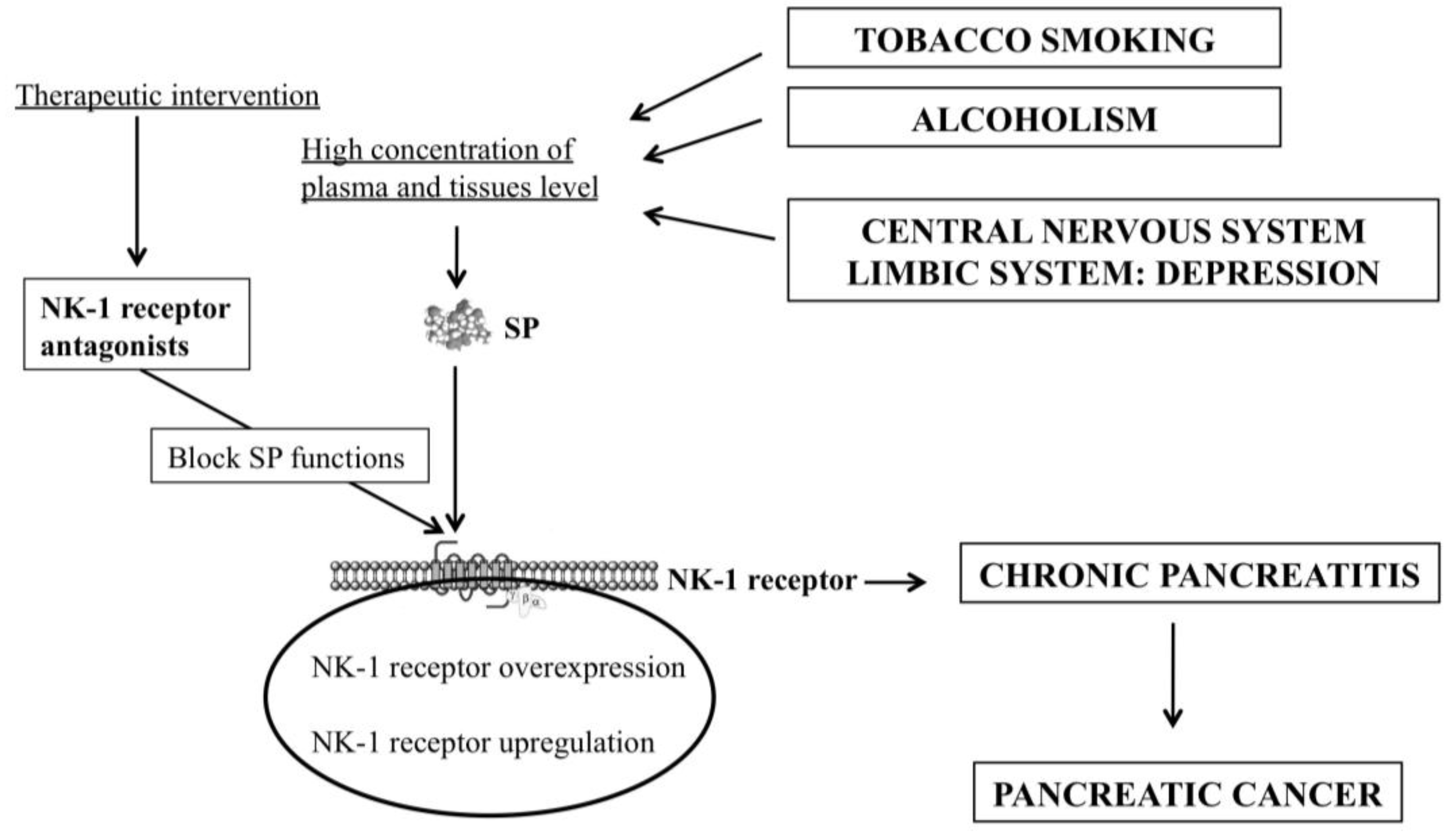
3. Smoking, Pancreatitis and the SP/NK-1 Receptor System
4. Alcoholism, Pancreatitis and the SP/NK-1 Receptor System
5. Depression, Pancreatic Cancer and the SP/NK-1 Receptor System
6. Chronic Pancreatitis and the SP/NK-1 Receptor System
7. Pancreatic Cancer and the SP/NK-1 Receptor System
8. NK-1 Receptor Antagonists for the Prevention and Treatment of Pancreatic Cancer
9. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in pancreatic cancer. World J. Gastroenterol. 2014, 20, 2312–2334. [Google Scholar] [CrossRef] [PubMed]
- Momi, N.; Kaur, S.; Krishn, S.R.; Batra, S.K. Discovering the route from inflammation to pancreatic cancer. Minerva Gastroenterol. Dietol. 2012, 58, 283–297. [Google Scholar] [PubMed]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 2014, 46, 1727–1750. [Google Scholar] [CrossRef] [PubMed]
- Rosato, V.; Polesel, J.; Bosseti, C.; Serrano, D.; Negri, E.; La Vecchia, C. Population attributable risk for pancreatic cancer in northern Italy. Pancreas 2015, 44, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Howes, N.; Neoptolemos, J.P. Risk of pancreatic ductal adenocarcinoma in chronic pancreatitis. Gut 2002, 51, 765–766. [Google Scholar] [CrossRef] [PubMed]
- Binker, M.G.; Binker-Cosen, A.A.; Richards, D.; Gaisano, H.Y.; de Cosen, R.H.; Cosen-Binker, L.I. Chronic stress sensitizes rats to pancreatitis induced by cerulein: Role of TNF-α. World J. Gastroenterol. 2010, 16, 5565–5581. [Google Scholar] [CrossRef] [PubMed]
- Kim-Fuchs, C.; Le, C.P.; Pimentel, M.A.; Shackleford, D.; Ferrari, D.; Angst, E.; Hollande, F.; Sloan, E.K. Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav. Immun. 2014, 40, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, F.D.; Coveñas, R. Targeting opioid and neurokinin-1 receptors to treat alcoholism. Curr. Med. Chem. 2011, 18, 4321–4334. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, X.; Lin, Y. Cigarette smoke synergizes lipopolysaccharide-induced interleukin-1β and tumor necrosis factor-α secretion from macrophages via substance P-mediated nuclear factor-κB activation. Am. J. Respir. Cell Mol. Biol. 2011, 44, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Quartara, L.; Maggi, C.A. The tachykinin NK-1 receptor. Part I: Ligands and mechanisms of cellular activation. Neuropeptides 1997, 31, 537–563. [Google Scholar] [CrossRef]
- Coveñas, R.; Muñoz, M. Cancer progression and substance P. Histol. Histopathol. 2014, 29, 881–890. [Google Scholar] [PubMed]
- Luo, W.; Sharif, T.R.; Sharif, M. Substance P-induced mitogenesis in human astrocytoma cells correlates with activation of the mitogen-activated protein kinase signaling pathway. Cancer Res. 1996, 56, 4983–4991. [Google Scholar] [PubMed]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Déry, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta-arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Yang, J.; Tong, L.; Yuan, S.; Tian, Y.; Hong, L.; Wang, W.; Zhang, H. Substance P immunoreactive nerve fibres are related to gastric cancer differentiation status and could promote proliferation and migration of gastric cancer cells. Cell Biol. Int. 2011, 35, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, L.E.; Kwatra, M.M. NK1 (substance P) receptor. UCSD Mol. Pages 2012, 2012, 1, 10–16. [Google Scholar]
- Medrano, S.; Gruenstein, E.; Dimlich, R.V. Substance P receptors on human astrocytoma cells are linked to glycogen breakdown. Neurosci. Lett. 1994, 167, 14–18. [Google Scholar] [CrossRef]
- Patel, H.J.; Ramkissoon, S.H.; Patel, P.S.; Rameshwar, P. Transformation of breast cells by truncated neurokinin-1 receptor is secondary to activation by preprotachykinin-A peptides. Proc. Natl. Acad. Sci. USA 2005, 102, 17436–17441. [Google Scholar] [CrossRef] [PubMed]
- Moharita, A.; Harrison, J.S.; Rameshwar, P. Neurokinin receptors and subtypes as potential targets in breast cancer: Relevance to bone marrow metastasis. Drug Res. Rev. 2004, 1, 1–6. [Google Scholar] [CrossRef]
- Peng, S.L. Transcription factors in the pathogenesis of autoimmunity. Clin. Immunol. 2004, 110, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, E.; Leeman, S.E.; Watts, L.A.; Coukos, J.A.; O’Brien, M.J.; Cerda, S.R.; Farraye, F.A.; Stucchi, A.F. Truncated neurokinin-1 receptor is increased in colonic epithelial cells from patients with colitis-associated cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 17420–17425. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.M.; Hecht, S.S.; Kovatch, R.M.; Amin, S.; Hoffmann, D.; Rice, J.M. Tumorigenicity of the tobacco-specific carcinogen 4-(m-(m-(methyl-nitrosamino)-)-1-(-(3-pyridyl)-)-1-b-butanone in infant mice. Cancer Lett. 1991, 58, 177–181. [Google Scholar] [CrossRef]
- Hoffmann, D.; Hecht, S.S. Nicotine-derived N-nitrosamines and tobacco-r-related cancer: Current status and future directions. Cancer Res. 1985, 45, 935–944. [Google Scholar] [PubMed]
- Hoffmann, D.; Lavoie, E.J.; Hecht, S.S. Nicotine: A precursor for carcinogens. Cancer Lett. 1985, 26, 67–75. [Google Scholar] [CrossRef]
- Chowdhury, P.; Doi, R.; Tangoku, A.; Rayford, P.L. Structural and functional changes of rat exocrine pancreas exposed to nicotine. Int. J. Pancreatol. 1995, 18, 257–264. [Google Scholar] [PubMed]
- Sliwińska, M.; Milnerowicz, H. Influence of tobacco smoking on lipase activity in patients with pancreatitis. Przegl. Lek. 2005, 62, 1058–1061. [Google Scholar]
- Brown, P. The influence of smoking on pancreatic function in man. Med. J. Aust. 1976, 2, 290–293. [Google Scholar] [PubMed]
- Bynum, T.E.; Solomon, T.E.; Johnson, L.R.; Jacobson, E.D. Inhibition of pancreatic secretion in man by cigarette smoking. Gut 1972, 13, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Milnerowicz, H.; Sliwińska, M.; Jabłonowska, M.; Milnerowicz, S. Effect of tobacco smoking on amylase activity in patients with pancreatitis. Przegl. Lek. 2004, 61, 1071–1072. [Google Scholar] [PubMed]
- Wittel, U.A.; Pandey, K.K.; Andrianifahanana, M.; Johansson, S.L.; Cullen, D.M.; Akhter, M.P.; Brand, R.E.; Prokopczyk, B.; Batra, S.K. Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am. J. Gastroenterol. 2006, 101, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Geppetti, P. Substance P. Int. J. Biochem. Cell Biol. 2001, 33, 555–576. [Google Scholar] [CrossRef]
- Holzer, P. Local effector functions of capsaicin-sensitive sensory nerve endings: Involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience 1988, 24, 739–768. [Google Scholar] [CrossRef]
- Lieb, K.; Fiebich, B.L.; Berger, M.; Bauer, J.; Schulze-Osthoff, K. The neuropeptide substance P activates transcription factor NF-kappa β and kappa β-dependent gene expression in human astrocytoma cells. J. Immunol. 1997, 159, 4952–4958. [Google Scholar] [PubMed]
- Lotz, M.; Vaughan, J.H.; Carson, D.A. Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science 1988, 241, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, S.L.; Chowdhury, P. The genetics of nicotine dependence: Relationship to pancreatic cancer. World J. Gastroenterol. 2006, 12, 7433–7439. [Google Scholar] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P. Risk factors for pancreatic cancer. J. Cell. Biochem. 2005, 95, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L. Pancreatitis and the risk of pancreatic cancer. International pancreatitis study group. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Lowenfels, A.B.; Müllhaupt, B.; Cavallini, G.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L.; Frulloni, L.; et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 2005, 54, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Durbec, J.P.; Sarles, H. Multicenter survey of the etiology of pancreatic diseases. Relationship between the relative risk of developing chronic pancreatitis and alcohol, protein and lipid consumption. Digestion 1978, 18, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Pezzilli, R.; Morselli-Labate, A.M. Alcoholic pancreatitis: Pathogenesis, incidence and treatment with special reference to the associated pain. Int. J. Environ. Res. Public Health 2009, 6, 2763–2782. [Google Scholar] [CrossRef] [PubMed]
- Gullo, L.; Barbara, L.; Labò, G. Effect of cessation of alcohol use on the course of pancreatic dysfunction in alcoholic pancreatitis. Gastroenterology 1988, 95, 1063–1068. [Google Scholar] [PubMed]
- Gukovsky, I.; Lugea, A.; Shahsahebi, M.; Cheng, J.H.; Hong, P.P.; Jung, Y.J.; Deng, Q.G.; French, B.A.; Lungo, W.; French, S.W.; et al. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G68–79. [Google Scholar] [CrossRef] [PubMed]
- Wick, E.C.; Hoge, S.G.; Grahn, S.W.; Kim, E.; Divino, L.A.; Grady, E.F.; Bunnett, N.W.; Kirkwood, K.S. Transient receptor potential vanilloid 1, calcitonin gene-related peptide, and substance P mediate nociception in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G959–969. [Google Scholar] [CrossRef] [PubMed]
- Vera-Portocarrero, L.; Westlund, K.N. Role of neurogenic inflammation in pancreatitis and pancreatic pain. Neurosignals 2005, 14, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Shrikhande, S.V.; Friess, H.; di Mola, F.F.; Tempia-Caliera, A.; Conejo-García, J.R.; Zhu, Z.; Zimmermann, A.; Büchler, M.W. NK-1 receptor gene expression is related to pain in chronic pancreatitis. Pain 2001, 91, 209–217. [Google Scholar] [CrossRef]
- Spiegel, D.; Giese-Davis, J. Depression and cancer: Mechanisms and disease progression. Biol. Psychiatry 2003, 54, 269–282. [Google Scholar] [CrossRef]
- Hilakivi-Clarke, L.; Rowland, J.; Clarke, R.; Lippman, M.E. Psychosocial factors in the development and progression of breast cancer. Breast Cancer Res. Treat. 1994, 29, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Humpel, C.; Knaus, G.A.; Auer, B.; Knaus, H.G.; Haring, C.; Theodorsson, E.; Saria, A. Effects of haloperidol and clozapine on preprotachykinin mRNA tachykinin tissue level release and NK-1 receptors in the striatonigral system. Synapse 1990, 6, 1–9. [Google Scholar] [PubMed]
- Shibata, K.; Haverstick, D.M.; Bannon, M.J. Tachykinin gene expression in rat limbic nuclei: Modulation by dopamine antagonists. J. Pharmacol. Exp. Ther. 1990, 255, 388–392. [Google Scholar] [PubMed]
- Shirayama, Y.; Mitsushio, H.; Takashima, M.; Ichikawa, H.; Takayashi, K. Reduction of substance P after chronic antidepressant treatment in the striatum, substantia nigra and amygdala of the rat. Brain Res. 1996, 739, 70–78. [Google Scholar] [CrossRef]
- Kramer, M.S.; Cutler, N.; Feighner, J.; Shrivastava, R.; Carman, J.; Sramek, J.J.; Reines, S.A.; Liu, G.; Snavely, D.; Wyatt-Knowles, E.; et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 1998, 281, 1640–1645. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Montgomery, S.; Ball, W.; Morrison, M.; Snavely, D.; Liu, G.; Hargreaves, R.; Hietala, J.; Lines, C.; Beebe, K.; et al. Lack of efficacy of the substance P (neurokinin-1 receptor) antagonist aprepitant in the treatment of major depressive disorder. Biol. Psychiatry 2006, 59, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Rosso, M.; Coveñas, R. The NK-1 receptor is involved in the antitumoural action of L-733,060 and in the mitogenic action of substance P on human pancreatic cancer cell lines. Lett. Drug Des. Discov. 2006, 3, 323–329. [Google Scholar] [CrossRef]
- Muñoz, M.; Rosso, M. The NK-1 receptor antagonist aprepitant as a broad spectrum antitumor drug. Invest. New Drugs 2010, 28, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Fehder, W.P. Alterations in immune response associated with anxiety in surgical patients. CNRA 1999, 10, 124–129. [Google Scholar]
- De Vane, C.L. Substance P: A new era, a new role. Pharmacotherapy 2001, 21, 1061–1069. [Google Scholar] [CrossRef]
- Lang, K.; Drell, T.L.; Lindecke, A.; Niggemann, B.; Kaltschmidt, C.; Zaenker, K.S.; Entschladen, F. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int. J. Cancer 2004, 112, 231–238. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.M.; O’Connell, J.; O’Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance P in inflammatory disease. J. Cell Physiol. 2004, 201, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Gold, L.S. Too many rodent carcinogens: Mitogenesis increases mutagenesis. Science 1990, 249, 970–971. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Fazio, M.; Williams, G.M. Cell cycle-specific mutagenesis at the hypoxanthine phosphoribosyltransferase locus in adult rat liver epithelial cells. Proc. Natl. Acad. Sci. USA 1980, 77, 7377–7379. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Zhu, Z.; Liard, V.; Shi, X.; Shrikhande, S.V.; Wang, L.; Lieb, K.; Korc, M.; Palma, C.; Zimmermann, A.; et al. Neurokinin-1 receptor expression and its potential effects on tumor growth in human pancreatic cancer. Lab. Invest. 2003, 83, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and their receptors: Contributions to physiological control and the mechanisms of disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Tsuchida, K.; Negishi, M.; Ito, S.; Nakanishi, S. Direct linkage of three tachykinin receptors to stimulation of both phosphatidylinositol hydrolysis and cAMP cascades in transfected Chinese hamster ovary cells. J. Biol. Chem. 1992, 267, 2437–2442. [Google Scholar] [PubMed]
- Takeda, Y.; Blount, P.; Sachais, B.S.; Hershey, A.D.; Raddatz, R.; Krause, J.E. Ligand binding kinetics of substance P and neurokinin A receptors stably expressed in Chinese hamster ovary cells and evidence for differential stimulation of inositol 1,4,5-triphosphate and cyclic AMP second messenger responses. J. Neurochem. 1992, 59, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Tung, W.L.; Lee, C.M. Effects of tachykinins on [3H]taurine release from human astrocytoma cells (U-373 MG). Brain Res. 1991, 549, 171–173. [Google Scholar] [CrossRef]
- Johnson, C.L.; Johnson, C.G. Substance P regulation of glutamate and cystine transport in human astrocytoma cells. Recept. Channels 1993, 1, 53–59. [Google Scholar] [PubMed]
- Gitter, B.D.; Regoli, D.; Howbert, J.J.; Glasebrook, A.L.; Waters, D.C. Interleukin-6 secretion from human astrocytoma cells induced by substance P. J. Neuroimmunol. 1994, 51, 101–108. [Google Scholar] [CrossRef]
- Tansky, M.F.; Pothoulakis, C.H.; Leeman, S.E. Functional consequences of alteration of N-linked glycosylation sites on the neurokinin 1 receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 10691–10696. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B. The war on cancer. Lancet 1996, 347, 1377–1381. [Google Scholar] [CrossRef]
- Fackler, O.T.; Grosse, R. Cell motility through plasma membrane blebbing. J. Cell Biol. 2008, 181, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Meshki, J.; Douglas, S.D.; Lai, J.P.; Schwartz, L.; Kilpatrick, L.E.; Tuluc, F. Neurokinin 1 receptor mediates membrane blebbing in HEK293 cells through a Rho/Rho-associated coiled-coil kinase-dependent mechanism. J. Biol. Chem. 2009, 284, 9280–9289. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, G.; Ma, Q.; Li, W.; Liu, J.; Han, L.; Duan, W.; Xu, Q.; Liu, H.; Wang, Z.; et al. Neurotransmitter substance P mediates pancreatic cancer perineural invasion via NK-1R in cancer cells. Mol. Cancer Res. 2013, 11, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Eibl, G.; Kisfalvi, K.; Fan, R.S.; Burdick, M.; Reber, H.; Hines, O.J.; Strieter, R.; Rozengurt, E. Broad-spectrum G proteincoupled receptor antagonist, [d-Arg1, d-Trp5,7,9, Leu11]SP: A dual inhibitor of growth and angiogenesis in pancreatic cancer. Cancer Res. 2005, 65, 2738–2745. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Morbidelli, L.; Pacini, M.; Gepetti, P.; Alessandri, G.; Maggi, C.A. Substance P stimulates neovascularization in vivo and proliferation of cultured endothelial cells. Microvasc. Res. 1990, 40, 264–278. [Google Scholar] [CrossRef]
- Hennig, I.M.; Laissue, J.A.; Horisberger, U.; Reubi, J.C. Substance-P receptors in human primary neoplasms: Tumoral and vascular localization. Int. J. Cancer 1995, 61, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Rosso, M.; Coveñas, R. The NK-1 receptor: A new target in cancer therapy. Curr. Drug Targets 2011, 12, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Reeve, J.G.; Bleehen, N.M. [d-Arg1, d-Phe5, d-Trp7,9, Leu11] Substance P induces apoptosis in lung cancer cells in vitro. Biochem. Biophys. Res. Commun. 1994, 199, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.J.; Rozengurt, E. [d-Arg1, d-Phe5, d-Trp7,9, Leu11]substance P, a potent bombesin antagonist in murine Swiss 3T3 cells, inhibits the growth of human small cell lung cancer cells in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 1859–1863. [Google Scholar] [CrossRef] [PubMed]
- Seckl, M.J.; Higgins, T.; Rozengurt, E. [d-Arg1, d-Trp5,7,9, Leu11]Substance P coordinately and reversibly inhibits bombesin- and vasopressin-induced signal transduction pathways in Swiss 3T3 cells. J. Biol. Chem. 1996, 271, 29453–29460. [Google Scholar] [PubMed]
- Seckl, M.J.; Higgins, T.; Widmer, F.; Rozengurt, E. [d-Arg1, d-Trp5,7,9, Leu11]substance P: A novel potent inhibitor of signal transduction and growth in vitro and in vivo in small cell lung cancer cells. Cancer Res. 1997, 57, 51–54. [Google Scholar] [PubMed]
- Hökfelt, T.; Pernow, B.; Wahren, J. Substance P: A pioneer amongst neuropeptides. J. Intern. Med. 2001, 249, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Coveñas, R. Safety of neurokinin-1 receptor antagonists. Expert Opin. Drug Saf. 2013, 12, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, R.; Ferreira, J.C.; Hughes, D.; Brands, J.; Hale, J.; Mattson, B.; Mills, S. Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Ann. NY Acad. Sci. 2011, 1222, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S.; Siepmann, D.; Herrgott, I.; Sunderkötter, C.; Luger, T.A. Targeting the neurokinin receptor 1 with aprepitant: A novel antipruritic strategy. PLoS ONE 2010, 5, e10968. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz, M.; Coveñas, R. Targeting NK-1 Receptors to Prevent and Treat Pancreatic Cancer: a New Therapeutic Approach. Cancers 2015, 7, 1215-1232. https://doi.org/10.3390/cancers7030832
Muñoz M, Coveñas R. Targeting NK-1 Receptors to Prevent and Treat Pancreatic Cancer: a New Therapeutic Approach. Cancers. 2015; 7(3):1215-1232. https://doi.org/10.3390/cancers7030832
Chicago/Turabian StyleMuñoz, Miguel, and Rafael Coveñas. 2015. "Targeting NK-1 Receptors to Prevent and Treat Pancreatic Cancer: a New Therapeutic Approach" Cancers 7, no. 3: 1215-1232. https://doi.org/10.3390/cancers7030832