Highlights of the Latest Advances in Research on CDK Inhibitors

 ,
, 


Abstract
:1. Overview of CDKs and CDK Inhibitors
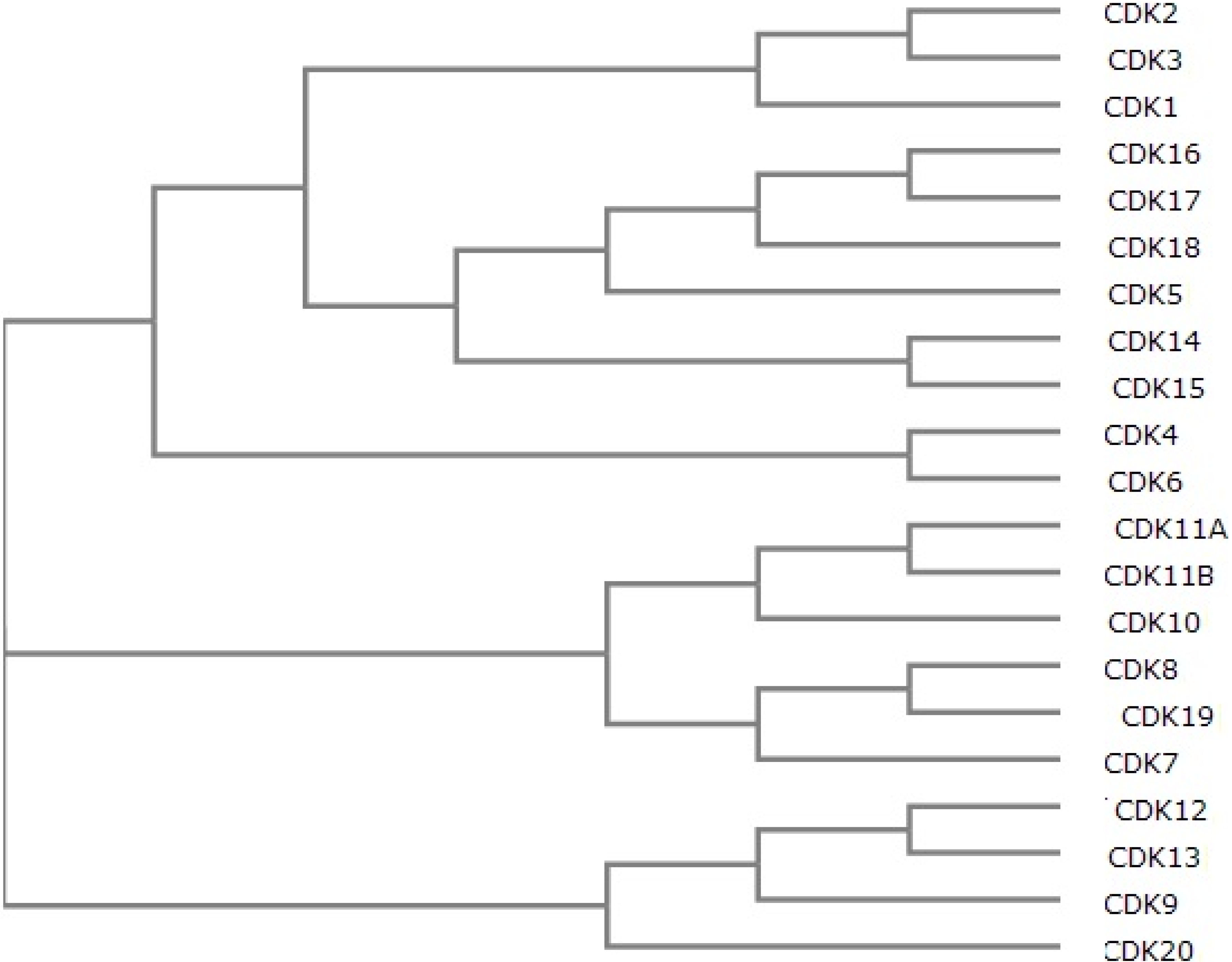
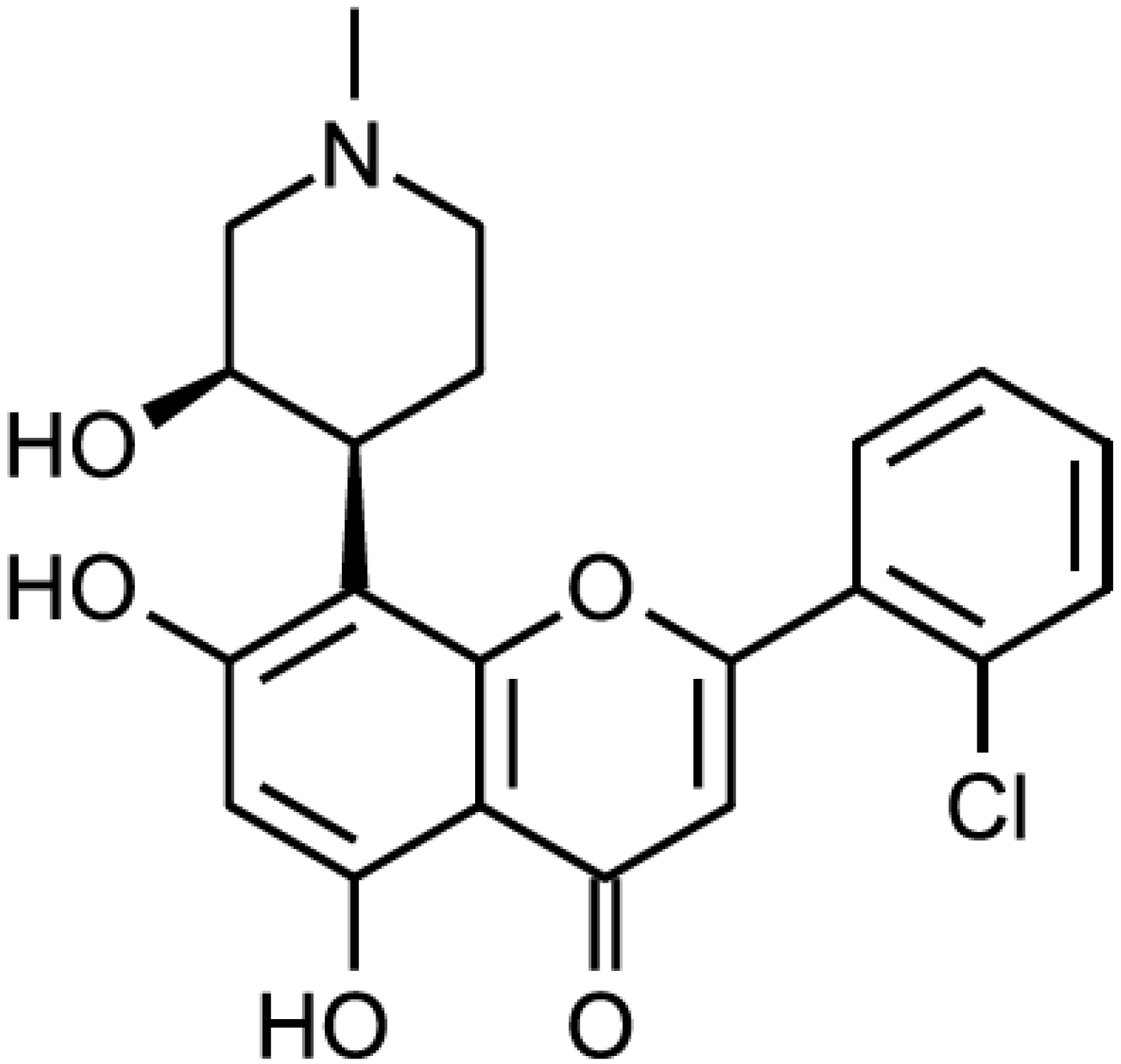

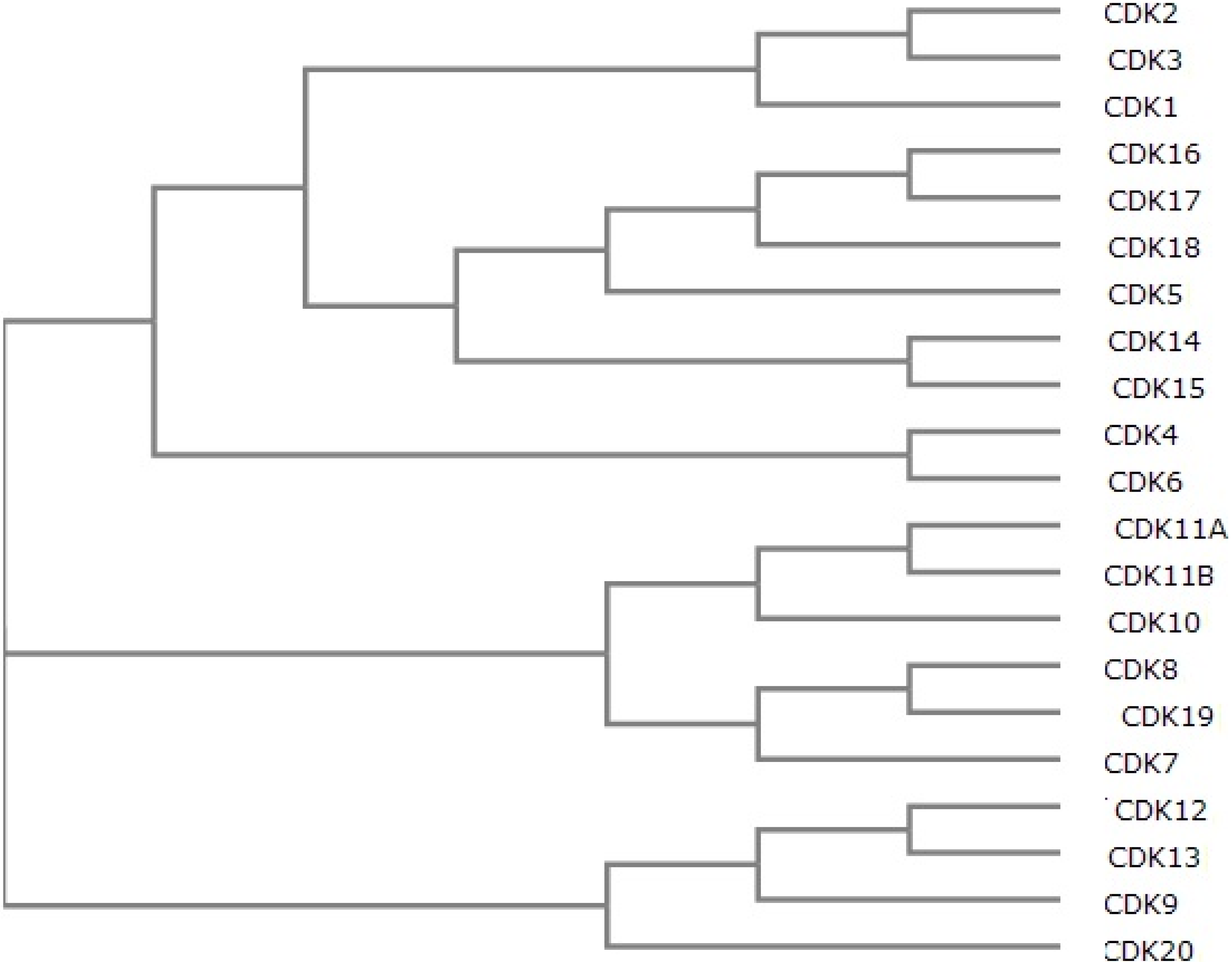{kind=link}
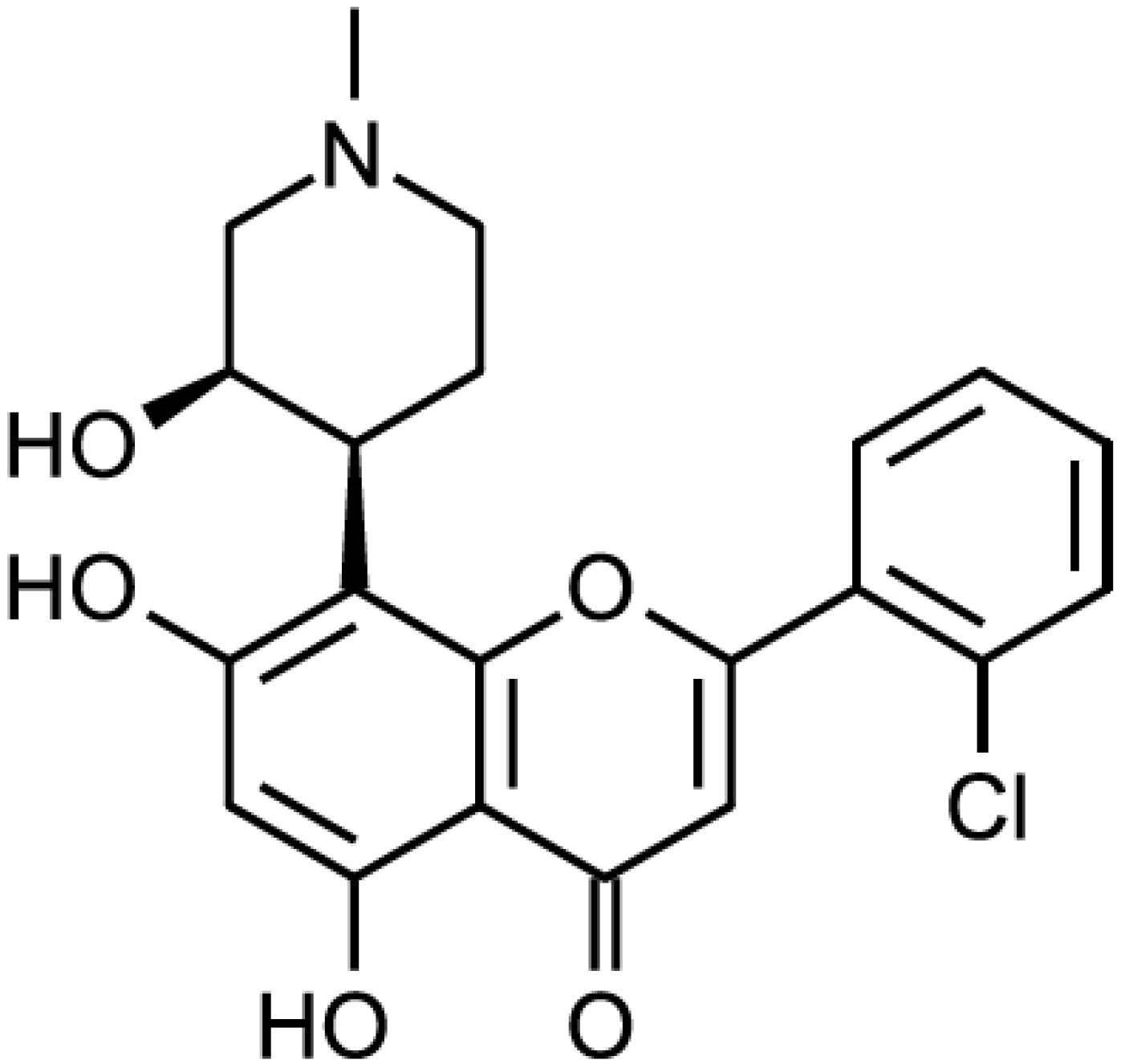{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Alternative Names | Kinases Inhibited | In Clinical Development Yes/No | Refs. |
|---|---|---|---|---|
| 3α-Amino-5α-androstane | CDK5 | No | [36] | |
| 7x | CDK4, ARK5 | No | [37] | |
| AG-024322 | CDK1, CDK2, CDK4 | Yes | [4] | |
| AMG 925 | CDK4, FLT3 | No | [38] | |
| AT7519 | CDK1, CDK2 | Yes | [39] | |
| AZD5438 | CDK1, CDK2, CDK4, CDK5, CDK6, CDK9 | Yes | [4] | |
| BAY 1000394 | CDK1, CDK2, CDK3, CDK4, CDK7, CDK9 | No | [40] | |
| BML-259 | CDK2, CDK5 | No | [4] | |
| Compound 1 | CDK4, ABL, FGFR1, FYN, KDR, LCK, LYN, SRC | No | [41] | |
| Compound 530 | CDK4, CDK4 | No | [42] | |
| CR8 | CDK2, CDK5 | No | [43] | |
| Dinaciclib | MK-7965, SCH 727965 | CDK1, CDK2, CDK5, CDK9 | Yes | [44] |
| F07#13 | CDK2, CDK9 | No | [45] | |
| Fascaplysin | CDK4, CDK6 | No | [4] | |
| Flavopiridol | L-868275, HMR-1275, alvocidib, NSC-649890 | CDK1, CDK2, CDK4, CDK7 | Yes | [4] |
| Kenpaullone | NSC 664704, 9-bromopaullone | CDK1, CDK2, CDK5 | No | [4] |
| LY2835219 | abemaciclib | CDK4, CDK6 | Yes | [46] |
| NBI1 | CDK2 | No | [47] | |
| NU2058 | CDK1, CDK2 | No | [4] | |
| Olomoucine | CDK1, CDK2, CDK5 | No | [4] | |
| P276-00 | CDK1 | Yes | [4] | |
| PD-0332991 | CDK4, CDK6 | Yes | [4] | |
| PHA-793887 | CDK1, CDK2, CDK4 | Yes (Stopped) | [48] | |
| Purvalanol A/B | CDK1, CDK2, CDK5 | No | [4] | |
| R547 | Ro-4584820 | CDK1, CDK2, CDK4 | Yes | [4] |
| RGB-286638 | Pan-CDK | No | [49] | |
| Roscovitine | CY-202, (R)-roscovitine, seliciclib | CDK2, CDK5 | Yes | [4] |
| Ryuvidine | CDK4 | No | [4] | |
| SNS-032 | BMS-387032 | CDK2, CDK7, CDK9 | Yes | [4] |
| SU 9516 | CDK1, CDK2 | No | [4] | |
| VMY-1-101 | CDK2, CDK5, CDK7 | No | [50] | |
| VMY-1-103 | CDK2, CDK5, CDK7 | No | [50] |
2. Advances in Preclinical Studies
| Inhibitor | Kinases Inhibited | Tested | Disease(s) | Refs. |
|---|---|---|---|---|
| Compound 1 | CDK4, ABL, FGFR1, FYN, KDR, LCK, LYN, SRC | In vitro | n.a. | [41] |
| NBI1 | CDK2 | In vitro, cell lines HCT116, HT29, T98G, A2780, MDA-MB-468, SKBr3, MCF-7, A549, Jurkat, HL60 and Saos-2 | colon carcinoma, glioblastoma, ovarian carcinoma, breast carcinoma, acute myeloid leukemia, lung carcinoma, osteosarcoma | [47,51] |
| Compound 530 | CDK2, CDK4 | In vitro | n.a. | [42] |
| F07#13 | CDK2, CDK9 | In vitro, mouse models | AIDS | [45] |
| 3α-Amino-5α-androstane | CDK5 | In vitro, Saccharomyces cerevisiae | n.a. | [36] |
| VMY-1-101 | CDK2, CDK5, CDK7 | In vitro, cell lines MDA-MB-231, MCF-7 | breast carcinoma | [50] |
| VMY-1-103 | CDK2, CDK5, CDK7 | In vitro, cell lines MDA-MB-231, MCF-7 | breast carcinoma | [50] |
| AMG 925 | CDK4, FLT3 | xenograft mouse model | acute myeloid leukemia | [38] |
| 7x | CDK4, ARK5 | In vitro, panel of human tumor cell lines, xenograft mouse model | various human cancers (cell lines), breast carcinoma (mouse model) | [37] |
| PD 0332991 | CDK4, CDK6 | mouse model | brainstem glioma | [52] |
| Flavopiridol | CDK1, CDK2, CDK4, CDK7 | xenograft mouse model | glioma | [53] |
| RGB-286638 | pan-CDK | xenograft mouse model | multiple myeloma | [49] |
| BAY 1000394 | CDK1, CDK2, CDK3, CDK4, CDK7, CDK9 | mouse model | breast carcinoma | [40] |
| Roscovitine | CDK2, CDK5 | mouse model | radiation-induced salivary gland dysfunction, renal and hepatic cystogenesis, pain | [54,55,57] |
| CR8 | CDK2, CDK5 | mouse model | renal and hepatic cystogenesis, traumatic brain injury | [43,55,58] |
| AZD5438 | CDK1, CDK2 | xenograft mouse model | non-small cell lung carcinoma | [39] |
| P276 | CDK4 | xenograft mouse model | pancreatic carcinoma | [56] |
3. Advances in clinical studies
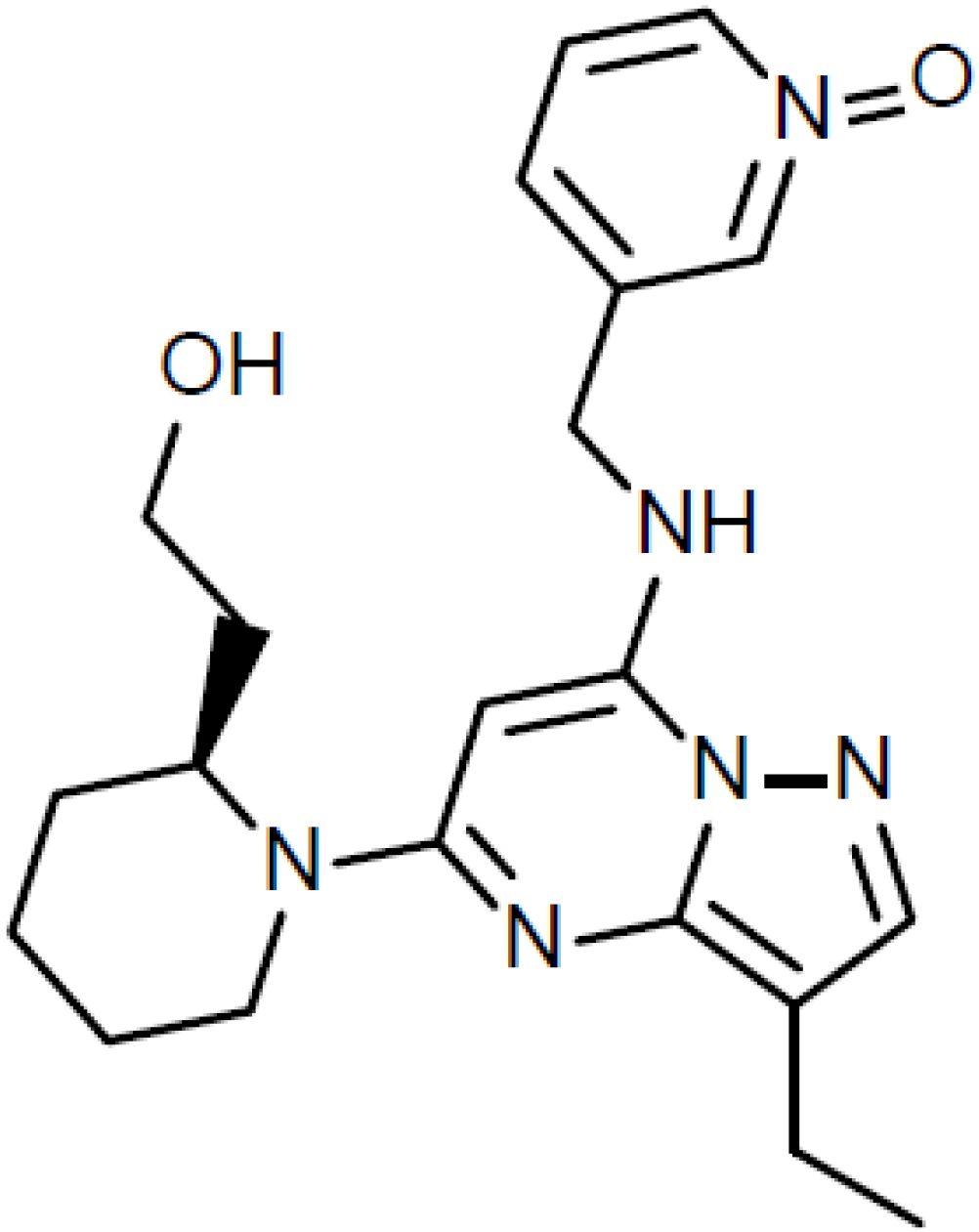
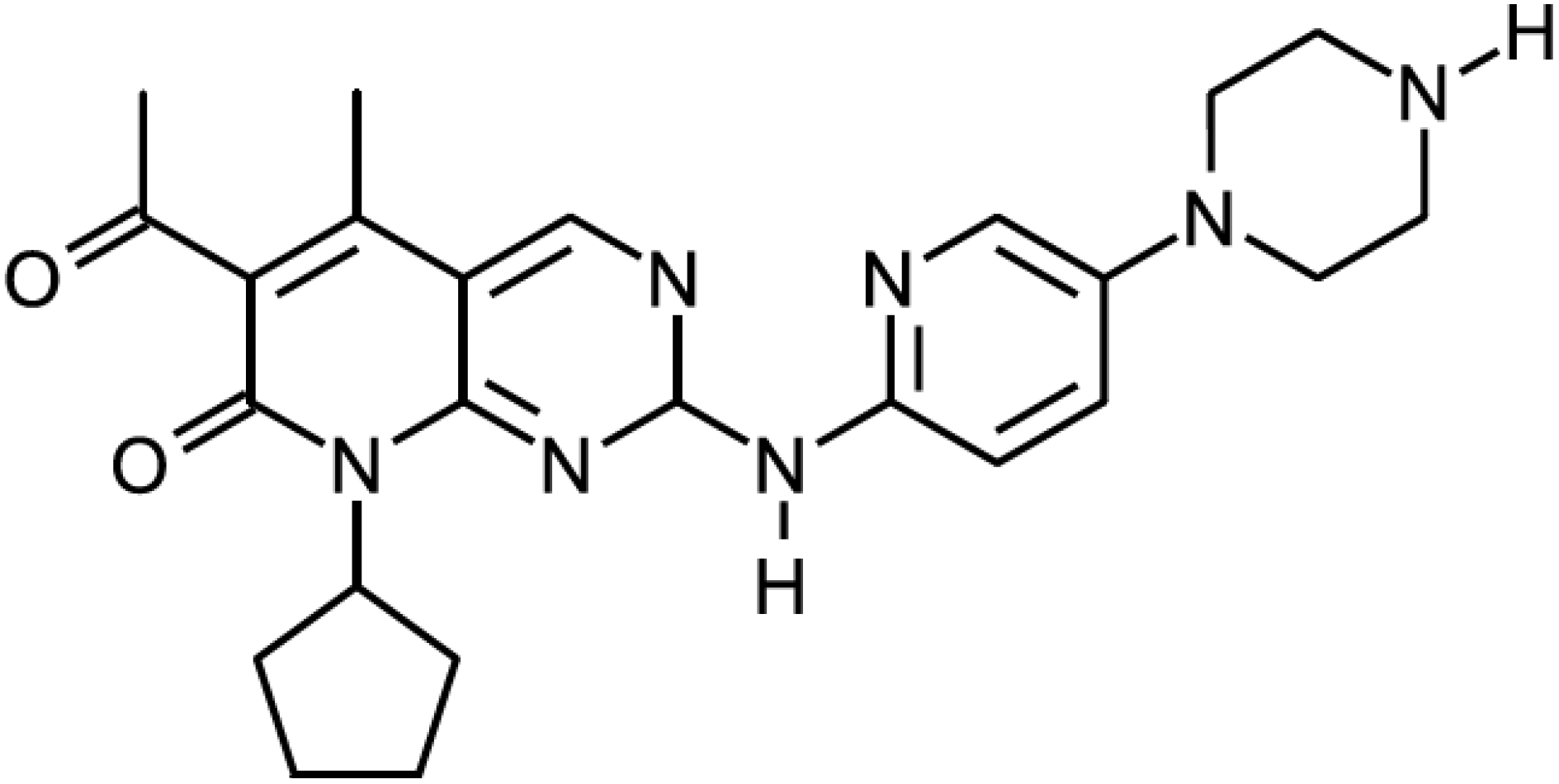
4. Perspectives
| Treatment | Clinical Trial Phase | Disease(s) | Response Rate | Adverse Effects | Refs. |
|---|---|---|---|---|---|
| Flavopiridol | Phase I | relapsed myeloma | 1/50 (2%) | diarrhea, cytopenias, transaminase elevation | [59] |
| Flavopiridol in combination with doxorubicin | Phase I | advanced sarcomas | 19/28 (68%) | neutropenia, leukopenia, febrile neutropenia | [60] |
| Flavopiridol in combination with imatinib mesylate | Phase I | Bcr-Abl + chronic myelogenous leukemia | 5/21 (24%) | anemia, leukopenia, lymphopenia, thrombocytopenia | [61] |
| Flavopiridol in combination with cisplatin | Phase II | platin-resistant ovarian and primary peritoneal carcinoma | 17/40 (43%) platin-resistant patients; 4/5 (80%) platin-sensitive patients | neutropenia, nausea, vomiting, fatigue, thrombosis, anemia | [62] |
| Flavopiridol in combination with cyclophosphamide and rituximab | Phase I | chronic lymphocytic leukemia | 7/9 (78%) | fatigue, electrolyte disturbances, diarrhea, abdominal discomfort, nausea/vomiting, liver dysfunction, anemia, leukopenia, neutropenia, thrombocytopenia | [63] |
| Flavopiridol | Phase I/II | chronic lymphocytic leukemia | 112/52 (46%) | n.a. | [64] |
| Flavopiridol | ? | chronic lymphocytic leukemia | 41/95 (43%) ≥70 years old; 10/21 (47%) <70 years old | tumor lysis syndrome, cytokine release syndrome, neutropenia, diarrhea, fatigue | [65] |
| Flavopiridol in combination with bortezomib | Phase I | refractory B-cell neoplasms | 7/16 (44%) | neutropenia, lymphopenia, and thrombocytopenia | [66] |
| Flavopiridol in combination with bortezomib | Phase I | Refractory indolent B-cell neoplasms | 13/39 (33%) | leukopenia, lymphopenia, neutropenia, thrombocytopenia, diarrhea, fatigue, sensory neuropathy | [67] |
| Dinaciclib in combination with aprepitant | Phase I | advanced malignancies | n.a. | no change in safety profile of dinaciclib | [44] |
| Dinaciclib | Phase I | advanced malignancies | 10/48 (21%) | nausea, anemia, decreased appetite and fatigue | [68] |
| Dinaciclib | Phase I | relapsed and/or refractory acute myeloid leukemia | 12/20 (60%) | diarrhea, fatigue, transaminitis, manifestations of tumor lysis syndrome; one patient deceased of acute renal failure | [69] |
| Dinaciclib vs. erlotinib | Phase II | non-small cell lung cancer | Not successful | neutropenia, leukopenia, vomiting, diarrhea | [70] |
| Dinaciclib vs. capecitabine | Phase II | advanced breast cancer | 2/7 (29%) (not superior to capecitabine) | neutropenia, leukopenia, increase in aspartate aminotransferase, febrile neutropenia | [71] |
| Dinaciclib vs. capecitabine | Phase I | chronic lymphocytic leukemia | 5/6 (83%) | hematological, digestive and metabolic; no dose-limiting toxicities | [72] |
| Dinaciclib | Phase I/II | relapsed multiple myeloma | 3/27 (11%) | leukopenia, thrombocytopenia, gastrointestinal symptoms, alopecia, fatigue | [74] |
| PD 0332991 | Phase I | advanced cancer | 10/37 (27%) | neutropenia, anemia, leukopenia, fatigue, nausea, diarrhea | [75] |
| PD 0332991 | Phase I | advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma | 19/29 (66%) | anemia, thrombocytopenia, neutropenia, febrile neutropenia | [76] |
| PD 0332991 | Phase II | advanced breast cancer | 20/37 (%) | neutropenia, leucopenia, lymphopenia, thrombocytopenia | [77] |
| PD 0332991 in combination with letrozole vs. letrozole alone | Phase II | advanced breast cancer | 87% vs. 57% (66 patients) | neutropenia, leukopenia, and fatigue | [78] |
| LY2835219 | Phase I | metastatic breast cancer | 33/47 (70%) | diarrhea, nausea, fatigue, neutropenia, vomiting, decreased platelet and white-blood cell counts | [46] |
| PHA-793887 | Phase I | solid tumors | n.a. | severe, dose-related hepatic toxicity | [48] |
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Diallo, A.; Prigent, C. The serine/threonine kinases that control cell cycle progression as therapeutic targets. Bull. Cancer 2011, 98, 1335–1345. [Google Scholar]
- Doonan, J.H.; Kitsios, G. Functional evolution of cyclin-dependent kinases. Mol. Biotechnol. 2009, 42, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Pines, J. Cyclins and cyclin-dependent kinases: Take your partners. Trends Biochem. Sci. 1993, 18, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Valius, M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [PubMed]
- Hengstschläger, M.; Braun, K.; Soucek, T.; Miloloza, A.; Hengstschläger-Ottnad, E. Cyclin-dependent kinases at the G1-S transition of the mammalian cell cycle. Mutat. Res. 1999, 436, 1–9. [Google Scholar] [CrossRef]
- Dhariwala, F.A.; Rajadhyaksha, M.S. An unusual member of the CDK family: CDK5. Cell. Mol. Neurobiol. 2008, 28, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, V.; Léopold, P. The cyclin C/CDK8 kinase. Prog. Cell Cycle Res. 1996, 2, 197–204. [Google Scholar]
- Napolitano, G.; Majello, B.; Lania, L. Role of cyclinT/CDK9 complex in basal and regulated transcription (review). Int. J. Oncol. 2002, 21, 171–177. [Google Scholar] [PubMed]
- Kasten, M.; Giordano, A. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene 2001, 20, 1832–1838. [Google Scholar] [CrossRef] [PubMed]
- Loyer, P.; Trembley, J.H.; Grenet, J.A.; Busson, A.; Corlu, A.; Zhao, W.; Kocak, M.; Kidd, V.J.; Lahti, J.M. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: Influence of cyclin L isoforms on splice site selection. J. Biol. Chem. 2008, 283, 7721–7732. [Google Scholar] [CrossRef]
- Duan, Y.; He, X.; Yang, H.; Ji, Y.; Tao, T.; Chen, J.; Hu, L.; Zhang, F.; Li, X.; Wang, H.; et al. Cyclin D3/CDK11(p58) complex involved in Schwann cells proliferation repression caused by lipopolysaccharide. Inflammation 2010, 33, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Kohoutek, J.; Blazek, D. Cyclin K goes with CDK12 and CDK13. Cell Div. 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, M.; Wang, W.; Chen, J. 14-3-3 Binding to Cyclin Y contributes to cyclin Y/CDK14 association. Acta Biochim. Biophys. Sin. Shanghai 2014, 46, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Kim, S.Y.; Kim, Y.J.; Chung, Y.H. ALS2CR7 (CDK15) attenuates TRAIL induced apoptosis by inducing phosphorylation of survivin Thr34. Biochem. Biophys. Res. Commun. 2014, 450, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Raymond, E. Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Acc. Chem. Res. 2003, 36, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Benson, C.; White, J.; de Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 daysevery 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, J.A.; Krailo, M.; Reid, J.M.; Ruben, S.L.; Ames, M.M.; Owen, W.; Reaman, G. Phase I clinical and pharmacokinetic study of Flavopiridol in children with refractory solid tumors: A children’s oncology group study. J. Clin. Oncol. 2005, 23, 9179–9186. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.A.; Lin, T.S.; Johnson, A.J.; Hurh, E.; Rozewski, D.M.; Farley, K.L.; Wu, D.; Blum, K.A.; Fischer, B.; Mitchell, S.M.; Moran, M.E.; et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009, 113, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Phelps, M.A.; Klisovic, R.B.; Rozewski, D.M.; Ni, W.; Albanese, K.A.; Rovin, B.; Kefauver, C.; Devine, S.M.; Lucas, D.M.; et al. Phase I clinical and pharmacokinetic study of a novel schedule of flavopiridol in relapsed or refractory acute leukemias. Haematologica 2010, 95, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- El-Rayes, B.F.; Gadgeel, S.; Parchment, R.; Lorusso, P.; Philip, P.A. A phase I study of Flavopiridol and docetaxel. Investig. New Drugs 2006, 24, 305–310. [Google Scholar] [CrossRef]
- Fornier, M.N.; Rathkopf, D.; Shah, M.; Patil, S.; O’Reilly, E.; Tse, A.N.; Hudis, C.; Lefkowitz, R.; Kelsen, D.P.; Schwartz, G.K. Phase I dose-Wnding study of weekly docetaxel followed by Flavopiridol for patients with advanced solid tumors. Clin. Cancer Res. 2007, 13, 5841–5846. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Kasimis, B.S.; Cogswell, J.; Schwarzenberger, P.; Shapiro, G.I.; Fidias, P.; Bukowski, R.M. Phase I study of flavopiridol in combination with paclitaxel and carboplatin in patients with nonsmall-cell lung cancer. Clin. Lung Cancer 2008, 9, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.; Dickson, M.A.; Feldman, D.R.; Carvajal, R.D.; Shah, M.A.; Wu, N.; Lefkowitz, R.; Gonen, M.; Cane, L.M.; Dials, H.J.; et al. Phase I study of Flavopiridol with oxaliplatin and Fluorouracil/leucovorin in advanced solid tumors. Clin. Cancer Res. 2009, 15, 7405–7411. [Google Scholar] [CrossRef] [PubMed]
- Fekrazad, H.M.; Verschraegen, C.F.; Royce, M.; Smith, H.O.; Chyi Lee, F.; Rabinowitz, I. A phase I study of flavopiridol in combination with gemcitabine and irinotecan in patients with metastatic cancer. Am. J. Clin. Oncol. 2010, 33, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Rathkopf, D.E.; Carvajal, R.D.; Grant, S.; Roberts, J.D.; Reid, J.M.; Ames, M.M.; McGovern, R.M.; Lefkowitz, R.A.; Gonen, M.; et al. A phase I pharmacokinetic study of pulse-dose vorinostat with flavopiridol in solid tumors. Investig. New Drugs 2011, 29, 1004–1012. [Google Scholar] [CrossRef]
- Karp, J.E.; Smith, B.D.; Resar, L.S.; Greer, J.M.; Blackford, A.; Zhao, M.; Moton-Nelson, D.; Alino, K.; Levis, M.J.; Gore, S.D.; et al. Phase 1 and pharmacokinetic study of bolus-infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood 2011, 117, 3302–3310. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Ruppert, A.S.; Johnson, A.J.; Fischer, B.; Heerema, N.A.; Andritsos, L.A.; Blum, K.A.; Flynn, J.M.; Jones, J.A.; Hu, W.; et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009, 27, 6012–6018. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Tse, A.; Shah, M.A.; Lefkowitz, R.A.; Gonen, M.; Gilman-Rosen, L.; Kortmansky, J.; Kelsen, D.P.; Schwartz, G.K.; O’Reilly, E.M. A phase II study of flavopiridol (Alvocidib) in combination with docetaxel in refractory, metastatic pancreatic cancer. Pancreatology 2009, 9, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and biology of fascaplysin, a potent marine-derived CDK-4 inhibitor. Mini Rev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [PubMed]
- Ryu, C.K.; Kang, H.Y.; Lee, S.K.; Nam, K.A.; Hong, C.Y.; Ko, W.G.; Lee, B.H. 5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents. Bioorg. Med. Chem. Lett. 2000, 10, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.A.; Pentimalli, F.; Esposito, L.; Giordano, A. ATP-noncompetitive CDK inhibitors for cancer therapy: An overview. Expert Opin. Investig. Drugs 2013, 22, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Corbel, C.; Wang, Q.; Bousserouel, H.; Hamdi, A.; Zhang, B.; Lozach, O.; Ferandin, Y.; Tan, V.B.; Guéritte, F.; Colas, P.; et al. First BRET-based screening assay performed in budding yeast leads to the discovery of CDK5/p25 interaction inhibitors. Biotechnol. J. 2011, 6, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Akula, B.; Cosenza, S.C.; Athuluridivakar, S.; Mallireddigari, M.R.; Pallela, V.R.; Billa, V.K.; Subbaiah, D.R.; Bharathi, E.V.; Vasquez-Del Carpio, R.; et al. Discovery of 8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile (7x) as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and AMPK-related kinase 5 (ARK5). J. Med. Chem. 2014, 57, 578–599. [Google Scholar] [CrossRef] [PubMed]
- Keegan, K.; Li, C.; Li, Z.; Ma, J.; Ragains, M.; Coberly, S.; Hollenback, D.; Eksterowicz, J.; Liang, L.; Weidner, M.; et al. Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Mol. Cancer Ther. 2014, 13, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, P.; Tumati, V.; Yu, L.; Chan, N.; Tomimatsu, N.; Burma, S.; Bristow, R.G.; Saha, D. AZD5438, an inhibitor of CDK1, 2, and 9, enhances the radiosensitivity of non-small cell lung carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e507–e514. [Google Scholar] [CrossRef] [PubMed]
- Lücking, U.; Jautelat, R.; Krüger, M.; Brumby, T.; Lienau, P.; Schäfer, M.; Briem, H.; Schulze, J.; Hillisch, A.; Reichel, A.; et al. The lab oddity prevails: Discovery of pan-CDK inhibitor (R)-S-cyclopropyl-S-(4-{[4-{[(1R,2R)-2-hydroxy-1-methylpropyl]oxy}-5-(trifluorome thyl)pyrimidin-2-yl]amino}phenyl)sulfoximide (BAY 1000394) for the treatment of cancer. Chem. Med. Chem. 2013, 8, 1067–1085. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.C.; Ngo, R.; Dai, K.; Li, C.; Liang, L.; Lee, J.; Emkey, R.; Eksterowicz, J.; Ventura, M.; Young, S.W.; et al. Development of a time-resolved fluorescence resonance energy transfer assay for cyclin-dependent kinase 4 and identification of its ATP-noncompetitive inhibitors. Anal. Biochem. 2012, 421, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Premnath, P.N.; Liu, S.; Perkins, T.; Abbott, J.; Anderson, E.; McInnes, C. Fragment based discovery of arginine isosteres through REPLACE: Towards non-ATP competitive CDK inhibitors. Bioorg. Med. Chem. 2014, 22, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Kabadi, S.V.; Stoica, B.A.; Hanscom, M.; Loane, D.J.; Kharebava, G.; Murray Ii, M.G.; Cabatbat, R.M.; Faden, A.I. CR8, a selective and potent CDK inhibitor, provides neuroprotection in experimental traumatic brain injury. Neurotherapeutics 2012, 9, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Mita, M.; Shapiro, G.I.; Poon, J.; Small, K.; Tzontcheva, A.; Kantesaria, B.; Zhu, Y.; Bannerji, R.; Statkevich, P. Effect of aprepitant on the pharmacokinetics of the cyclin-dependent kinase inhibitor dinaciclib in patients with advanced malignancies. Cancer Chemother. Pharmacol. 2012, 70, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Van Duyne, R.; Guendel, I.; Jaworski, E.; Sampey, G.; Klase, Z.; Chen, H.; Zeng, C.; Kovalskyy, D.; El Kouni, M.H.; Lepene, B.; et al. Effect of mimetic CDK9 inhibitors on HIV-1-activated transcription. J. Mol. Biol. 2013, 425, 812–829. [Google Scholar] [CrossRef] [PubMed]
- Metastatic breast cancer responds to CDK4/6 inhibitor. Available online: http://cancerdiscovery.aacrjournals.org/content/early/2014/04/22/2159-8290.CD-NB2014-062.full (accessed on 2 May 2014).
- Canela, N.; Orzáez, M.; Fucho, R.; Mateo, F.; Gutierrez, R.; Pineda-Lucena, A.; Bachs, O.; Pérez-Payá, E. Identification of an hexapeptide that binds to a surface pocket in cyclin A and inhibits the catalytic activity of the complex cyclin-dependent kinase 2-cyclin A. J. Biol. Chem. 2006, 281, 35942–35953. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Soria, J.C.; Anthoney, D.A.; Proctor, A.; Scaburri, A.; Pacciarini, M.A.; Laffranchi, B.; Pellizzoni, C.; Kroemer, G.; Armand, J.P.; et al. A first in man, phase I dose-escalation study of PHA-793887, an inhibitor of multiple cyclin-dependent kinases (CDK2, 1 and 4) reveals unexpected hepatotoxicity in patients with solid tumors. Cell Cycle 2011, 10, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Cirstea, D.; Hideshima, T.; Santo, L.; Eda, H.; Mishima, Y.; Nemani, N.; Hu, Y.; Mimura, N.; Cottini, F.; Gorgun, G.; et al. Small-molecule multi-targeted kinase inhibitor RGB-286638 triggers P53-dependent and -independent anti-multiple myeloma activity through inhibition of transcriptional CDKs. Leukemia 2013, 27, 2366–2375. [Google Scholar] [CrossRef] [PubMed]
- Yenugonda, V.M.; Deb, T.B.; Grindrod, S.C.; Dakshanamurthy, S.; Yang, Y.; Paige, M.; Brown, M.L. Fluorescent cyclin-dependent kinase inhibitors block the proliferation of human breast cancer cells. Bioorg. Med. Chem. 2011, 19, 2714–2725. [Google Scholar] [CrossRef] [PubMed]
- Orzáez, M.; Guevara, T.; Sancho, M.; Pérez-Payá, E. Intrinsic caspase-8 activation mediates sensitization of erlotinib-resistant tumor cells to erlotinib/cell-cycle inhibitors combination treatment. Cell Death Dis. 2012, 3, e415. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.L.; Misuraca, K.; Cordero, F.; Dobrikova, E.; Min, H.D.; Gromeier, M.; Kirsch, D.G.; Becher, O.J. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One 2013, 8, e77639. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Adachi, K.; Ohba, S.; Hirose, Y. The CDK inhibitor flavopiridol enhances temozolomide-induced cytotoxicity in human glioma cells. J. Neurooncol. 2013, 115, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.L.; Hill, G.A.; Klein, R.R.; Arnett, D.G.; Burd, R.; Limesand, K.H. Prevention of radiation-induced salivary gland dysfunction utilizing a CDK inhibitor in a mouse model. PLoS One 2012, 7, e51363. [Google Scholar] [CrossRef] [PubMed]
- Bukanov, N.O.; Moreno, S.E.; Natoli, T.A.; Rogers, K.A.; Smith, L.A.; Ledbetter, S.R.; Oumata, N.; Galons, H.; Meijer, L.; Ibraghimov-Beskrovnaya, O. CDK inhibitors R-roscovitine and S-CR8 effectively block renal and hepatic cystogenesis in an orthologous model of ADPKD. Cell Cycle 2012, 11, 4040–4046. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, D.; Periyasamy, G.; Ponnurangam, S.; Chakrabarti, D.; Sugumar, A.; Padigaru, M.; Weir, S.J.; Balakrishnan, A.; Sharma, S.; Anant, S. CDK-4 inhibitor P276 sensitizes pancreatic cancer cells to gemcitabine-induced apoptosis. Mol. Cancer Ther. 2012, 11, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gu, X.; Zhang, W.; Zhang, J.; Ma, Z. CDK5 inhibitor roscovitine alleviates neuropathic pain in the dorsal root ganglia by downregulating N-methyl-d-aspartate receptor subunit 2A. Neurol. Sci. 2014, 35, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Kabadi, S.V.; Stoica, B.A.; Loane, D.J.; Luo, T.; Faden, A.I. CR8, a novel inhibitor of CDK, limits microglial activation, astrocytosis, neuronal loss, and neurologic dysfunction after experimental traumatic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, C.C.; Poi, M.; Bowers, M.A.; Zhao, W.; Phelps, M.A.; Benson, D.M.; Kraut, E.H.; Farag, S.; Efebera, Y.A.; Sexton, J.; et al. A phase I trial of flavopiridol in relapsed multiple myeloma. Cancer Chemother. Pharmacol. 2014, 73, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; D’Adamo, D.R.; Dickson, M.A.; Keohan, M.L.; Carvajal, R.D.; Maki, R.G.; de Stanchina, E.; Musi, E.; Singer, S.; Schwartz, G.K. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: Preclinical investigations and results of a phase I dose-escalation clinical trial. Clin. Cancer Res. 2012, 18, 2638–2647. [Google Scholar] [CrossRef]
- Bose, P.; Perkins, E.B.; Honeycut, C.; Wellons, M.D.; Stefan, T.; Jacobberger, J.W.; Kontopodis, E.; Beumer, J.H.; Egorin, M.J.; Imamura, C.K.; et al. Phase I trial of the combination of flavopiridol and imatinib mesylate in patients with Bcr-Abl+ hematological malignancies. Cancer Chemother. Pharmacol. 2012, 69, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Peethambaram, P.P.; Oberg, A.L.; Maples, W.; Groteluschen, D.L.; Boente, M.; Burton, J.K.; Gomez Dahl, L.C.; Tibodeau, J.D.; Isham, C.R.; et al. A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol. Oncol. 2012, 127, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.M.; Ruppert, A.S.; Maddocks, K.; Andritsos, L.; Baiocchi, R.; Jones, J.; Johnson, A.J.; Smith, L.L.; Zhao, Y.; Ling, Y.; et al. Cyclophosphamide, alvocidib (flavopiridol), and rituximab, a novel feasible chemoimmunotherapy regimen for patients with high-risk chronic lymphocytic leukemia. Leuk. Res. 2013, 37, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Lozanski, G.; Ruppert, A.S.; Lozanski, A.; Blum, K.A.; Jones, J.A.; Flynn, J.M.; Johnson, A.J.; Grever, M.R.; Heerema, N.A.; et al. Outcome of patients with relapsed or refractory chronic lymphocytic leukemia treated with flavopiridol: Impact of genetic features. Leukemia 2012, 26, 1442–1444. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.M.; Ruppert, A.S.; Blum, K.; Jones, J.; Flynn, J.M.; Johnson, A.J.; Ji, J.; Phelps, M.A.; Grever, M.R.; Byrd, J.C. Flavopiridol treatment of patients aged 70 or older with refractory or relapsed chronic lymphocytic leukemia is a feasible and active therapeutic approach. Haematologica 2012, 97, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Holkova, B.; Perkins, E.B.; Ramakrishnan, V.; Tombes, M.B.; Shrader, E.; Talreja, N.; Wellons, M.D.; Hogan, K.T.; Roodman, G.D.; Coppola, D.; et al. Phase I trial of bortezomib (PS-341; NSC 681239) and alvocidib (flavopiridol; NSC 649890) in patients with recurrent or refractory B-cell neoplasms. Clin. Cancer Res. 2011, 17, 3388–3397. [Google Scholar] [CrossRef] [PubMed]
- Holkova, B.; Kmieciak, M.; Perkins, E.B.; Bose, P.; Baz, R.; Roodman, G.D.; Stuart, R.K.; Ramakrishnan, V.; Wan, W.; Peer, C.J.; et al. Phase I trial of bortezomib and “non-hybrid”(bolus) infusion schedule of alvocidib (flavopiridol) in patients with recurrent or refractory indolent B-cell neoplasms. Clin. Cancer Res. 2014. [Google Scholar] [PubMed]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.M.; Statkevich, P.; Yao, S.L.; Bannerji, R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11. [Google Scholar] [CrossRef]
- Gojo, I.; Sadowska, M.; Walker, A.; Feldman, E.J.; Iyer, S.P.; Baer, M.R.; Sausville, E.A.; Lapidus, R.G.; Zhang, D.; Zhu, Y.; et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother. Pharmacol. 2013, 72, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.J.; Nemunaitis, J.; Joy, A.A.; Martin, J.C.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Yao, S.L.; Zhu, Y.; Zhou, H.; et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer 2014, 83, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.M.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.L.; Small, K.; et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Fabre, C.; Gobbi, M.; Ezzili, C.; Zoubir, M.; Sablin, M.P.; Small, K.; Im, E.; Shinwari, N.; Zhang, D.; Zhou, H.; et al. Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients. Cancer Chemother. Pharmacol. 2014. [Google Scholar] [PubMed]
- Guha, M. Cyclin-dependent kinase inhibitors move into Phase III. Nat. Rev. Drug Discov. 2012, 11, 892–894. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.R.; Chng, W.J.; Zonder, J.A.; Callander, N.; Roy, V.; Furth, B.; Erlichman, C.; Stewart, K. Phase 1/2 Trial of a novel CDK inhibitor dinaciclib (SCH727965) in patients with relapsed multiple myeloma demonstrates encouraging single agent activity. In Proceedings of the 54th ASH Meeting, Atlanta, GA, USA, 9 December 2012.
- Flaherty, K.T.; Lorusso, P.M.; Demichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Tap, W.D.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Antonescu, C.R.; Landa, J.; Qin, L.X.; Rathbone, D.D.; Condy, M.M.; et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 2013, 31, 2024–2028. [Google Scholar] [CrossRef] [PubMed]
- DeMichele, A.; Sanders Clark, A.; Heitjan, D.; Randolph, S.; Gallagher, M.; Lal, P.; Feldman, M.D.; Zhang, P.J.; Schnader, A.; Zafman, K.; et al. A phase II trial of an oral CDK 4/6 inhibitor, PD0332991, in advanced breast cancer. In Proceedings of the 2013 ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2013.
- Finn, R.S.; Crown, J.P.; Boer, K.; Lang, I.; Parikh, R.J.; Breazna, A.; Ho, S.N.; Kim, S.T.; Randolph, S.; Slamon, D.J. Results of a randomized phase 2 study of PD 0332991, a Cyclin-Dependent Kinase (CDK) 4/6 inhibitor, in combination with letrozole vs. letrozole alone for first-line treatment of ER+, HER2– advanced breast cancer (TRIO-18). In Proceedings of the AACR Annual Meeting 2014, San Diego, CA, USA, 6 April 2014.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers 2014, 6, 2224-2242. https://doi.org/10.3390/cancers6042224
Cicenas J, Kalyan K, Sorokinas A, Jatulyte A, Valiunas D, Kaupinis A, Valius M. Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers. 2014; 6(4):2224-2242. https://doi.org/10.3390/cancers6042224
Chicago/Turabian StyleCicenas, Jonas, Karthik Kalyan, Aleksandras Sorokinas, Asta Jatulyte, Deividas Valiunas, Algirdas Kaupinis, and Mindaugas Valius. 2014. "Highlights of the Latest Advances in Research on CDK Inhibitors" Cancers 6, no. 4: 2224-2242. https://doi.org/10.3390/cancers6042224
APA StyleCicenas, J., Kalyan, K., Sorokinas, A., Jatulyte, A., Valiunas, D., Kaupinis, A., & Valius, M. (2014). Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers, 6(4), 2224-2242. https://doi.org/10.3390/cancers6042224