Drug Resistance in Cancer: An Overview


{kind=link}
{kind=link}
Abstract
:1. Introduction
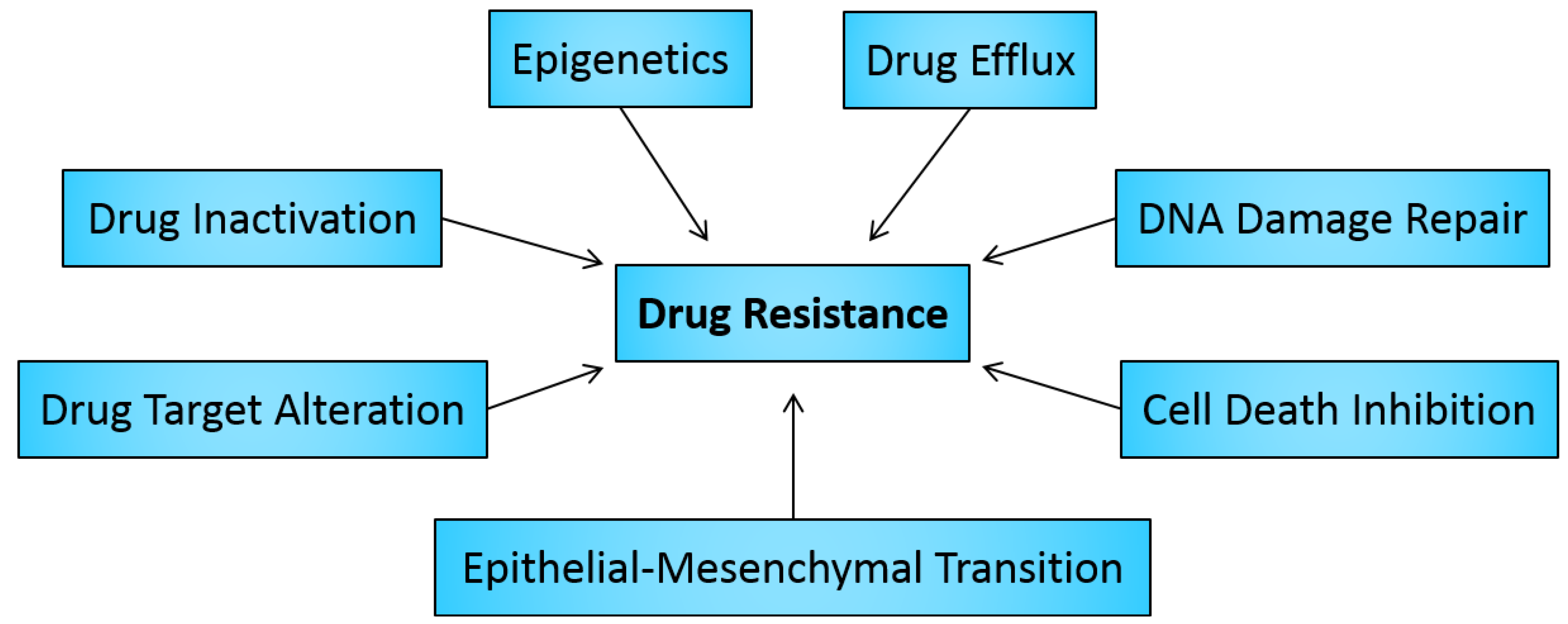
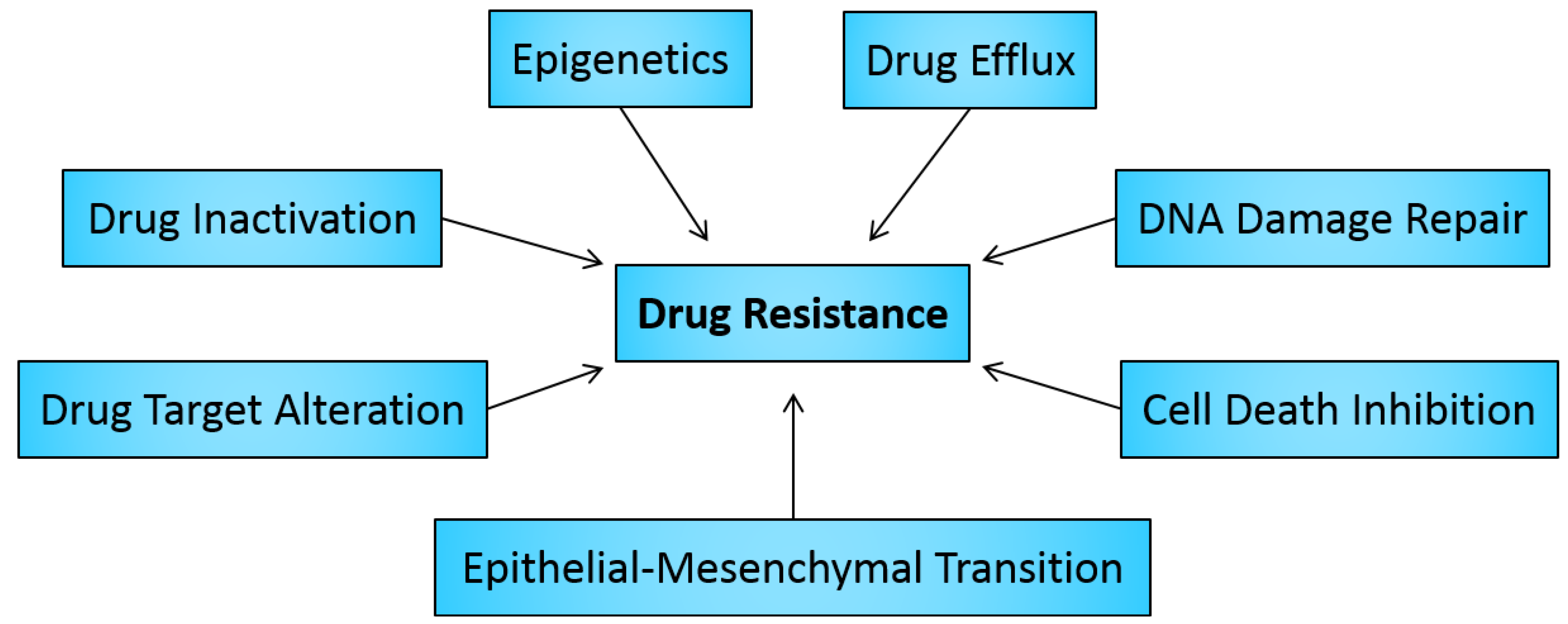
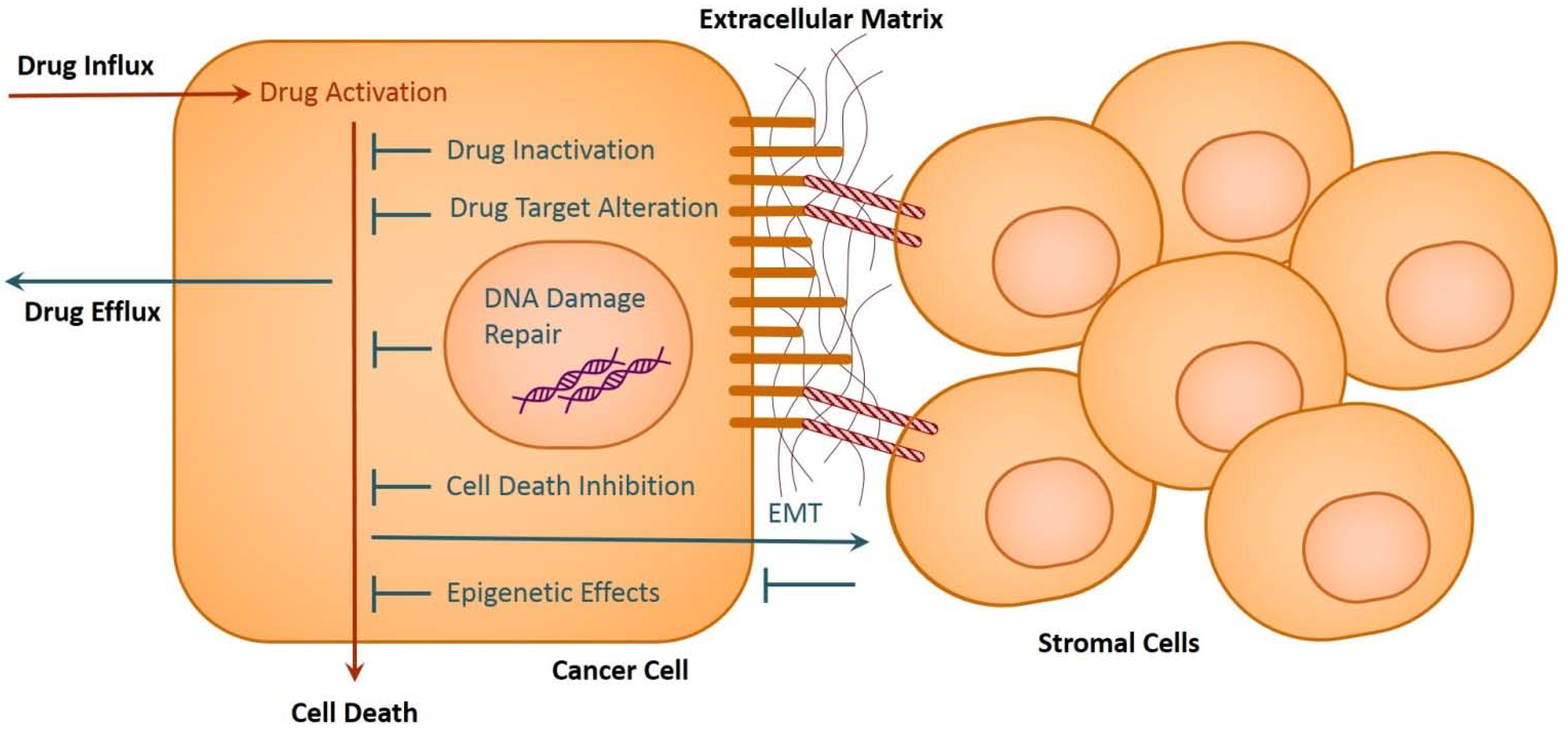
2. Drug Resistance in Cancer
2.1. Drug Inactivation
2.2. Alteration of Drug Targets
2.3. Drug Efflux
2.4. DNA Damage Repair
2.5. Cell Death Inhibition
2.6. Epithelial-Mesenchymal Transition and Metastasis

2.7. Cancer Cell Heterogeneity
3. Role of Epigenetics in Cancer Drug Resistance
4. Conclusions
Abbreviations
| EMT | epithelial-mesenchymal transition |
| AraC | cytarabine |
| CYP | cytochrome p450 |
| GST | glutathione-S-transferase |
| UGT | uridine diphospho-glucuronosyltransferase |
| TP53 | tumor protein p53 |
| Apaf-1 | apoptotic protease activating factor 1 |
| MAPK | mitogen-activated protein kinase |
| EGFR | epidermal growth factor receptor |
| HER2 | human epidermal growth factor receptor 2 |
| TS | thymidylate synthase |
| FdUMP | fluorodeoxyuridine monophosphate |
| CH2THF | 5,10-methylenetetrahydrofolate |
| PTEN | phosphatase and tensin homolog |
| IGF1R | insulin-like growth factor 1 receptor |
| CML | chronic myeloid leukemia |
| BCR-ABL | break point cluster-Abelson |
| Pgp | P-glycoprotein |
| ER | estrogen receptor |
| ABC | ATP-binding cassette |
| MDR1 | multidrug resistance protein 1 |
| MRP1 | multidrug resistance-associated protein 1 |
| BCRP | breast cancer resistance protein |
| ERK | extracellular signal-regulated kinases |
| DDR | DNA damage response |
| MGMT | O6-methylguanine DNA methyltransferase |
| BCL-2 | B-cell lymphoma 2 |
| TRAIL | tumor necrosis factor related apoptosis-inducing ligand |
| HDACi | histone deacetylase inhibitors |
| hMLH1 | human mutL homolog 1 |
| DAC | 2'-deoxy-5-azacytiding |
| RFC | reduced folate carrier |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Sampath, D.; Cortes, J.; Estrov, Z.; Du, M.; Shi, Z.; Andreeff, M.; Gandhi, V.; Plunkett, W. Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood 2006, 107, 2517–2574. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.; Doherty, M.M. Tumoral drug metabolism: Overview and its implications for cancer therapy. J. Clin. Oncol. 2005, 23, 205–229. [Google Scholar] [CrossRef] [PubMed]
- Plastaras, J.; Guengerich, F.; Nebert, D.; Marnett, L. Xenobiotic-metabolizing cytochromes P450 convert prostaglandin endoperoxide to hydroxyheptadecatrienoic acid and the mutagen, malondialdehyde. J. Biol. Chem. 2000, 275, 11784–11790. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; He, M.; Liu, H.; Wrighton, S.; Wang, L.; Guo, B.; Li, C. Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab. Dispos. 2007, 35, 1292–1300. [Google Scholar]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar]
- Mehta, K.; Fok, J.Y. Targeting transglutaminase-2 to overcome chemoresistance in cancer cells. In Drug Resistance in Cancer Cells; Mehta, K., Bates, S.E., Siddik, Z.H., Eds.; Springer: New York, NY, USA, 2009; pp. 95–114. [Google Scholar]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 tumor suppressor gene. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Aas, T.; Børresen, A.; Geisler, S.; Smith-Sørensen, B.; Johnsen, H.; Varhaug, J.; Akslen, L.; Lønning, P. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996, 2, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Soengas, M.S.; Alarcón, R.M.; Yoshida, H.; Giaccia, A.J.; Hakem, R.; Mak, T.W.; Lowe, S.W. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 1999, 284, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anticancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Manolitsas, T.P.; Englefield, P.; Eccles, D.M.; Campbell, I.G. No association of a 306 bp insertion polymorphism in the progesterone receptor gene with ovarian and breast cancer. Br. J. Cancer 1997, 75, 1397–1399. [Google Scholar] [CrossRef] [PubMed]
- Cumming, R.C.; Lightfoot, J.; Beard, K.; Youssoufian, H.; O’Brien, P.J.; Buchwald, M. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat. Med. 2001, 7, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. 2013, 13, 714–726. [Google Scholar]
- Gagnon, J.; Bernard, O.; Villeneuve, L.; Têtu, B.; Guillemette, C. Irinotecan inactivation is modulated by epigenetic silencing of UGT1A1 in colon cancer. Clin. Cancer Res. 2006, 12, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, A.A. Cellular mechanisms of multidrug resistance of tumor cells. Biochemistry (Mosc.) 2000, 65, 95–106. [Google Scholar]
- Hinds, M.; Deisseroth, K.; Mayes, J.; Altschuler, E.; Jansen, R.; Ledley, F.; Zwelling, L. Identification of a point mutation in the topoisomerase II gene from a human leukemia cell line containing an amsacrine resistant form of topoisomerase II. Cancer Res. 1991, 51, 4729–4731. [Google Scholar] [PubMed]
- Zwelling, L.; Hinds, M.; Chan, D.; Mayes, J.; Sie, K.; Parker, E.; Silberman, L.; Radcliffe, A.; Beran, M.; Blick, M. Characterization of an amsacrine-resistant line of human leukemia cells. Evidence for a drug resistant form of topoisomerase II. J. Biol. Chem. 1989, 264, 16411–16420. [Google Scholar]
- Slamon, D.; Godolphin, W.; Jones, L.; Holt, J.; Wong, S.; Keith, D.; Levin, W.; Stuart, S.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 4905, 707–712. [Google Scholar] [CrossRef]
- Slamon, D.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar]
- Bell, D.; Gore, I.; Okimoto, R.; Godin-Heymann, N.; Sordella, R.; Mulloy, R.; Sharma, S.; Brannigan, B.; Mohapatra, G.; Settleman, J.; et al. Inherited susceptibility to lung cancer may be associated with the T790 drug resistance mutation in EGFR. Nat. Genet. 2005, 37, 1315–1316. [Google Scholar] [PubMed]
- Kobyashi, S.; Boggon, T.; Dayaram, T.; Janne, P.; Kocher, O.; Meyerson, M.; Johnson, B.; Eck, M.; Tenen, D.; Halmos, B. EGFR mutation and resistance of non-small cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 7215, 971–974. [Google Scholar] [CrossRef]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Palmberg, C.; Koivisto, P.; Hyytinen, E.; Isola, J.; Visakorpi, T.; Kallioniemi, O.; Tammela, T. Androgen receptor gene amplification in a recurrent prostate cancer after monotherapy with the nonsteroidal potent antiandrogen Casodex (bicalutamide) with a subsequent favorable response to maximal androgen blockade. Eur. J. Urol. 1997, 31, 216–219. [Google Scholar]
- Dieras, V.; Vincent-Salomon, A.; Degeorges, A.; Beuzeboc, P.; Mignot, L.; de Cremoux, P. Trastuzumab (Herceptin) and breast cancer: Mechnisms of resistance. Bull Cancer 2007, 94, 259–266. [Google Scholar] [PubMed]
- Berns, K.; Horlings, H.; Hennessy, B.; Madiredjo, M.; Hijmans, M.; Beelen, K.; Linn, S.; Gonzalez-Angulo, A.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Browne, B.C.; Crown, J.; Venkatesan, N.; Duffy, M.J.; Clynes, M.; Slamon, D.; O’Donovan, N. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann. Oncol. 2011, 22, 68–73. [Google Scholar] [PubMed]
- Razis, E.; Bobos, M.; Kotoula, V.; Eleftheraki, A.G.; Kalofonos, H.P.; Pavlakis, K.; Papakostas, P.; Aravantinos, G.; Rigakos, G.; Efstratiou, I.; et al. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res. Treat. 2011, 128, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR–ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, H.A.; Asmaa, M.J.; Yong, A.C.; Asan, J.M.; Hassan, R.; Johan, M.F. Silencing of suppressor of cytokine signaling-3 due to methylation results in phosphorylation of STAT3 in imatinib resistant BCR-ABL positive chronic myeloid leukemia cells. Asian Pac. J. Cancer Prev. 2014, 15, 4555–4561. [Google Scholar]
- Hirayama, C.; Watanabe, H.; Nakashima, R.; Nanbu, T.; Hamada, A.; Kuniyasu, A.; Nakayama, H.; Kawaguchi, T.; Saito, H. Constitutive overexpression of P-glycoprotein, rather than breast cancer resistance protein or organic cation transporter 1, contributes to acquisition of imatinib-resistance in K562 cells. Pharm. Res. 2008, 25, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Nambu, T.; Araki, N.; Nakagawa, A.; Kuniyasu, A.; Kawaguchi, T.; Hamada, A.; Saito, H. Contribution of BCR-ABL-independent activation of ERK1/2 to acquired imatinib resistance in K562 chronic myeloid leukemia cells. Cancer Sci. 2010, 101, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Massarweh, S.; Osborne, C.K.; Creighton, C.J.; Qin, L.; Tsimelzon, A.; Huang, S.; Weiss, H.; Rimawi, M.; Schiff, R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Roth, C. Structure of MsbA from E. coli: A homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 2001, 293, 1793–1800. [Google Scholar]
- Sauna, Z.; Ambudkar, S. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J. Biol. Chem. 2001, 276, 11653–11661. [Google Scholar]
- Borst, P.; Elferink, O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.; Smit, J.; van Tellingen, O.; Beijnen, J.; Wagenaar, E.; van Deemter, L.; Mol, C.; van der Valk, M.; Robanus-Maandag, R.; te Riele, H.; et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Szakas, G.; Annereau, J.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.; Reinhold, W.; Guo, Y.; Kruh, G.; Reimers, M.; et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, C.; Ahlin, G.; Seithel, A.; Artursson, P.; Ungell, A.; Karlsson, J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab. Dispos. 2007, 35, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Abolhoda, A.; Wilson, A.; Ross, H.; Danenberg, P.V.; Burt, M.; Scotto, K.W. Rapid activation of MDR1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clin. Cancer Res. 1999, 5, 3352–3356. [Google Scholar] [PubMed]
- Haber, M.; Smith, J.; Bordow, S.; Flemming, C.; Cohn, S.; London, W.; Marshall, G.; Norris, M. Association of high-level MRP1 expression with poor clinical outcome in a large prospective study of neuroblastoma. J. Clin. Oncol. 2006, 24, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Yanase, K.; Tsukahara, S.; Asada, S.; Ishikawa, E.; Imai, Y.; Sugimoto, Y. Gefitinib reverses breast cancer resistance protein-mediated drug resistance. Mol. Cancer Ther. 2004, 3, 1119–1125. [Google Scholar] [PubMed]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Ishikawa, E.; Asada, S.; Sugimoto, Y. Estrogenmediated post transcriptional down-regulation of breast cancer resistance protein/ABCG2. Cancer Res. 2005, 65, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, K.; Tsukahara, S.; Mitsuhashi, J.; Katayama, K.; Sugimoto, Y. Estrogen-mediated post transcriptional downregulation of P-glycoprotein in MDR1-transduced human breast cancer cells. Cancer Sci. 2006, 97, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Yoshioka, S.; Tsukahara, S.; Mitsuhashi, J.; Sugimoto, Y. Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol. Cancer Ther. 2007, 6, 2092–2102. [Google Scholar] [PubMed]
- Fukuyo, Y.; Hunt, C.R.; Horikoshi, N. Geldanamycinand its anticancer activities. Cancer Lett. 2010, 290, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, L.; Favaretto, A.; Rosell, R. Platinum drugs and DNA repair mechanism in lung cancer. Anticancer Res. 2014, 34, 493–502. [Google Scholar] [PubMed]
- Olaussen, K.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Helmut, P.; et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Selvakumaran, M.; Pisarcik, D.; Bao, R.; Yeung, A.; Hamilton, T. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003, 63, 1311–1316. [Google Scholar] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetic lesions causing genetic lesions in human cancer: Promoter hypermethylation of DNA repair genes. Eur. J. Cancer 2000, 36, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Goode, E.; Ulrich, C.; Potter, J. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 1513–1530. [Google Scholar] [PubMed]
- Maier, P.; Spier, I.; Laufs, S.; Veldwijk, M.R.; Fruehauf, S.; Wenz, F.; Zeller, W.J. Chemoprotection of human hematopoietic stem cells by simultaneous lentiviral overexpression of multidrug resistance 1 and O(6)-methylguanine-DNA methyltransferase(P140K). Gene Ther. 2010, 17, 389–399. [Google Scholar] [CrossRef]
- Blanc, J.L.; Wager, M.; Guilhot, J.; Kusy, S.; Bataille, B.; Chantereau, T.; Lapierre, F.; Larsen, C.J.; Karayan-Tapon, L. Correlation of clinical features and methylation status of MGMT gene promoter in glioblastomas. J. Neurooncol. 2004, 68, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.; Fishel, M.; Holleran, J.; Kasza, K.; Kelley, M.; Egorin, M.; Dolan, M. Enhancement of cisplatin cytotoxicity by O6-benzylguanine involves endoplasmic reticulum stress. J. Pharmacol. Exp. Ther. 2008, 327, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Gegi, M.; Diserens, A.; Gorlia, T.; Hamou, M.; de Tribolet, N.; Weller, M.; Kros, J.; Hainfellner, J.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, R.; Chen, W. Correlation of promoter methylation in MGMT gene with glioma risk and prognosis: A meta-analysis. Mol. Neurobiol. 2014. [Google Scholar] [CrossRef]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl. Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.; Smit, E.; Khayat, D.; Besse, B.; Yang, X.; Hsu, C.; Reese, D.; Wiezorek, J.; Blackhall, F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumanb in patients with advanced non-squamous non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Mataga, M.; Rosenthal, S.; Heerboth, S.; Devalapalli, A.; Kokolus, S.; Evans, L.R.; Longacre, M.; Housman, G.; Sarkar, S. Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitors. Anticancer Res. 2012, 32, 2523–2530. [Google Scholar] [PubMed]
- Sarkar, S.; Faller, D.V. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleoties 2011, 21, 47–53. [Google Scholar] [CrossRef]
- Sarkar, S.; Faller, D.V. Telomere-homilogous G-rich oligonucleotides sensitize human ovarian cancer cells by combination therapy. Nucleic Acid Ther. 2013, 23, 167–174. [Google Scholar] [PubMed]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anticancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, e370. [Google Scholar]
- Cook, K.L.; Wärri, A.; Soto-Pantoja, D.R.; Clarke, P.A.G.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Cai, X.; Fan, D. Roles of epithelial-mesenchymal transition in cancer drug resistance. Curr. Cancer Drug Targets 2013, 13, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.; Brueckmann, I.; Scheel, C.; Kaestli, A.; Wiggins, P.; Rodrigues, L.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously covert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.; Weinberg, R. A perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar]
- Byler, S.; Goldgar, S.; Heerboth, S.; Leary, M.; Housman, G.; Moulton, K.; Sarkar, S. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014, 34, 1071–1077. [Google Scholar] [PubMed]
- Byler, S.; Sarkar, S. Do epigenetic drug treatments hold the key to killing cancer progenitor cells? Epigenomics 2014, 6, 161–165. [Google Scholar] [CrossRef]
- Lenisak, D.; Xu, Y.; Deschenes, J.; Lai, R.; Thoms, J.; Murray, D.; Gosh, S.; Mackey, J.R.; Sabri, S.; Abdulkarim, B. Beta1-integrin circumvents the antiproliferative effects of trastuzumab in human epidermal growth factor receptor-2-positive breast cancer. Cancer Res. 2009, 69, 8620–8628. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Svoboda, M.; de-Beaumont, R.; Freedman, A. The role of AKT and RAFTK in beta1 integrin mediated survival of precursor B-acute lymphoblastic leukemia cells. Leuk. Lymphoma 2002, 43, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L., 3rd; Sweeney, C. Co-opted integrin signaling in ErbB2-induced mammary tumor progression. Cancer Cell 2006, 10, 93–95. [Google Scholar]
- Bates, R.C.; Mercurio, A.M. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol. Ther. 2005, 4, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. β3 integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, R42. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [PubMed]
- Witz, I.P. The selectin-selectin ligand axis in tumor progression. Cancer Metastasis Rev. 2008, 27, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.A.; Canovas, D.; Bird, N.C. The role of cell adhesion molecules in the progression of colorectal cancer and the development of liver metastasis. Cell. Signal. 2009, 21, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Laubli, H.; Borsig, L. Selectins promote tumor metastasis. Semin. Cancer Biol. 2010, 20, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Gerger, A.; Zhang, W.; Hanna, D.L.; Yang, D.; Winder, T.; Wakatsuki, T.; Labonte, M.J.; Stintzing, S.; Volz, N.; et al. Plastin polymorphisms predict gender- and stage-specific colon cancer recurrence after adjuvant chemotherapy. Mol. Cancer Ther. 2014, 13, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Bégué, E.; Jean-Louis, F.; Bagot, M.; Jauliac, S.; Cayuela, J.M.; Laroche, L.; Parquet, N.; Bachelez, H.; Bensussan, A.; Courtois, G.; et al. Inducible expression and pathophysiologic functions of T-plastin in cutaneous T-cell lymphoma. Blood 2012, 120, 143–154. [Google Scholar]
- Staussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumor micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Parkin, B.; Ouillette, P.; Li, Y.; Keller, J.; Lam, C.; Roulston, D.; Li, C.; Shedden, K.; Malek, S.N. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood 2013, 121, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Krasnitz, A.; Rodgers, L.; Cook, K.; Meth, J.; Kendall, J.; Riggs, M.; Eberling, Y.; Troge, J.; Grubor, V.; et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010, 20, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.; Yachida, S.; Mudie, L.; Stephens, P.; Pleasance, E.; Stebbings, L.; Morsberger, L.; Latimer, C.; McLaren, S.; Lin, M.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; El-Osta, A. The rise of DNA methylation and the importance of chromatin on multidrug resistance in cancer. Exp. Cell Res. 2003, 290, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Kantharidis, P.; El-Oska, A.; de Silva, M.; Wall, D.M.; Hu, X.F.; Slater, A.; Nadalin, G.; Parkin, J.D.; Zalcberg, J.R. Altered methylation of the human MDR1 promoter is associated with acquired multidrug resistance. Clin. Cancer Res. 1997, 3, 2025–2032. [Google Scholar] [PubMed]
- Plumb, J.A.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2'-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044. [Google Scholar] [PubMed]
- Arnold, C.N.; Goel, A.; Boland, C.R. Role of MLH1 promoter hypermethylation in drug resistance to 5-flurouracil in colorectal cancer cell lines. Int. J. Cancer 2003, 106, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Bearzatto, A.; Szadkowski, M.; Macpherson, P.; Jiricny, J.; Karran, P. Epigenetic regulation of the MGMT and hMSH6 DNA repair genes in cells resistant to methylating agents. Cancer Res. 2000, 60, 3262–3270. [Google Scholar]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Worm, J.; Kirkin, A.F.; Dzhandzhugazyan, K.N.; Guldberg, P. Methylation-dependent silencing of the reduced folate carrier gene in inherently methotrexate-resistant human breast cancer cells. J. Biol. Chem. 2001, 276, 39990–40000. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.G.; Kim, S.J.; Chung, K.W.; Noh, D.Y.; Kwon, Y.; Lee, E.S.; Kang, H.S. Tamoxifen-resistant breast cancer show less frequent methylation of the estrogen receptor beta but not the estrogen receptor alpha gene. J. Mol. Med. 2005, 83, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Pick, M.; Lage, H.; Schadendorf, D.; Kaina, B. Acquired resistance of melanoma cells to the antineoplastic agent fotemustine is caused by reactivation of the DNA repair gene MGMT. Int. J. Cancer 2001, 92, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Izbicka, E.; MacDonald, J.R.; Davidson, K.; Lawrence, R.A.; Gomez, L.; von Hoff, D.D. 5,6 Dihydro-5'-azacytidine (DHAC) restores androgen responsiveness in androgen-insensitive prostate cancer cells. Anticancer Res. 1999, 19, 1285–1291. [Google Scholar] [PubMed]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011, 31, 2723–2732. [Google Scholar] [PubMed]
- Housman, G.; Mataga, A.M.; Devalapalli, A.; Heerboth, S.; Evans, L.R.; Sarkar, S. Demethylation and re-expression of tumor suppressor genes by HDAC inhibitors and calpain inhibitors in cancer cells: A study related to synergistic type growth inhibition and reduction of motility. In The Epigenetics World Congress, MA, USA, April 2011. Abstract 206.
- Sarkar, S.; Goldgar, S.; Byler, S.; Rosenthal, S.; Heerboth, S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: Sensitization of cancer cells by combination therapy. Epigenomics 2013, 5, 87–94. [Google Scholar] [CrossRef]
- Juergens, R.; Wrangle, J.; Vendetti, F.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS One 2014, 9, e87455. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Longacre, M.; Tatur, N.; Heerboth, S.; Lapinska, K. Histone deacetylases (HDACs): Function, mechanism, & inhibition. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley: Chichester, UK, 2014; pp. 1–9. [Google Scholar]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. The use of epigenetic drugs in diseases: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769-1792. https://doi.org/10.3390/cancers6031769
Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug Resistance in Cancer: An Overview. Cancers. 2014; 6(3):1769-1792. https://doi.org/10.3390/cancers6031769
Chicago/Turabian StyleHousman, Genevieve, Shannon Byler, Sarah Heerboth, Karolina Lapinska, Mckenna Longacre, Nicole Snyder, and Sibaji Sarkar. 2014. "Drug Resistance in Cancer: An Overview" Cancers 6, no. 3: 1769-1792. https://doi.org/10.3390/cancers6031769
APA StyleHousman, G., Byler, S., Heerboth, S., Lapinska, K., Longacre, M., Snyder, N., & Sarkar, S. (2014). Drug Resistance in Cancer: An Overview. Cancers, 6(3), 1769-1792. https://doi.org/10.3390/cancers6031769