Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
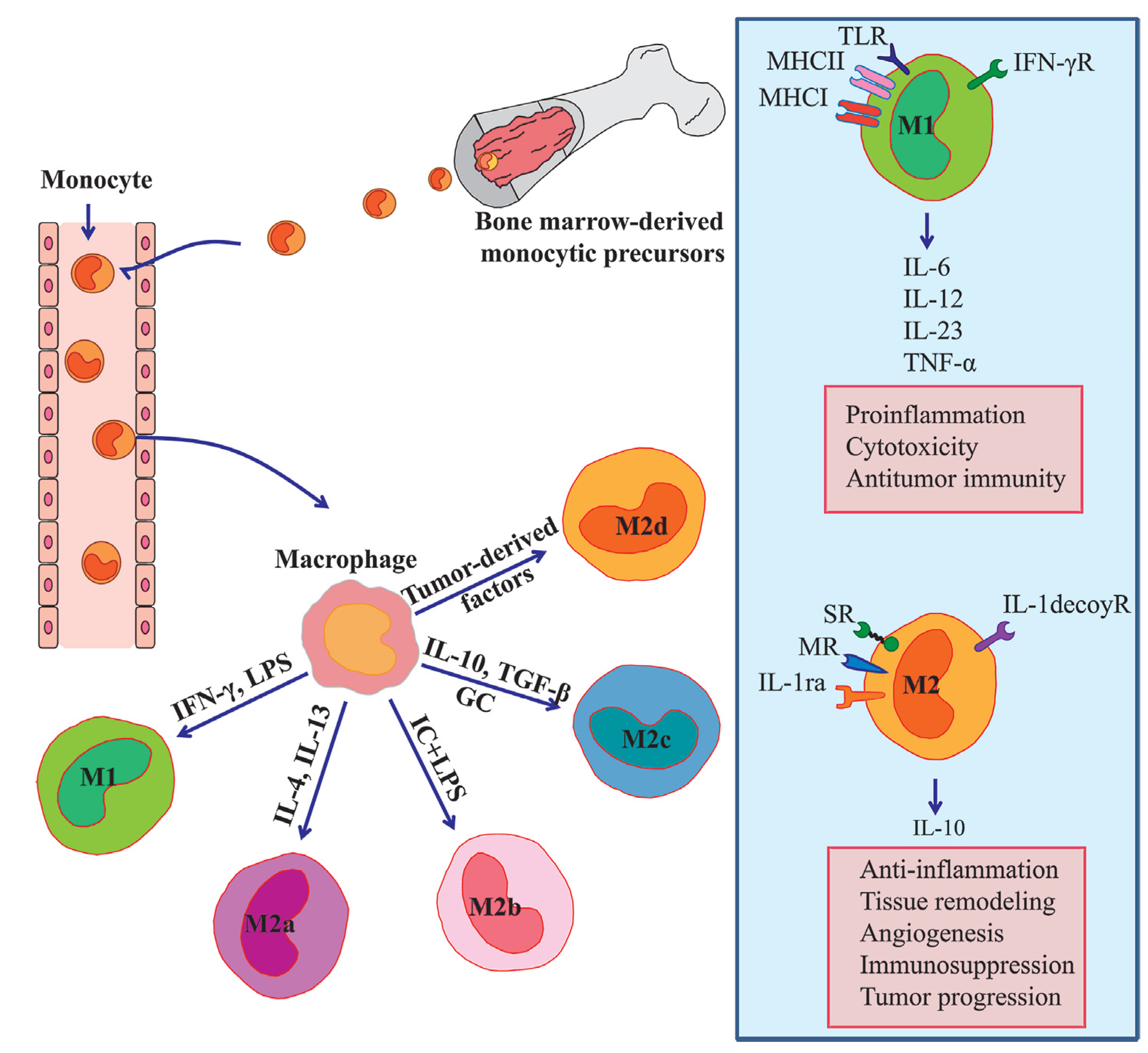
:1. Introduction

2. Molecular Mechanisms of Monocyte/Macrophage Mobilization
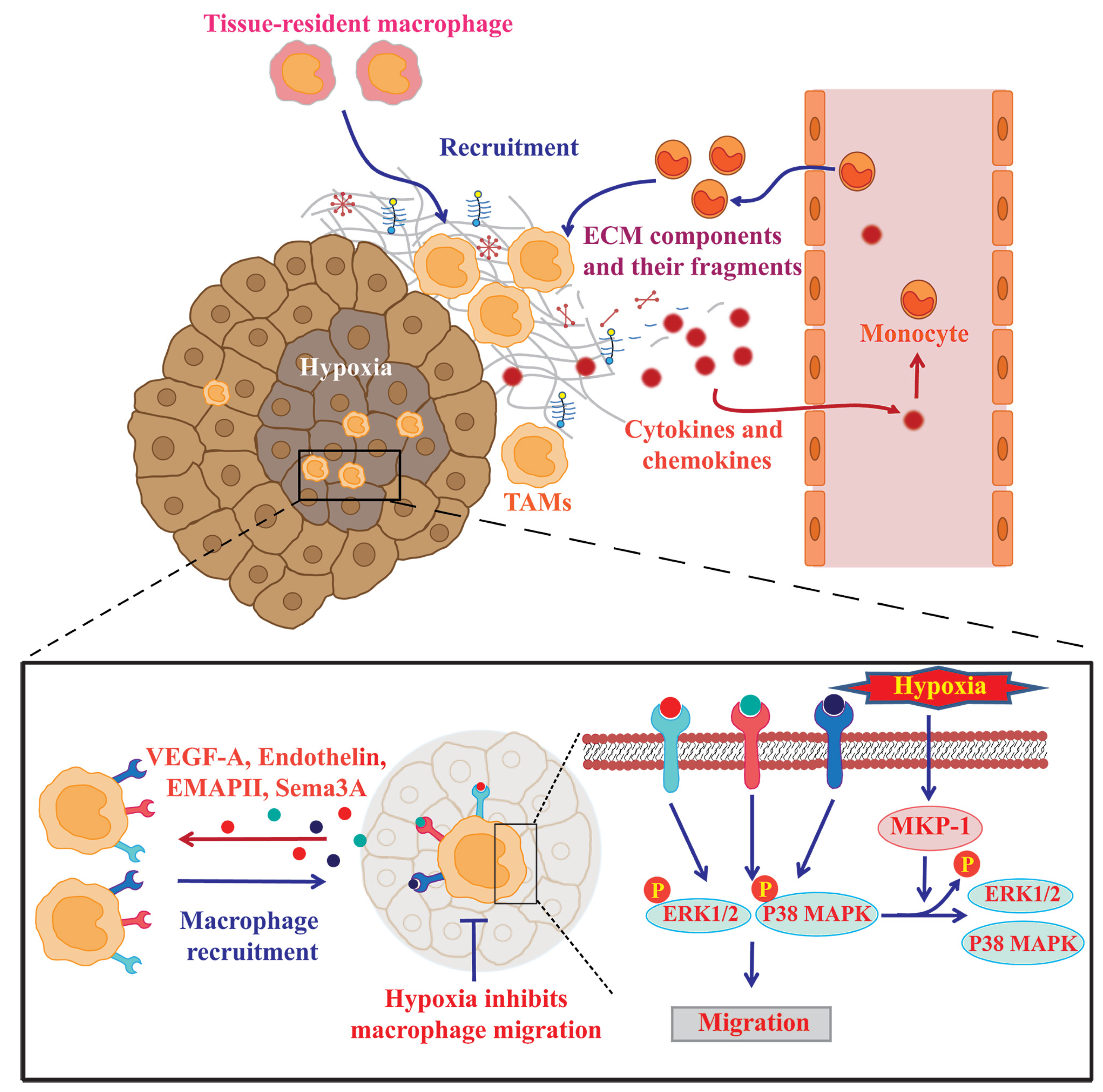
2.1. Soluble Factors Mediating Monocyte/Macrophage Mobilization into Tumors
2.2. Roles of ECM Components and Their Fragments in Controlling Macrophage Recruitment and Polarization
2.3. Hypoxia Promotes Macrophage Recruitment into Hypoxic Areas
3. Polarization of Macrophages toward the Pro-Angiogenic Phenotype
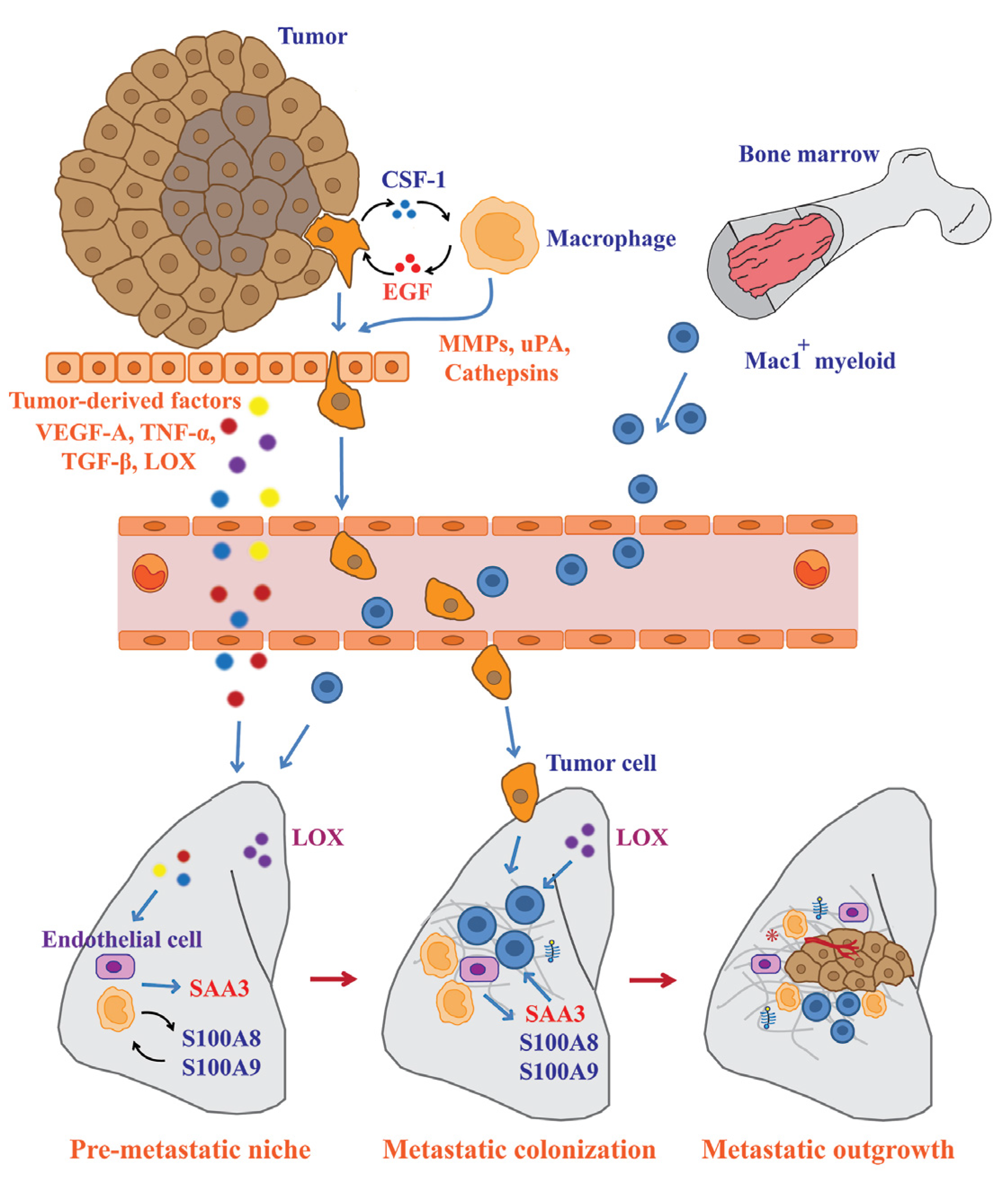
4. Macrophage-Dependent Promotion of Tumor Invasion and Metastasis
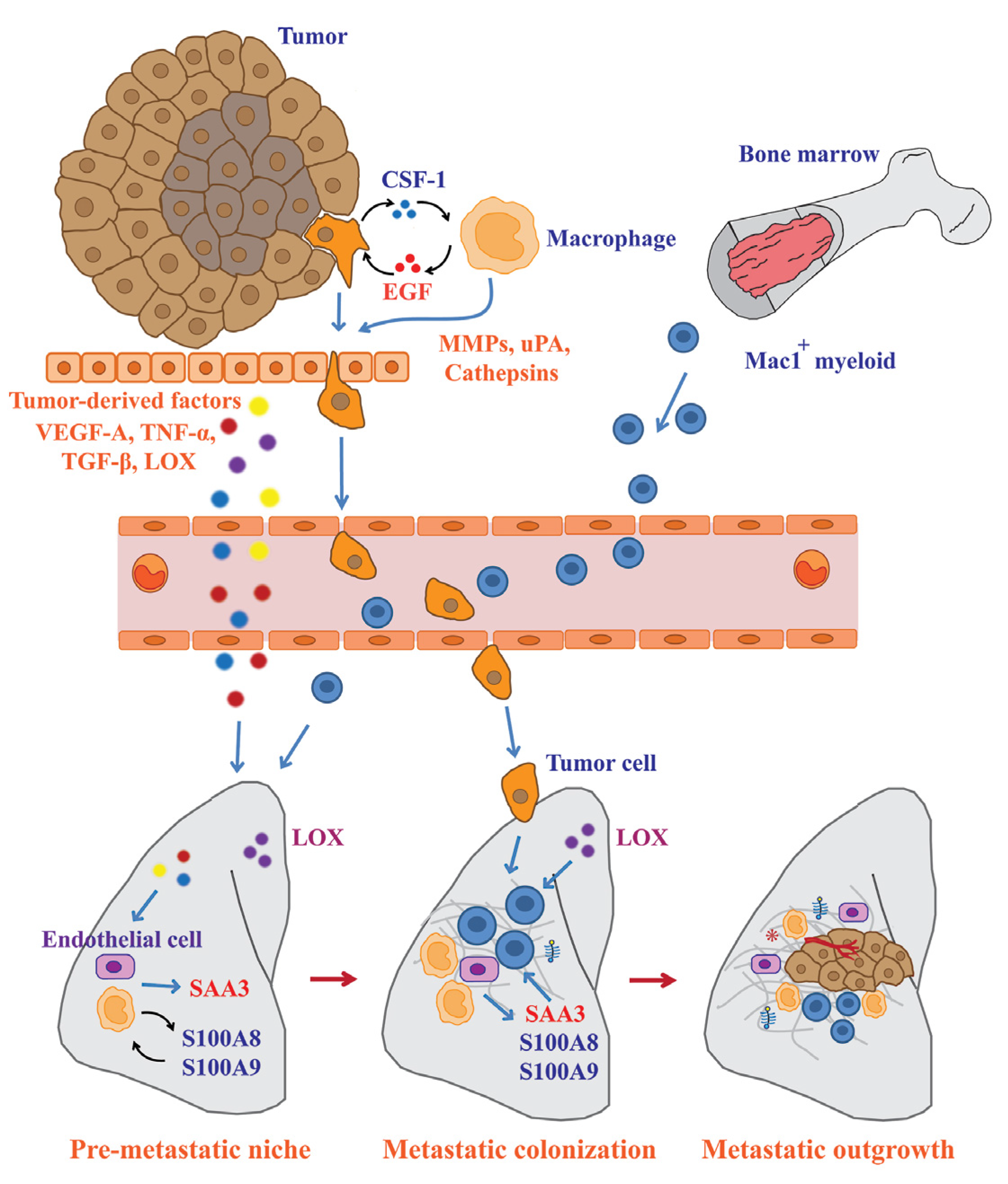
5. Significant Contribution of Macrophages to Pre-metastatic Niche Formation
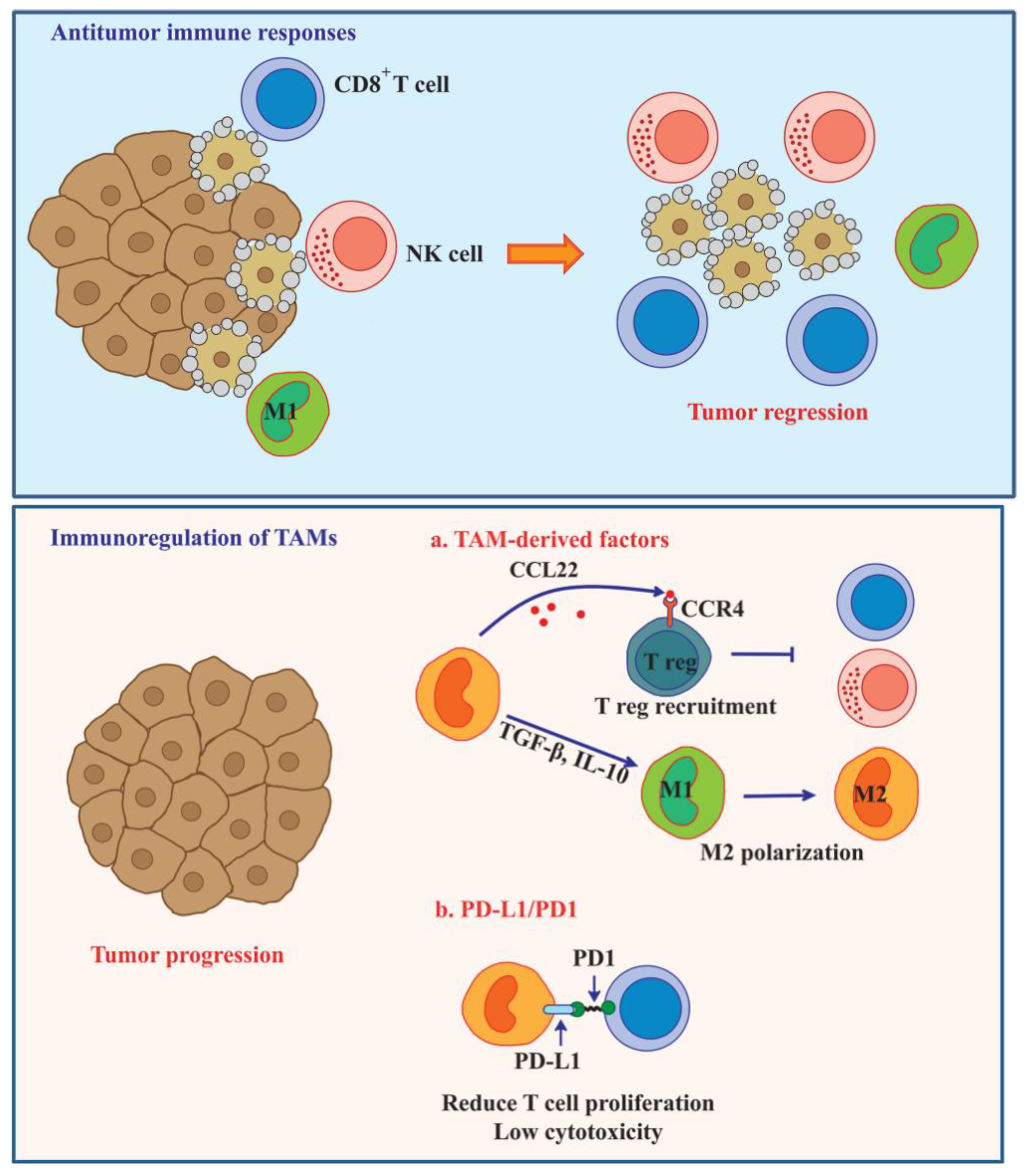
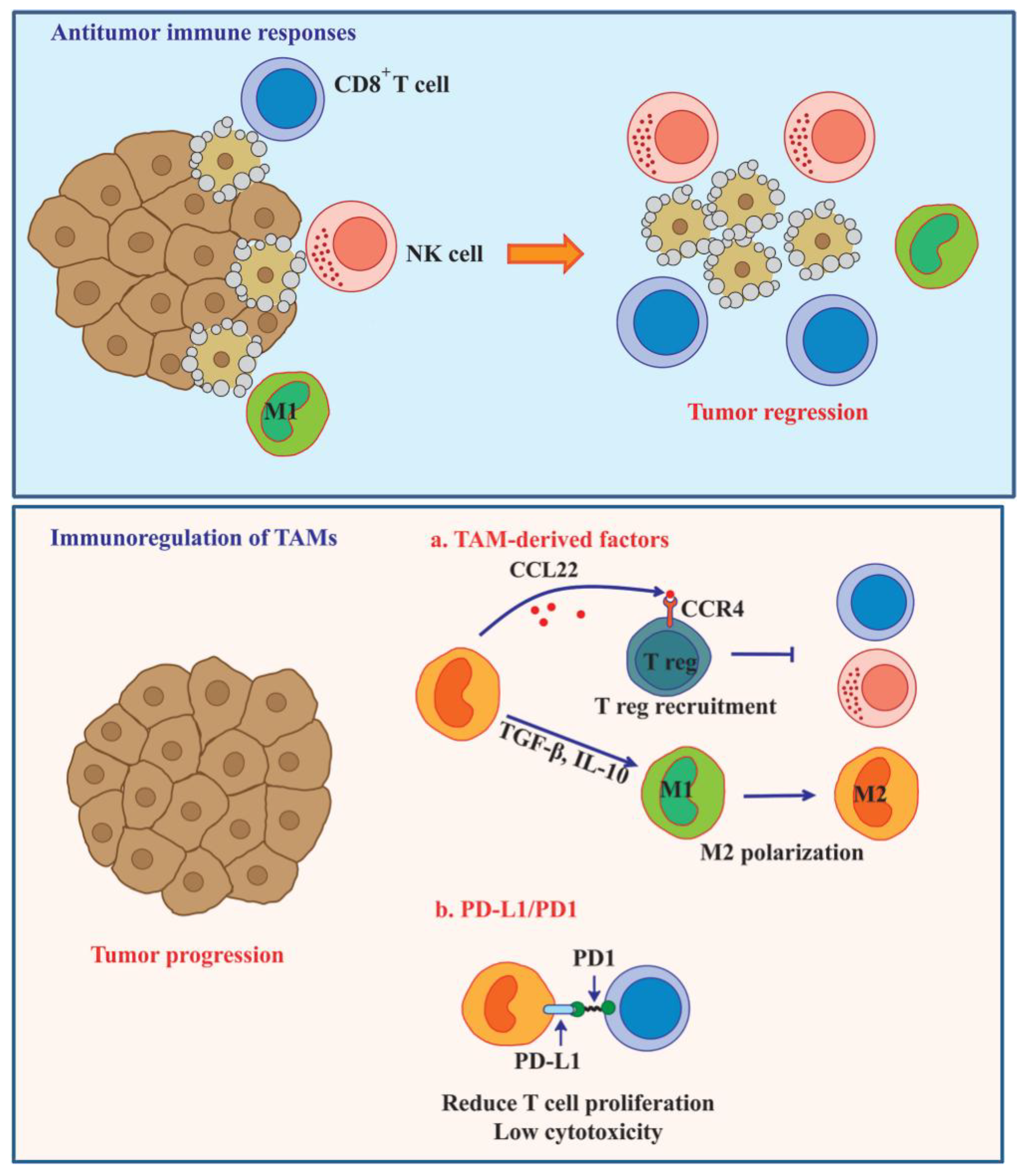
6. TAM-Mediated Immunosuppression in Tumor Microenvironment
7. Roles of TAMs in Self-Renewal and Chemotherapeutic Resistance of Cancer Stem Cells
8. Future Prospects in TAM-Targeting Cancer Therapy
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Van Ginderachter, J.A.; Movahedi, K.; Hassanzadeh Ghassabeh, G.; Meerschaut, S.; Beschin, A.; Raes, G.; de Baetselier, P. Classical and alternative activation of mononuclear phagocytes: Picking the best of both worlds for tumor promotion. Immunobiology 2006, 211, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Tal, A.O.; Scholz, A.; de Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; Lewis, C.E. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010, 70, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; van den Bossche, J.; Mack, M.; Pipeleers, D.; in’t Veld, P.; de Baetselier, P.; van Ginderachter, J.A. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Zeng, Z.; Guo, X.; Li, X.; Wei, F.; Zhang, W.; Chen, P.; Liang, F.; Xiang, B.; Ma, J.; et al. LPLUNC1 suppresses IL-6-induced nasopharyngeal carcinoma cell proliferation via inhibiting the Stat3 activation. Oncogene 2014, 33, 2098–2109. [Google Scholar] [CrossRef] [PubMed]
- Zijlmans, H.J.; Fleuren, G.J.; Baelde, H.J.; Eilers, P.H.; Kenter, G.G.; Gorter, A. The absence of CCL2 expression in cervical carcinoma is associated with increased survival and loss of heterozygosity at 17q11.2. J. Pathol. 2006, 208, 507–517. [Google Scholar] [CrossRef]
- Tsutsui, S.; Yasuda, K.; Suzuki, K.; Tahara, K.; Higashi, H.; Era, S. Macrophage infiltration and its prognostic implications in breast cancer: The relationship with VEGF expression and microvessel density. Oncol. Rep. 2005, 14, 425–431. [Google Scholar] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [PubMed]
- Dalton, H.J.; Armaiz-Pena, G.N.; Gonzalez-Villasana, V.; Lopez-Berestein, G.; Bar-Eli, M.; Sood, A.K. Monocyte subpopulations in angiogenesis. Cancer Res. 2014, 74, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, Y.; Pienta, K.J. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst. 2010, 102, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Sud, S.; McGregor, N.A.; Martinovski, G.; Rice, B.T.; Craig, M.J.; Varsos, Z.S.; Roca, H.; Pienta, K.J. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia 2009, 11, 1235–1242. [Google Scholar] [PubMed]
- Negus, R.P.; Stamp, G.W.; Relf, M.G.; Burke, F.; Malik, S.T.; Bernasconi, S.; Allavena, P.; Sozzani, S.; Mantovani, A.; Balkwill, F.R. The detection and localization of monocyte chemoattractant protein-1 (MCP-1) in human ovarian cancer. J. Clin. Invest. 1995, 95, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Arenberg, D.A.; Keane, M.P.; DiGiovine, B.; Kunkel, S.L.; Strom, S.R.; Burdick, M.D.; Iannettoni, M.D.; Strieter, R.M. Macrophage infiltration in human non-small-cell lung cancer: The role of CC chemokines. Cancer Immunol. Immunother. 2000, 49, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Filardi, E.; Nieto, C.; Dominguez-Soto, A.; Barroso, R.; Sanchez-Mateos, P.; Puig-Kroger, A.; Lopez-Bravo, M.; Joven, J.; Ardavin, C.; Rodriguez-Fernandez, J.L.; et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: Identification of CCL2/CCR2-dependent gene expression profile. J. Immunol. 2014, 192, 3858–3867. [Google Scholar]
- Gu, L.; Tseng, S.; Horner, R.M.; Tam, C.; Loda, M.; Rollins, B.J. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 2000, 404, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.Y.; Matsushima, K.; Baba, T.; Mukaida, N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J. Immunol. 2008, 181, 6384–6393. [Google Scholar] [CrossRef] [PubMed]
- Milliken, D.; Scotton, C.; Raju, S.; Balkwill, F.; Wilson, J. Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin. Cancer Res. 2002, 8, 1108–1114. [Google Scholar] [PubMed]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [PubMed]
- Kao, J.; Houck, K.; Fan, Y.; Haehnel, I.; Libutti, S.K.; Kayton, M.L.; Grikscheit, T.; Chabot, J.; Nowygrod, R.; Greenberg, S.; et al. Characterization of a novel tumor-derived cytokine. Endothelial-monocyte activating polypeptide II. J. Biol. Chem. 1994, 269, 25106–25119. [Google Scholar]
- Scholl, S.M.; Pallud, C.; Beuvon, F.; Hacene, K.; Stanley, E.R.; Rohrschneider, L.; Tang, R.; Pouillart, P.; Lidereau, R. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J. Natl. Cancer Inst. 1994, 86, 120–126. [Google Scholar] [PubMed]
- Dorsch, M.; Hock, H.; Kunzendorf, U.; Diamantstein, T.; Blankenstein, T. Macrophage colony-stimulating factor gene transfer into tumor cells induces macrophage infiltration but not tumor suppression. Eur. J. Immunol. 1993, 23, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Miyoshi, S.; Mikami, T.; Koyama, H.; Kitazawa, M.; Takeoka, M.; Sano, K.; Amano, J.; Isogai, Z.; Niida, S.; et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010, 70, 7073–7083. [Google Scholar] [CrossRef] [PubMed]
- De La Motte, C.A.; Hascall, V.C.; Calabro, A.; Yen-Lieberman, B.; Strong, S.A. Mononuclear leukocytes preferentially bind via CD44 to hyaluronan on human intestinal mucosal smooth muscle cells after virus infection or treatment with poly(I·C). J. Biol. Chem. 1999, 274, 30747–30755. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J.; Prestwich, G.D. Hyaluronan-binding proteins: Tying up the giant. J. Biol. Chem. 2002, 277, 4585–4588. [Google Scholar] [CrossRef] [PubMed]
- De la Motte, C.A.; Hascall, V.C.; Drazba, J.; Bandyopadhyay, S.K.; Strong, S.A. Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyinosinic acid:polycytidylic acid: Inter-alpha-trypsin inhibitor is crucial to structure and function. Am. J. Pathol. 2003, 163, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Chiodoni, C.; Colombo, M.P.; Sangaletti, S. Matricellular proteins: From homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2010, 29, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.M.; Quintero, P.A.; Perkins, D.L.; Kobayashi, D.K.; Kelley, D.G.; Marconcini, L.A.; Mecham, R.P.; Senior, R.M.; Shapiro, S.D. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Invest. 2006, 116, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Schiarea, S.; Liguori, M.; Fabbri, M.; Pesce, S.; Zammataro, L.; Pasqualini, F.; Nebuloni, M.; Chiabrando, C.; Mantovani, A.; et al. Tumor-conditioned macrophages secrete migration-stimulating factor: A new marker for M2-polarization, influencing tumor cell motility. J. Immunol. 2010, 185, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Gotte, M.; et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Invest. 2005, 115, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 2006, 177, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.M.; Wu, Y.; Chen, N.; Cheng, J.; Zhuang, S.M.; Zheng, L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007, 110, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S.; Landers, R.J.; Underwood, J.C.; Harris, A.L.; Lewis, C.E. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J. Pathol. 2000, 192, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, M.J.; Wilson, J.L.; Balkwill, F.R. Endothelin-2 is a macrophage chemoattractant: Implications for macrophage distribution in tumors. Eur. J. Immunol. 2002, 32, 2393–2400. [Google Scholar] [CrossRef] [PubMed]
- Matschurat, S.; Knies, U.E.; Person, V.; Fink, L.; Stoelcker, B.; Ebenebe, C.; Behrensdorf, H.A.; Schaper, J.; Clauss, M. Regulation of EMAP II by hypoxia. Am. J. Pathol. 2003, 162, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Korsisaari, N.; Kasman, I.M.; Forrest, W.F.; Pal, N.; Bai, W.; Fuh, G.; Peale, F.V.; Smits, R.; Ferrara, N. Inhibition of VEGF-A prevents the angiogenic switch and results in increased survival of Apc+/min mice. Proc. Natl. Acad. Sci. USA 2007, 104, 10625–10630. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Tang, N.; Corke, K.P.; Tazzyman, D.; Ameri, K.; Wells, M.; Lewis, C.E. Expression of HIF-1alpha by human macrophages: Implications for the use of macrophages in hypoxia-regulated cancer gene therapy. J. Pathol. 2002, 196, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Roda, J.M.; Liu, H.; O’Neil, T.; Marsh, C.B. Opposing roles for HIF-1alpha and HIF-2alpha in the regulation of angiogenesis by mononuclear phagocytes. Blood 2011, 117, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, M.J.; Balkwill, F.R. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation—a potential mechanism. Eur. J. Immunol. 2001, 31, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Saccani, A.; Bottazzi, B.; Bernasconi, S.; Allavena, P.; Gaetano, B.; Fei, F.; LaRosa, G.; Scotton, C.; Balkwill, F.; et al. Defective expression of the monocyte chemotactic protein-1 receptor CCR2 in macrophages associated with human ovarian carcinoma. J. Immunol. 2000, 164, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Zaynagetdinov, R.; Sherrill, T.P.; Polosukhin, V.V.; Han, W.; Ausborn, J.A.; McLoed, A.G.; McMahon, F.B.; Gleaves, L.A.; Degryse, A.L.; Stathopoulos, G.T.; et al. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J. Immunol. 2011, 187, 5703–5711. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Mantovani, A. Diversity and plasticity of mononuclear phagocytes. Eur. J. Immunol. 2011, 41, 2470–2472. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Kuwano, T.; Tsutsumi-Miyahara, C.; Ueda, S.; Kimura, Y.N.; Hamano, S.; Sonoda, K.H.; Saijo, Y.; Nukiwa, T.; Strieter, R.M.; et al. Infiltration of COX-2-expressing macrophages is a prerequisite for IL-1 beta-induced neovascularization and tumor growth. J. Clin. Invest. 2005, 115, 2979–2991. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Shibuta, K.; Barnard, G.F.; Kitano, S.; Mori, M. Angiogenin expression in human colorectal cancer: The role of focal macrophage infiltration. Clin. Cancer Res. 2000, 6, 3545–3551. [Google Scholar] [PubMed]
- Lin, E.Y.; Li, J.F.; Bricard, G.; Wang, W.; Deng, Y.; Sellers, R.; Porcelli, S.A.; Pollard, J.W. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol. Oncol. 2007, 1, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [PubMed]
- Laoui, D.; van Overmeire, E.; di Conza, G.; Aldeni, C.; Keirsse, J.; Morias, Y.; Movahedi, K.; Houbracken, I.; Schouppe, E.; Elkrim, Y.; et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014, 74, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Bingle, L.; Lewis, C.E.; Corke, K.P.; Reed, M.W.; Brown, N.J. Macrophages promote angiogenesis in human breast tumour spheroids in vivo. Br. J. Cancer 2006, 94, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Guruvayoorappan, C. Tumor versus tumor-associated macrophages: How hot is the link? Integr. Cancer Ther. 2008, 7, 90–95. [Google Scholar] [CrossRef]
- Murdoch, C.; Lewis, C.E. Macrophage migration and gene expression in response to tumor hypoxia. Int. J. Cancer 2005, 117, 701–708. [Google Scholar] [PubMed]
- Burke, B.; Giannoudis, A.; Corke, K.P.; Gill, D.; Wells, M.; Ziegler-Heitbrock, L.; Lewis, C.E. Hypoxia-induced gene expression in human macrophages: Implications for ischemic tissues and hypoxia-regulated gene therapy. Am. J. Pathol. 2003, 163, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Tazzyman, S.; Webster, S.; Lewis, C.E. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J. Immunol. 2007, 178, 7405–7411. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi Sergi, L.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Venneri, M.A.; Biziato, D.; Nonis, A.; Moi, D.; Sica, A.; di Serio, C.; Naldini, L.; de Palma, M. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 2009, 114, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; di Carlo, E.; Gariboldi, S.; Miotti, S.; Cappetti, B.; Parenza, M.; Rumio, C.; Brekken, R.A.; Chiodoni, C.; Colombo, M.P. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008, 68, 9050–9059. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Almholt, K.; Lund, L.R.; Rygaard, J.; Nielsen, B.S.; Dano, K.; Romer, J.; Johnsen, M. Reduced metastasis of transgenic mammary cancer in urokinase-deficient mice. Int. J. Cancer 2005, 113, 525–532. [Google Scholar] [PubMed]
- Gil-Bernabe, A.M.; Ferjancic, S.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [PubMed]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [PubMed]
- Hiratsuka, S.; Ishibashi, S.; Tomita, T.; Watanabe, A.; Akashi-Takamura, S.; Murakami, M.; Kijima, H.; Miyake, K.; Aburatani, H.; Maru, Y. Primary tumours modulate innate immune signalling to create pre-metastatic vascular hyperpermeability foci. Nat. Commun. 2013, 4, 1853. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Goel, S.; Kamoun, W.S.; Maru, Y.; Fukumura, D.; Duda, D.G.; Jain, R.K. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. USA 2011, 108, 3725–3730. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Gobert, M.; Treilleux, I.; Bendriss-Vermare, N.; Bachelot, T.; Goddard-Leon, S.; Arfi, V.; Biota, C.; Doffin, A.C.; Durand, I.; Olive, D.; et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009, 69, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ishii, T.; Inagaki, A.; Yano, H.; Komatsu, H.; Iida, S.; Inagaki, H.; Ueda, R. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res. 2006, 66, 5716–5722. [Google Scholar] [CrossRef] [PubMed]
- Iellem, A.; Mariani, M.; Lang, R.; Recalde, H.; Panina-Bordignon, P.; Sinigaglia, F.; d’Ambrosio, D. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J. Exp. Med. 2001, 194, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y.; Kono, K.; Kawaguchi, Y.; Akaike, H.; Kamimura, K.; Sugai, H.; Fujii, H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int. J. Cancer 2008, 122, 2286–2293. [Google Scholar] [PubMed]
- Clarke, M.F. Neurobiology: At the root of brain cancer. Nature 2004, 432, 281–282. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Xiao, H.; Xu, M.; Ye, X.; Hu, J.; Li, F.; Li, M.; Luo, C.; Yu, S.; Bian, X.; et al. Glioma-initiating cells: A predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 2011, 232, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K.; et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells. Cancer Res. 2012, 72, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; de Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 131, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhu, X.D.; Sun, H.C.; Xiong, Y.Q.; Zhuang, P.Y.; Xu, H.X.; Kong, L.Q.; Wang, L.; Wu, W.Z.; Tang, Z.Y. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 2010, 16, 3420–3430. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Invest. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with Anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.G.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Gadea, B.B.; Wang, H.W.; Gocheva, V.; Hunter, K.E.; Tang, L.H.; Joyce, J.A. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene 2012, 31, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Welford, A.F.; Biziato, D.; Coffelt, S.B.; Nucera, S.; Fisher, M.; Pucci, F.; di Serio, C.; Naldini, L.; de Palma, M.; Tozer, G.M.; et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J. Clin. Invest. 2011, 121, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Mo, C.; Wang, Y.; Wei, D.; Xiao, H. Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 2013, 138, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Shime, H.; Matsumoto, M.; Oshiumi, H.; Tanaka, S.; Nakane, A.; Iwakura, Y.; Tahara, H.; Inoue, N.; Seya, T. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc. Natl. Acad. Sci. USA 2012, 109, 2066–2071. [Google Scholar] [CrossRef] [PubMed]
- Coscia, M.; Quaglino, E.; Iezzi, M.; Curcio, C.; Pantaleoni, F.; Riganti, C.; Holen, I.; Monkkonen, H.; Boccadoro, M.; Forni, G.; et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J. Cell Mol. Med. 2010, 14, 2803–2815. [Google Scholar] [PubMed]
- Zhang, X.; Tian, W.; Cai, X.; Wang, X.; Dang, W.; Tang, H.; Cao, H.; Wang, L.; Chen, T. Hydrazinocurcumin encapsuled nanoparticles “re-educate” tumor-associated macrophages and exhibit anti-tumor effects on breast cancer following STAT3 suppression. PLoS One 2013, 8, e65896. [Google Scholar] [CrossRef] [PubMed]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zavaljevski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers 2014, 6, 1670-1690. https://doi.org/10.3390/cancers6031670
Chanmee T, Ontong P, Konno K, Itano N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers. 2014; 6(3):1670-1690. https://doi.org/10.3390/cancers6031670
Chicago/Turabian StyleChanmee, Theerawut, Pawared Ontong, Kenjiro Konno, and Naoki Itano. 2014. "Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment" Cancers 6, no. 3: 1670-1690. https://doi.org/10.3390/cancers6031670
APA StyleChanmee, T., Ontong, P., Konno, K., & Itano, N. (2014). Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers, 6(3), 1670-1690. https://doi.org/10.3390/cancers6031670