BRCA1 and Oxidative Stress

Abstract
:1. Introduction
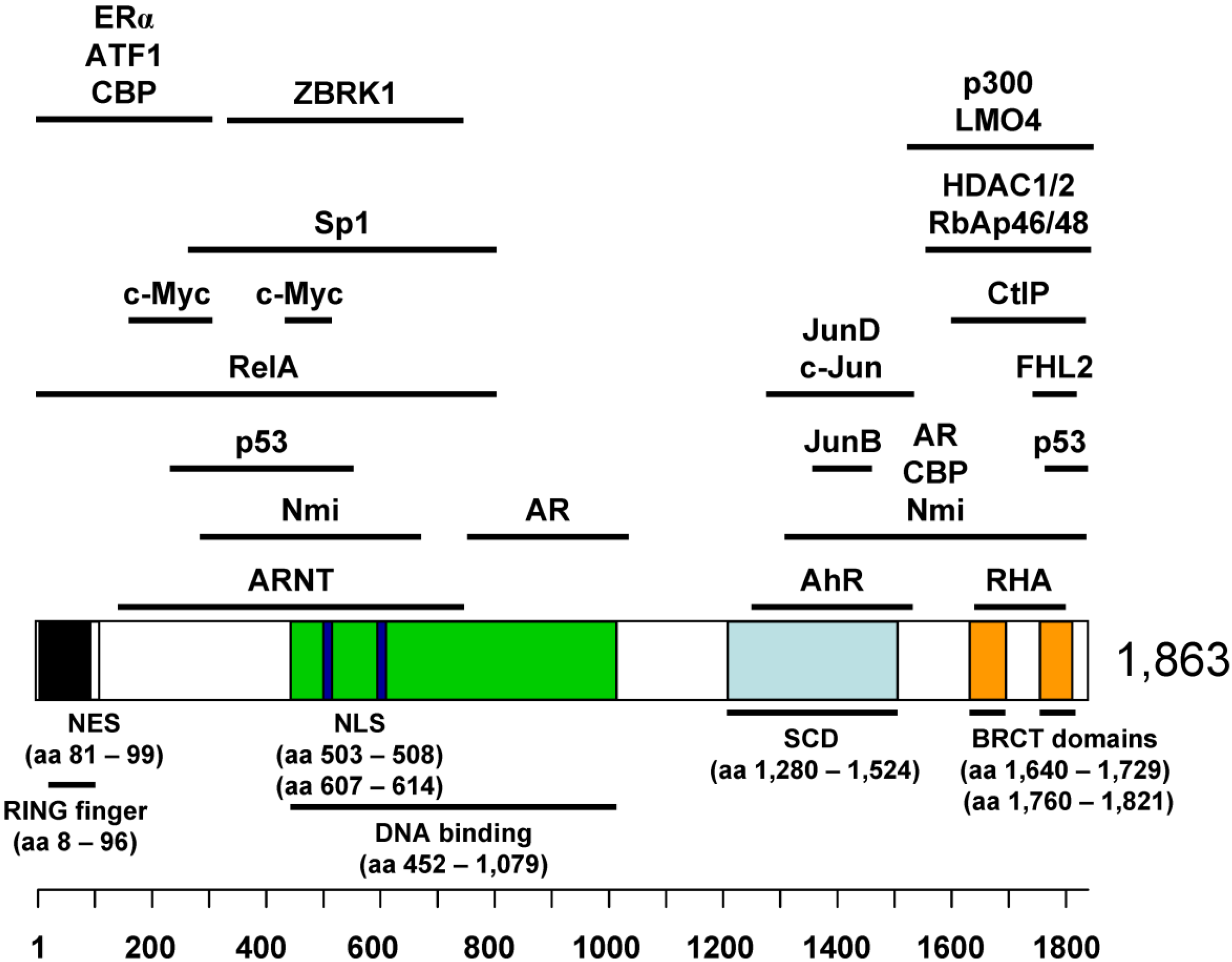
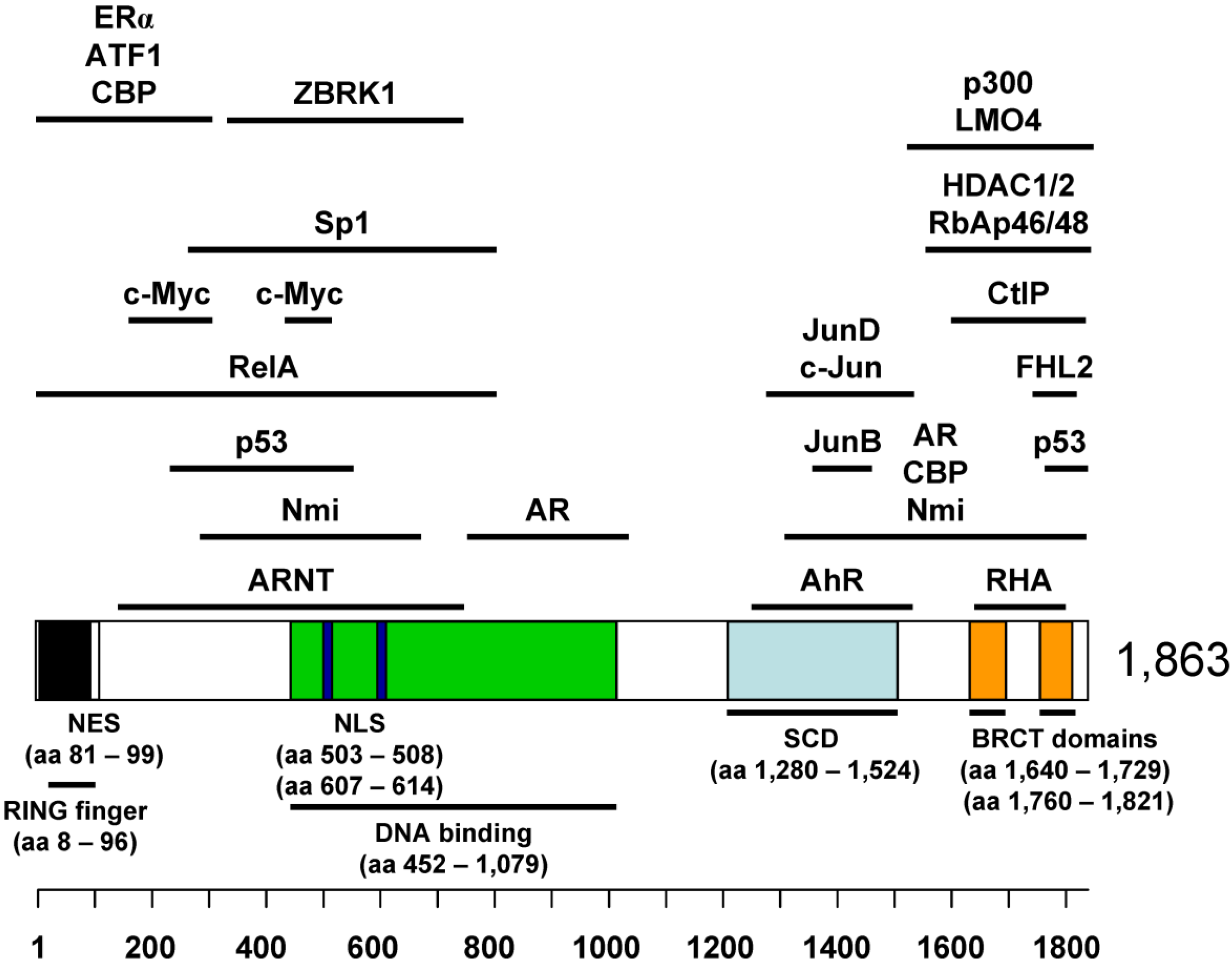
2. BRCA1 as a Tumor Suppressor
2.1. Genome Stability, Oxidative Stress and Tumor Suppression
2.2. Mouse Model: Genome Stability, Oxidative Stress and Tumor Suppression
2.3. Cell Studies: Genome Stability, Oxidative Stress and Tumor Suppression
2.4. BRCA1 and p53: Genome Stability, Oxidative Stress and Tumor Suppression
3. BRCA1 as an E3 Ubiquitin Ligase
3.1. BRCA1 E3 Ligase Activity and Tumor Suppression
3.2. BRCA1: Oxidative Stress and Ubiquitination
4. BRCA1 as a Transcriptional Regulator
4.1. Binding Partners in BRCA1 Transcriptional Regulation

{kind=link}
{kind=link}
| Transcription factor | Region in BRCA1 (aa) | Assays | References |
|---|---|---|---|
| AhR | 1241–1530 | Co-IP/GST | [167] |
| AR (androgen receptor) | 758–1064 & 1314–1863 | GST/M2H | [168] |
| ARNT | 131–757 | Co-IP/GST/GAL4 | [169] |
| ATF1 | 1–304 | Co-IP/GST/Y2H/GAL4 | [165] |
| BRF1 | N/D | AP/GST | [132] |
| CBP | 1–300 & 1314–1863 | GST | [129] |
| CtIP | 1602–1863 | Co-IP/GST/Y2H | [10] |
| E2F1 | N/D | GST | [170] |
| E2F4 | N/D | GST | [170] |
| ERα | 1–300 | Co-IP/GST | [135] |
| FHL2 | 1756–1852 | GST/GAL4/Y2H | [166] |
| HDAC1 | 1553–1863 | GST | [8] |
| HDAC2 | 1553–1863 | GST | [8] |
| HIF-1α | N/D | Co-IP | [157] |
| JunB | 1343–1440 | Co-IP/GST/GAL4/Y2H | [171] |
| JunD | AD1 | Co-IP/GST/GAL4/Y2H | [171] |
| c-Jun | AD1 | Co-IP/GST/GAL4 | [171] |
| LMO4 | 1528–1863 | Co-IP/GST/Y2H | [172] |
| c-Myc | 175–303 & 443–511 | Co-IP/GST/Y2H | [141] |
| NF-YA | N/D | Co-IP | [151] |
| Nmi | 298–693 & 1301–1863 | Co-IP/GST/Y2H | [142] |
| NRF2 | N/D | Co-IP | [117] |
| OCT-1 | N/D | Co-IP | [151] |
| p300 | 1528–1863 | Co-IP/GAL4 | [129] |
| p53 | 224–550 & 1760–1863 | Co-IP/GST | [86,87] |
| RbAp46 | 1553–1863 | Co-IP/GST/GAL4/Y2H | [8,131] |
| RbAp48 | 1553–1863 | Co-IP/GST | [8] |
| RelA | 1–802 | Co-IP/GST | [154] |
| RHA | 1650–1800 | GST/GAL4/Y2H | [128] |
| Sp1 | 260–802 | Co-IP/GST | [163] |
| STAT1 | N/D | Co-IP/GST/M2H | [111] |
| STAT5A | N/D | Co-IP | [173] |
| USF2 | N/D | Co-IP | [174] |
| ZBRK1 | 341–748 | Co-IP/GST/Y2H | [147] |
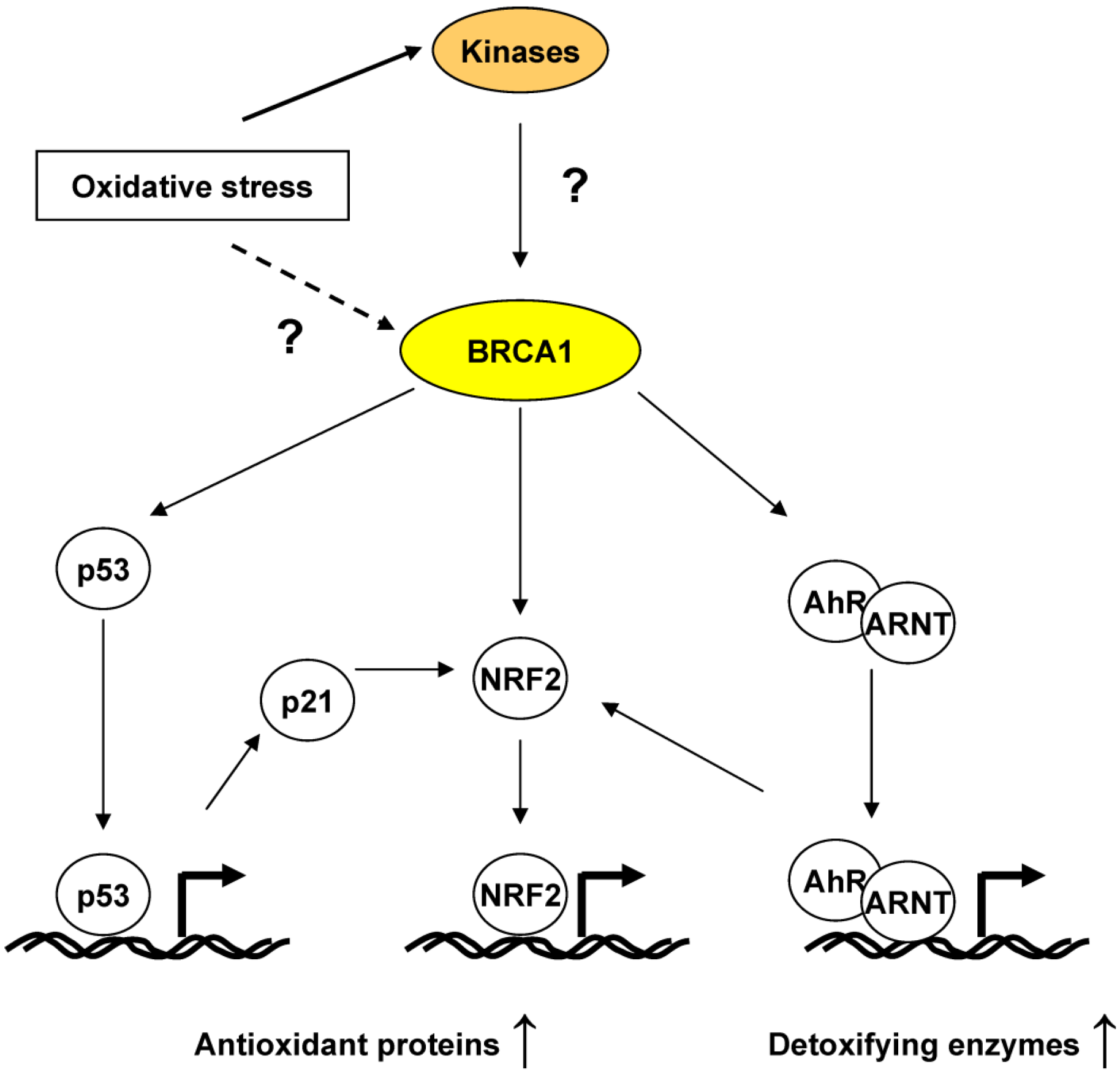
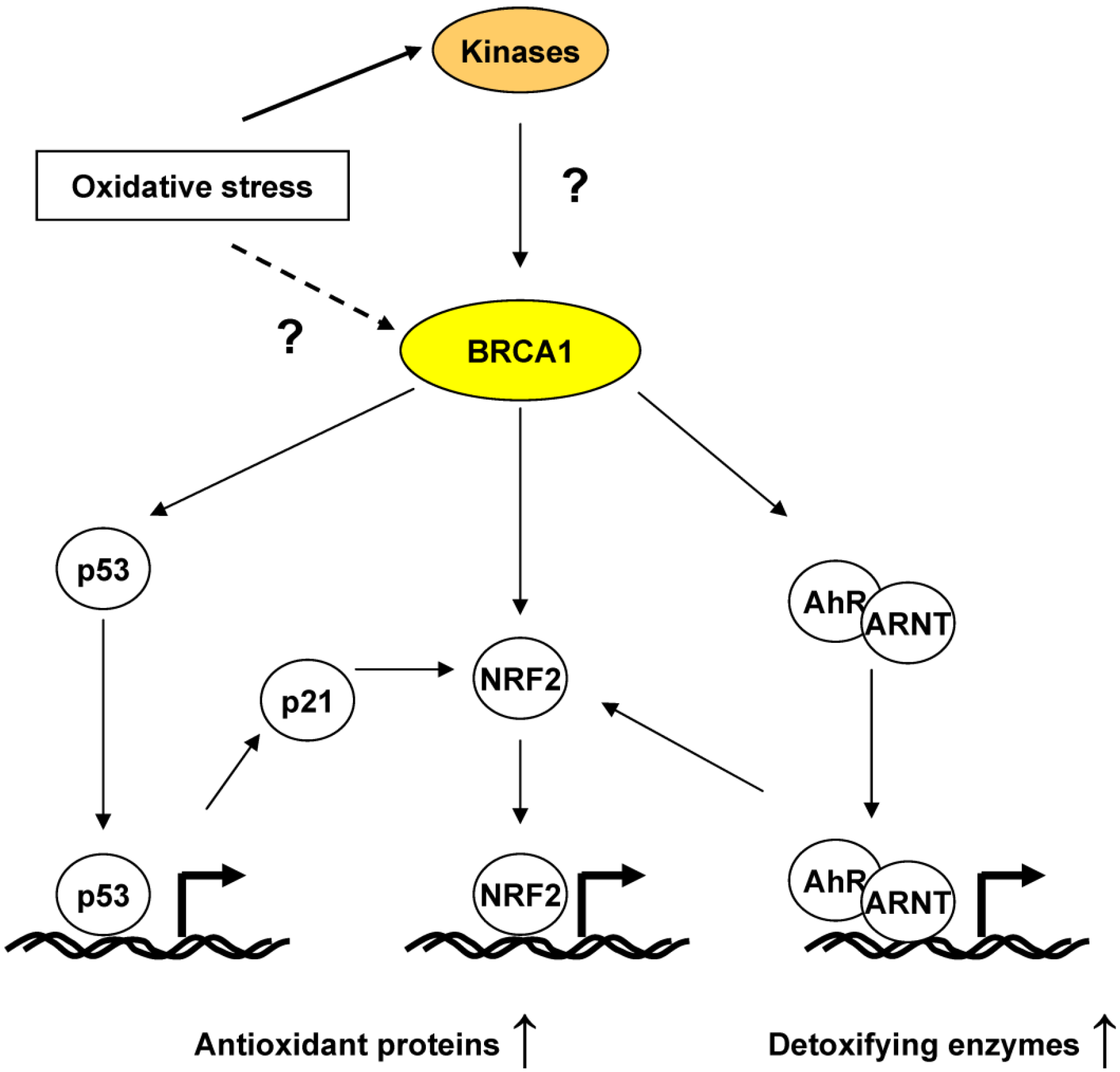
4.2. BRCA1 Regulation of Oxidative Stress through Transcriptional Activities
5. BRCA1, an ROS Sensor?
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Futreal, P.A.; Liu, Q.; Shattuck-Eidens, D.; Cochran, C.; Harshman, K.; Tavtigian, S.; Bennett, L.M.; Haugen-Strano, A.; Swensen, J.; Miki, Y.; et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science 1994, 266, 120–122. [Google Scholar]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar]
- Easton, D.F.; Bishop, D.T.; Ford, D.; Crockford, G.P. Genetic linkage analysis in familial breast and ovarian cancer: Results from 214 families. The breast cancer linkage consortium. Am. J. Hum. Genet. 1993, 52, 678–701. [Google Scholar]
- Miki, Y.; Swensen, J.; Stattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtiqian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar]
- Smith, T.M.; Lee, M.K.; Szabo, C.I.; Jerome, N.; McEuen, M.; Taylor, M.; Hood, L.; King, M.C. Complete genomic sequence and analysis of 117 kb of human DNA containing the gene BRCA1. Genome Res. 1996, 6, 1029–1049. [Google Scholar] [CrossRef]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef]
- Jensen, D.E.; Proctor, M.; Marquis, S.T.; Gardner, H.P.; Ha, S.I.; Chodosh, L.A.; Ishov, A.M.; Tommerup, N.; Vissing, H.; Sekido, Y.; et al. BAP1: A novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 1998, 16, 1097–1112. [Google Scholar]
- Yarden, R.I.; Brody, L.C. BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 4983–4988. [Google Scholar] [CrossRef]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vito with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar]
- Li, S.; Chen, P.L.; Subramanian, T.; Chinnadurai, G.; Tomlinson, G.; Osborne, C.K.; Sharp, Z.D.; Lee, W.H. Binding of CtIP to the BRCT repeats of BRCA1 involved in the transcription regulation of p21 is disrupted upon DNA damage. J. Biol. Chem. 1999, 274, 11334–11338. [Google Scholar] [CrossRef]
- Wong, A.K.; Ormonde, P.A.; Pero, R.; Chen, Y.; Lian, L.; Salada, G.; Berry, S.; Lawrence, Q.; Dayananth, P.; Ha, P.; et al. Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene 1998, 17, 2279–2285. [Google Scholar]
- Yu, X.; Baer, R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J. Biol. Chem. 2000, 275, 18541–18549. [Google Scholar]
- Paull, T.T.; Cortez, D.; Bowers, B.; Elledge, S.J.; Gellert, M. Direct DNA binding by Brca1. Proc. Natl. Acad. Sci. USA 2001, 98, 6086–6091. [Google Scholar] [CrossRef]
- Simons, A.M.; Horwitz, A.A.; Starita, L.M.; Griffin, K.; Williams, R.S.; Glover, J.N.; Parvin, J.D. BRCA1 DNA-binding activity is stimulated by BARD1. Cancer Res. 2006, 66, 2012–2018. [Google Scholar]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.F.; Riley, D.J.; Allred, D.C.; Chen, P.L.; von Hoff, D.; Osborne, C.K.; Lee, W.H. Aberrant subcellular localization of BRCA1 in breast cancer. Science 1995, 270, 789–791. [Google Scholar]
- Chen, C.F.; Li, S.; Chen, Y.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. The nuclear localization sequences of the BRCA1 protein interact with the importin-alpha subunit of the nuclear transport signal receptor. J. Biol. Chem. 1996, 271, 32863–32868. [Google Scholar]
- Rodríguez, J.A.; Henderson, B.R. Identification of a functional nuclear export sequence in BRCA1. J. Biol. Chem. 2000, 275, 38589–38596. [Google Scholar] [CrossRef]
- Fabbro, M.; Rodríguez, J.A.; Baer, R.; Henderson, B.R. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J. Biol. Chem. 2002, 277, 21315–21324. [Google Scholar] [CrossRef]
- Feng, Z.; Kachnic, L.; Zhang, J.; Powell, S.N.; Xia, F. DNA damage induces p53-dependent BRCA1 nuclear export. J. Biol. Chem. 2004, 279, 28574–28584. [Google Scholar]
- Bennett, L.M.; Haugen-Strano, A.; Cochran, C.; Brownlee, H.A.; Fiedorek, F.T., Jr.; Wiseman, R.W. Isolation of the mouse homologue of BRCA1 and genetic mapping to mouse chromosome 11. Genomics 1995, 29, 576–581. [Google Scholar] [CrossRef]
- Jin, H.; Selfe, J.; Whitehouse, C.; Morris, J.R.; Solomon, E.; Roberts, R.G. Structural evolution of the BRCA1 genomics region in primates. Genomics 2004, 84, 1071–1082. [Google Scholar] [CrossRef]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. The BRCA1 gene in breast cancer. J. Cell. Physiol. 2003, 196, 19–41. [Google Scholar] [CrossRef]
- Silver, D.P.; Livingston, D.M. Mechanism of BRCA1 tumor suppression. Cancer Discov. 2012, 2, 679–684. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Ma, Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol. Ther. 2010, 125, 376–393. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Jennings, P.; Limonciel, A.; Felice, L.; Leonard, M.O. An overview of transcriptional regulation in response to toxicological insult. Arch. Toxicol. 2013, 87, 49–72. [Google Scholar] [CrossRef]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell. Logev. 2010, 3, 23–34. [Google Scholar] [CrossRef]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakila, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef]
- Caputo, F.; Vegliante, R.; Ghibelli, L. Redox modulation of the DNA damage response. Biochem. Pharmacol. 2012, 84, 1292–1306. [Google Scholar] [CrossRef]
- Storr, S.J.; Woolston, C.M.; Zhang, Y.; Martin, S.G. Redox environment, free radical, and oxidative DNA damage. Antioxid. Redox Signal. 2013, 18, 2399–2408. [Google Scholar] [CrossRef]
- Saeidnia, S.; Abdollahi, M. Antioxidants: Friends or foe in prevention or treatment of cancer: The debate of the century. Toxicol. Appl. Pharmacol. 2013, 271, 49–63. [Google Scholar] [CrossRef]
- Kakehashi, A.; Wei, M.; Fukushima, S.; Wanibuchi, H. Oxidative stress in the carcinogenicity of chemical carcinogens. Cancers 2013, 5, 1332–1354. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Schwarzenbacher, R.; Lipton, S.A. Molecular pathways to neurodegeneration. Nat. Med. 2004, 10, S2–S9. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Jimenez-Del-Rio, M.; Velez-Pardo, C. The bad, the good, and the ugly about oxidative stress. Oxid. Med. Cell. Logev. 2012. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risk due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar]
- Wilson, C.A.; Ramos, L.; Villaseñor, M.R.; Anders, K.H.; Press, M.F.; Clarke, K.; Karlan, B.; Chen, J.J.; Scully, R.; Livingston, D.; et al. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat. Genet. 1999, 21, 236–240. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matisa-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 95, 564–569. [Google Scholar]
- Thompson, D.; Easton, D.; Breast Cancer Linkage Consortium. Variation in BRCA1 cancer risks by mutation position. Cancer Epidemiol. Biomark. Prev. 2002, 11, 329–336. [Google Scholar]
- Leongamornlert, D.; Mahmud, N.; Tymrakiewicz, M.; Saunders, E.; Dadaev, T.; Castro, E.; Goh, C.; Govindasami, K.; Guy, M.; O’Brien, L.; et al. Germline BRCA1 mutations increase prostate cancer risk. Br. J. Cancer 2012, 106, 1697–1701. [Google Scholar] [CrossRef]
- Li, D.; Kumaraswamy, E.; Harlan-Williams, L.M.; Jensen, R.A. The role of BRCA1 and BRCA2 in prostate cancer. Front. Biosci. 2013, 18, 1145–1159. [Google Scholar]
- Johannsson, O.; Ostermeyer, E.A.; Håkansson, S.; Friedman, L.S.; Johansson, U.; Sellberg, G.; Brøndum-Nielsen, K.; Sele, V.; Olsson, H.; King, M.C.; et al. Founding BRCA1 mutations in hereditary breast and ovarian cancer in southern Sweden. Am. J. Hum. Genet. 1996, 58, 441–450. [Google Scholar]
- Lal, G.; Liu, G.; Schmocker, B.; Kaurah, P.; Ozcelik, H.; Narod, S.A.; Redston, M.; Gallinger, S. Inherited predisposition to pancreatic adenocarcinoma: Role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000, 60, 409–416. [Google Scholar]
- Thompson, D.; Easton, D.F.; Breast Cancer Linkage Consortium. Cancer incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef]
- Lynch, H.T.; Deters, C.A.; Snyder, C.L.; Lynch, J.F.; Villeneuve, P.; Silberstein, J.; Martin, H.; Narod, S.A.; Brand, R.E. BRCA1 and pancreatic cancer: Pedigree findings and their causal relationships. Cancer Genet. Cytogenet. 2005, 158, 119–125. [Google Scholar] [CrossRef]
- Dagan, E. Predominant Ashkenazi BRCA1/2 mutations in families with pancreatic cancer. Genet. Test. 2008, 12, 267–271. [Google Scholar] [CrossRef]
- Al-Sukhni, W.; Rothenmund, H.; Borgida, A.E.; Zogopoulos, G.; O’Shea, A.M.; Pollett, A.; Gallinger, S. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Hum. Genet. 2008, 124, 271–278. [Google Scholar] [CrossRef]
- Kim, D.H.; Crawford, B.; Ziegler, J.; Beattie, M.S. Prevalence and characteristic cancer in families with BRCA1 and BRCA2 mutations. Fam. Cancer 2009, 8, 153–158. [Google Scholar] [CrossRef]
- Stadler, Z.K.; Salo-Mullen, E.; Patil, S.M.; Pietanza, M.C.; Vijai, J.; Saloustros, E.; Hansen, N.A.; Kauff, N.D.; Kurtz, R.C.; Kelsen, D.P.; et al. Prevalence of BRCA1 and BRCA2 mutations in Ashekenazi Jewish families with breast and pancreatic cancer. Cancer 2012, 118, 493–499. [Google Scholar] [CrossRef]
- Igbal, J.; Ragone, A.; Lubinski, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; Neuhausen, S.L.; et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012, 107, 2005–2009. [Google Scholar] [CrossRef]
- Brodie, S.G.; Deng, C.X. BRCA1-associated tumorigenesis: What have we learned from knockout mice? Trends Genet. 2001, 17, S18–S22. [Google Scholar] [CrossRef]
- Deng, C.X.; Brodie, S.G. Knockout mouse models and mammary tumorigenesis. Semin. Cancer Biol. 2001, 11, 387–394. [Google Scholar] [CrossRef]
- Drost, R.; Bouwman, P.; Rottenberg, S.; Boon, U.; Schut, E.; Klarenbeek, S.; Klijin, C.; van der Heijden, I.; van der Gulden, H.; Wientjens, E.; et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 2011, 20, 797–809. [Google Scholar] [CrossRef]
- Shakya, R.; Reid, L.J.; Reczek, C.R.; Cole, F.; Egli, D.; Lin, C.S.; deRooij, D.G.; Hirsch, S.; Ravi, K.; Hicks, J.B.; et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 2011, 334, 525–528. [Google Scholar] [CrossRef]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactived by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar]
- Ruffner, H.; Joazeiro, C.A.; Hemmati, D.; Hunter, T.; Verma, I.M. Cancer-predisposing mutations within the RING domain of BRCA1: Loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc. Natl. Acad. Sci. USA 2001, 98, 5134–5139. [Google Scholar] [CrossRef]
- Mallery, D.L.; Vandenberg, C.J.; Hiom, K. Activation of the E3 ligase function of the BRCA1/BARD complex by polyubiquitin chains. EMBO J. 2002, 21, 6755–6762. [Google Scholar] [CrossRef]
- Baer, R.; Ludwig, T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002, 12, 86–91. [Google Scholar] [CrossRef]
- Lee, J.S.; Chung, J.H. Diverse functions of BRCA1 in the DNA damage response. Expert Rev. Mol. Med. 2001, 2001, 1–11. [Google Scholar]
- Yun, M.H.; Hiom, K. Understanding the functions of BRCA1 in the DNA-damage response. Biochem. Soc. Trans. 2009, 37, 597–604. [Google Scholar] [CrossRef]
- Dunnick, J.K.; Elwell, M.R.; Hugg, J.; Barrett, J.C. Chemically induced mammary gland cancer in the National Toxicology Program’s carcinogenesis bioassay. Carcinogenesis 1995, 16, 173–179. [Google Scholar] [CrossRef]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet. 2001, 28, 266–271. [Google Scholar] [CrossRef]
- Bachelier, R.; Xu, X.; Wang, X.; Li, W.; Naramura, M.; Gu, H.; Deng, C.X. Normal lymphocyte development and thymic lymphoma formation in Brca1 exon-11-deficient mice. Oncogene 2003, 22, 528–537. [Google Scholar] [CrossRef]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senesence, aging, and maliganant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003, 17, 201–213. [Google Scholar] [CrossRef]
- Cao, L.; Kim, S.; Xiao, C.; Wang, R.H.; Coumoul, X.; Wang, X.; Li, W.M.; Xu, X.L.; de Soto, J.A.; Takai, H.; et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 2006, 25, 2167–2177. [Google Scholar] [CrossRef]
- Cao, L.; Xu, X.; Cao, L.L.; Wang, R.H.; Coumoul, X.; Kim, S.S.; Deng, C.X. Absence of full-length Brca1 sensitizes mice to oxidative stress and carcinogen-induced tumorigenesis in the esophagus and forestomach. Carcinogenesis 2007, 28, 1401–1407. [Google Scholar] [CrossRef]
- Francisco, D.C.; Peddi, P.; Hair, J.M.; Flood, B.A.; Cecil, A.M.; Kalogerinis, P.T.; Sigounas, G.; Georgakilas, A.G. Induction and processing of complex DNA damage in human breast cancer cells MCF-7 and nonmalignant MCF-10A cells. Free Radic. Biol. Med. 2008, 44, 558–569. [Google Scholar] [CrossRef]
- Hair, J.M.; Terzoudi, G.I.; Hatzi, V.I.; Lehockey, K.A.; Srivastava, D.; Wang, W.; Pantelias, G.E.; Georgakilas, A.G. BRCA1 role in the mitigation of radiotoxicity and chromosomal instability through repair of clustered DNA lesions. Chem. Biol. Interact. 2010, 188, 350–358. [Google Scholar] [CrossRef]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of abberant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar]
- Le Page, F.; Randrianarison, V.; Marot, D.; Cabannes, J.; Perricaudet, M.; Feunteun, J.; Sarasin, A. BRCA1 and BRCA2 are necessary for the transcription-coupled repair of the oxidative 8-oxoguanine lesion in human cells. Cancer Res. 2000, 60, 5548–5552. [Google Scholar]
- Cadet, J.; Berger, M.; Douki, T.; Ravanat, J.L. Oxidative damage to DNA: Formation, measurement and biological significance. Rev. Physiol. Biochem. Pharmacol. 1997, 131, 1–87. [Google Scholar]
- Dziaman, T.; Huzarski, T.; Gackowski, D.; Rozalski, R.; Siomek, A.; Szpila, A.; Jalanta, G.; Lubinski, J.; Olinski, R. Elevated level of 8-oxi-7,8-dihydro-2'-deoxyguanosine in leukocytes of BRCA1 mutation carriers compared to healthy controls. Int. J. Cancer 2009, 125, 2209–2213. [Google Scholar] [CrossRef]
- Saha, T.; Rih, J.K.; Roy, R.; Ballal, R.; Rosen, E.M. Transcriptional regulation of the base excision repair pathway by BRCA1. J. Biol. Chem. 2010, 285, 19092–19105. [Google Scholar]
- Saha, T.; Rih, J.K.; Rosen, E.M. BRCA1 down-regulates cellular levels of reactive oxygen species. FEBS Lett. 2009, 583, 1535–1543. [Google Scholar] [CrossRef]
- Kang, H.J.; Hong, Y.B.; Kim, H.J.; Wang, A.; Bae, I. Bioactive food components prevent carcinogenic stress via Nrf2 activation in BRCA1 deficient breast epithelial cells. Toxicol. Lett. 2012, 209, 154–160. [Google Scholar] [CrossRef]
- Kang, H.J.; Hong, Y.B.; Yi, Y.W.; Cho, C.H.; Wang, A.; Bae, I. The correlation between BRCA1 defect and environmental factors in the risk of breast cancer. J. Toxicol. Sci. 2013, 38, 355–361. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.; Lin, Z.; Whitaker-Menez, D.; Birbe, R.C.; Bombonati, A.; Pavlides, S.; Lamb, R.; Sneddon, S.; Howell, A.; et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: Implications for breast cancer prevention with antioxidant therapies. Cell Cycle 2012, 11, 4402–4413. [Google Scholar]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Hereditary ovarian cancer and two-compartment tumor metabolism: Epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFκB activation in the tumor stroma. Cell Cycle 2012, 11, 1–15. [Google Scholar] [CrossRef]
- ovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Gupta, N.; Brezden-Masley, C.; Teoh, H.; Wheatcroft, M.D.; Al-Omaran, M.; Verma, S. BRCA1 shields vascular smooth muscle cells from oxidative stress. J. Thorac. Cardiovasc. Surg. 2013. [Google Scholar] [CrossRef]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interaction. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef]
- Somasundaram, K.; Zhang, H.; Zeng, Y.W.; Houvras, Y.; Peng, Y.; Zhang, H.; Wu, G.S.; Licht, J.D.; Wever, B.L.; El-Deiry, W.S. Tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature 1997, 389, 187–190. [Google Scholar] [CrossRef]
- Ouchi, T.; Monteiro, A.N.; August, A.; Aaronson, S.A.; Hanafusa, H. BRCA1 regulates p53-dependent gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 2302–2306. [Google Scholar] [CrossRef]
- Zhang, H.; Somasundaram, K.; Peng, Y.; Tian, H.; Zhang, H.; Bi, D.; Weber, B.L.; El-Deiry, W.S. BRCA1 physically associated with p53 and stimulates its transcriptional activity. Oncogene 1998, 16, 1713–1721. [Google Scholar]
- Chai, Y.L.; Cui, J.; Shao, N.; Shyam, E.; Reddy, P.; Rao, V.N. The second BRCT domain of BRCA1 protein interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter. Oncogene 1999, 18, 263–268. [Google Scholar] [CrossRef]
- MacLachlan, T.K.; Takimoto, R.; El-Deiry, W.S. BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol. Cell. Biol. 2002, 22, 4280–4292. [Google Scholar] [CrossRef]
- Somasundaram, K.; MacLachlan, T.K.; Burns, T.F.; Sgagias, M.; Cowan, K.H.; Weber, B.L.; El-Deiry, W.S. BRCA1 signals ARF-dependent stabilization and coactivation of p53. Oncogene 1999, 18, 6605–6614. [Google Scholar] [CrossRef]
- Fabbro, M.; Savage, K.; Hobson, K.; Deans, A.J.; Powell, S.N.; McArthur, G.A.; Khanna, K.K. BRCA1-BARD1 complexes are required for p53Ser−15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004, 279, 31251–31258. [Google Scholar]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef]
- Bedford, L.; Lowe, J.; Dick, L.R.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [CrossRef]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. A multiubituitin chain is confined to specific lysine in a targeted short-lived proteins. Science 1989, 243, 1576–1583. [Google Scholar]
- Wu-Baer, F.; Lagrazon, K.; Yuan, W.; Baer, R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003, 278, 34743–34746. [Google Scholar] [CrossRef]
- Nishikawa, H.; Ooka, S.; Sato, K.; Arima, K.; Okamoto, J.; Klevit, R.E.; Fukuda, M.; Ohta, T. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 2004, 279, 3916–3924. [Google Scholar]
- Morris, J.R.; Solomon, E. BRCA1:BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum. Mol. Genet. 2004, 13, 807–817. [Google Scholar] [CrossRef]
- Sato, K.; Hayami, R.; Wu, W.; Nishikawa, T.; Nishikawa, H.; Okuda, Y.; Ogata, H.; Fukuda, M.; Ohta, T. Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 2004, 279, 30919–30922. [Google Scholar] [CrossRef]
- Yu, X.; Fu, S.; Lai, M.; Baer, R.; Chen, J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20, 1721–1726. [Google Scholar] [CrossRef]
- Wu, W.; Nishikawa, H.; Hayami, R.; Sato, K.; Honda, A.; Aratani, S.; Nakajima, T.; Fukuda, M.; Ohta, T. BRCA1 ubiquitinates RPB8 in response to DNA damage. Cancer Res. 2007, 67, 951–958. [Google Scholar] [CrossRef]
- Horwitz, A.A.; Affar, E.B.; Heine, G.F.; Shi, Y.; Parvin, J.D. A mechanism for transcriptional repression dependent on the BRCA1 E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2007, 104, 6614–6619. [Google Scholar]
- Chen, A.; Kleiman, F.E.; Manley, J.L.; Ouchi, T.; Pan, Z.Q. Autoubiquitination of the BRCA1-BARD1 RING ubiquitin ligase. J. Biol. Chem. 2002, 277, 22085–22092. [Google Scholar]
- Wu, W.; Koike, A.; Takeshita, T.; Ohta, T. The ubiquitin E3 ligase activity of BRCA1 and its biological functions. Cell Division 2008, 3, 1. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.M. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar]
- Shabbeer, S.; Omer, D.; Berneman, D.; Weitzman, O.; Alpaugh, A.; Pietraszkiewicz, A.; Metsuyanim, S.; Shinskaya, A.; Papa, M.Z.; Yarden, R.I. BRCA1 targets G2/M cell cycle proteins for ubiquitination and proteasomal degradation. Oncogene 2013, 32, 5005–5016. [Google Scholar] [CrossRef]
- Starita, L.M.; Machida, Y.; Sankaran, S.; Elias, J.E.; Griffin, K.; Schlegel, B.P.; Gygi, S.P.; Parvin, J.D. BRCA1-dependent ubiquitination of γ-tubulin regulates centrosome number. Mol. Cell. Biol. 2004, 24, 8457–8466. [Google Scholar] [CrossRef]
- Eakin, C.M.; Maccoss, M.J.; Finney, F.L.; Klevit, R.E. Estrogen receptor α is a putative substrate for the BRCA1 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2007, 104, 5794–5799. [Google Scholar] [CrossRef]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumor suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef]
- Ohta, T.; Sato, K.; Wu, W. The BRCA1 ubiquitin ligase and homologous recombination repair. FEBS Lett. 2011, 585, 2836–2844. [Google Scholar] [CrossRef]
- Caestecker, K.W.; van de Walle, G.R. The role of BRCA1 in DNA double-strand repair: Past and present. Exp. Cell Res. 2013, 319, 575–587. [Google Scholar] [CrossRef]
- Rosen, E.M. BRCA1 in the DNA damage response and at telomeres. Front. Genet. 2013, 4, 85. [Google Scholar] [CrossRef]
- Ouchi, T.; Lee, S.W.; Ouchi, M.; Aaronson, S.A.; Horvath, C.M. Collaboration of signal transducer and activator of transcription 1 (STAT1) and BRCA1 in differential regulation of IFN-γ target genes. Proc. Natl. Acad. Sci. USA 2000, 97, 5208–5213. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21Cip1/WAF1 upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Mitshishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defense pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Takai, J.; Ebina, M.; Yamamoto, M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. [Google Scholar]
- Mullan, P.B.; Quinn, J.E.; Harkin, D.P. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 2006, 25, 5854–5863. [Google Scholar] [CrossRef]
- Chapman, M.S.; Verma, I.M. Transcriptional activation by BRCA1. Nature 1996, 382, 678–679. [Google Scholar] [CrossRef]
- Monteiro, A.N.; August, A.; Hanafusa, H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc. Natl. Acad. Sci. USA 1996, 93, 13595–13599. [Google Scholar] [CrossRef]
- Monteiro, A.N.; August, A.; Hanafusa, H. Common BRCA1 variants and transcriptional activation. Am. J. Hum. Genet. 1997, 61, 761–762. [Google Scholar] [CrossRef]
- Haile, D.T.; Parvin, J.D. Activation of transcription in vitro by the the BRCA1 carboxyl-terminal domain. J. Biol. Chem. 1999, 274, 2113–2117. [Google Scholar] [CrossRef]
- Hu, Y.F.; Hao, Z.L.; Li, R. Chromatin remodeling and activation of chromosomal DNA replication by an acidic transcriptional activation domain from BRCA1. Genes Dev. 1999, 13, 637–642. [Google Scholar] [CrossRef]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef]
- Schlegel, B.P.; Green, J.V.; Ladias, J.A.; Parvin, J.D. BRCA1 interaction with RNA polymerase II reveals a role for hRPB2 and hRPB10α in activated transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 3148–3153. [Google Scholar]
- Anderson, S.F.; Schlegel, B.P.; Nakajima, T.; Wolpin, E.S.; Parvin, J.D. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998, 19, 254–256. [Google Scholar] [CrossRef]
- Pao, G.M.; Janknecht, R.; Ruffner, H.; Hunter, T.; Verma, I.M. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. USA 2000, 97, 1020–1025. [Google Scholar]
- Moisan, A.; Larochelle, C.; Guillemette, B.; Gaudreau, L. BRCA1 can modulate RNA polymerase II carboxy-terminal domain phosphorylation levels. Mol. Cell. Biol. 2004, 24, 6947–6956. [Google Scholar] [CrossRef]
- Chen, G.C.; Guan, L.S.; Yu, J.H.; Li, G.C.; Choi Kim, H.R.; Wang, Z.Y. Rb-associated protein 46 (RbAp46) inhibits transcriptional transactivation mediated by BRCA1. Biochem. Biophys. Res. Commun. 2001, 284, 507–514. [Google Scholar] [CrossRef]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef]
- Fagan, D.H.; Yee, D. Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 423–429. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.; Yuan, R.; Ma, Y.; Meng, Q.; Erdos, M.R.; Pestell, R.G.; Yuan, F.; Auborn, K.J.; Goldberg, I.D.; et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 1999, 284, 1354–1356. [Google Scholar] [CrossRef]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.Q.; Meng, Q.; Wang, J.A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001, 20, 77–87. [Google Scholar] [CrossRef]
- Rosen, E.M.; Fan, S.; Ma, Y. BRCA1 regulation of transcription. Cancer Lett. 2006, 236, 175–185. [Google Scholar] [CrossRef]
- Zheng, L.; Annab, L.A.; Afshari, C.A.; Lee, W.H.; Boyer, T.G. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9587–9592. [Google Scholar]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.Q.; Meng, Q.; Wang, J.A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. p300 modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res. 2002, 62, 141–151. [Google Scholar]
- Wang, C.; Fan, S.; Li, Z.; Fu, M.; Rao, M.; Ma, Y.; Lisanti, M.P.; Albanese, C.; Katzenellenbogen, B.S.; Kushner, P.J.; et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor α activity. Cancer Res. 2005, 65, 6557–6567. [Google Scholar] [CrossRef]
- Xu, J.; Fan, S.; Rosen, E.M. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology 2005, 146, 2031–2047. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, H.; Kajino, K.; Greene, M.I. BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene 1998, 17, 1939–1948. [Google Scholar]
- Li, H.; Lee, T.H.; Avraham, H. A novel tricomplex of BRCA1, Nmi, and c-Myc inhibits c-Myc-induced human telomerase reverse transcriptase gene (hTERT) promoter activity in breast cancer. J. Biol. Chem. 2002, 277, 20965–20973. [Google Scholar]
- Kennedy, R.D.; Gorski, J.J.; Quinn, J.E.; Stewart, G.E.; James, C.R.; Moore, S.; Mulligan, K.; Emberley, E.D.; Lioe, T.F.; Morrison, P.J.; et al. BRCA1 and c-Myc associate to transcriptionally repress psoriasin, a DNA damage-inducible gene. Cancer Res. 2005, 65, 10265–10272. [Google Scholar] [CrossRef]
- Schaeper, U.; Subramanian, T.; Lim, L.; Boyd, J.M.; Chinnadurai, G. Interaction between a cellular protein that binds to the C-terminal region of adenovirus E1A (CtBP) and a novel cellular protein is disrupted by E1A through a conserved PLDLS motif. J. Biol. Chem. 1998, 273, 8549–8552. [Google Scholar]
- Li, S.; Ting, N.S.; Zheng, L.; Chen, P.L.; Ziv, Y.; Shiloh, Y.; Lee, E.Y.; Lee, W.H. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000, 406, 210–215. [Google Scholar] [CrossRef]
- Wu-Baer, F.; Baer, R. Effect of DNA damage on a BRCA1 complex. Nature 2001, 414, 36. [Google Scholar] [CrossRef]
- Zheng, L.; Pan, H.; Li, S.; Flesken-Nikitin, A.; Chen, P.L.; Boyer, T.G.; Lee, W.H. Sequence-specific transcriptional corepressor function for BRCA1 through a novel zinc finger protein, ZBRK1. Mol. Cell 2000, 6, 757–768. [Google Scholar] [CrossRef]
- Tan, W.; Zheng, L.; Lee, W.H.; Boyer, T.G. Functional dissection of transcription factor ZBRK1 reveals zinc fingers with dual roles in DNA-binding and BRCA1-dependent transcriptional repression. J. Biol. Chem. 2004, 279, 6576–6587. [Google Scholar]
- Furuta, S.; Wang, J.M.; Wei, S.; Jeng, Y.M.; Jian, X.; Gu, B.; Chen, P.L.; Lee, E.Y.; Lee, W.H. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell 2006, 10, 13–24. [Google Scholar]
- Ahmed, K.M.; Tsai, C.Y.; Lee, W.H. Derepression of HMGA2 via removal of ZBRK1/BRCA1/CtIP complex enhances mammary tumorigenesis. J. Biol. Chem. 2010, 285, 4464–4471. [Google Scholar] [CrossRef]
- Fan, W.; Jin, S.; Tong, T.; Zhao, H.; Fan, F.; Antinore, M.J.; Rajasekaran, B.; Wu, M.; Zhan, Q. BRCA1 regulates GADD45 through its iteraction with the OCT-1 and CAAT motif. J. Biol. Chem. 2002, 277, 8061–8067. [Google Scholar]
- Andrews, H.N.; Mullan, P.B.; McWilliams, S.; Sebelova, S.; Quinn, J.E.; Gilmore, P.M.; McCabe, N.; Pace, A.; Koller, B.; Johnston, P.G.; et al. BRCA1 regulates the interferon γ-mediated apoptotic response. J. Biol. Chem. 2002, 277, 26225–26232. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar]
- Benezra, M.; Chevallier, N.; Morrison, D.J.; MacLachlan, T.K.; El-Deiry, W.S.; Licht, J.D. BRCA1 augments transcription by the NF-κB transcription factor by binding to the Rel domain of the p65/RelA subunit. J. Biol. Chem. 2003, 278, 26333–26341. [Google Scholar]
- Habraken, Y.; Jolois, O.; Piette, J. Differential involvement of the hMRE11/hRAD50/NBS1 complex, BRCA1 and MLH1 in NF-κB activation by camptothecin and X-ray. Oncogene 2003, 22, 6090–6099. [Google Scholar] [CrossRef]
- Volcic, M.; Karl, S.; Baumann, B.; Salles, D.; Daniel, P.; Fulda, S.; Wiesmüller, L. NF-κB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res. 2012, 40, 181–195. [Google Scholar] [CrossRef]
- Kang, H.J.; Kim, H.J.; Rih, J.K.; Mattson, T.L.; Kim, K.W.; Cho, C.H.; Isaacs, J.S.; Bae, I. BRCA1 plays a role in the hypoxic response by regulating HIF-1α stability and by modulating vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 13047–13056. [Google Scholar]
- Kawai, H.; Li, H.; Chun, P.; Avraham, S.; Avraham, H.K. Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene 2002, 21, 7730–7739. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signaling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Riedemann, J.; Macaulay, V.M. IGF1R signaling and its inhibition. Endocr. Relat. Cancer 2006, 13, S33–S43. [Google Scholar] [CrossRef]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.; Stratford, A.L.; Park, E.; Gee, J.M.; Finlay, P.; Jones, H.E.; et al. Phosphorylated insulin-like growth factor-I/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef]
- Maor, S.B.; Abramovitch, S.; Erdos, M.R.; Brody, L.C.; Werner, H. BRCA1 suppresses insulin-like growth factor-I receptor promoter activity: Potential interaction between BRCA1 and Sp1. Mol. Genet. Metab. 2000, 69, 130–136. [Google Scholar] [CrossRef]
- Abramovitch, S.; Glaser, T.; Ouchi, T.; Werner, H. BRCA1-Sp1 interactions in transcriptional regulation of the IGF-1R gene. FEBS Lett. 2003, 541, 149–154. [Google Scholar] [CrossRef]
- Kang, H.J.; Yi, Y.W.; Kim, H.J.; Hong, Y.B.; Seong, Y.S.; Bae, I. BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell Death Dis. 2012, 3, e336. [Google Scholar] [CrossRef]
- Houvras, Y.; Benezra, M.; Zhang, H.; Manfredi, J.J.; Weber, B.L.; Licht, J.D. BRCA1 physically and functionally interact with ATF1. J. Biol. Chem. 2000, 275, 36230–36237. [Google Scholar]
- Yan, J.; Zhu, J.; Zhong, H.; Lu, Q.; Huang, C.; Ye, Q. BRCA1 interacts with FHL2 and enhances FHL2 transactivation function. FEBS Lett. 2003, 553, 183–189. [Google Scholar]
- Kang, H.J.; Kim, H.J.; Cho, C.H.; Hu, Y.; Li, R.; Bae, I. BRCA1 transcriptional activity is enhanced by interactions between its AD1 domain and AhR. Cancer Chemother. Pharmacol. 2008, 62, 965–975. [Google Scholar] [CrossRef]
- Yeh, S.; Hu, Y.C.; Rahman, M.; Lin, H.K.; Hsu, C.L.; Ting, H.J.; Kang, H.Y.; Chang, C. Increase of androgen-induced cell death and androgen receptor transactivation by BRCA1 in prostate cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 11256–11261. [Google Scholar] [CrossRef]
- Kang, H.J.; Kim, H.J.; Kim, S.K.; Barouki, R.; Cho, C.H.; Khanna, K.K.; Rosen, E.M.; Bae, I. BRCA1 modulates xenobiotic stress-inducible gene expression by interacting with ARNT in human breast cancer cells. J. Biol. Chem. 2006, 281, 14654–14662. [Google Scholar]
- Wang, H.; Shao, N.; Ding, Q.M.; Cui, J.; Reddy, E.S.; Rao, V.N. BRCA1 proteins are transported to the nucleus in the absence of serum and splice variants BRCA1a, BRCA1b are tyrosine phosphoproteins that associate with E2F, cyclins and cyclin dependent kinases. Oncogene 1997, 15, 143–157. [Google Scholar]
- Hu, Y.F.; Li, R. JunB potentiates function of BRCA1 activation domain 1 (AD1) through a coiled-coil-mediated interaction. Genes Dev. 2002, 16, 1509–1517. [Google Scholar] [CrossRef]
- Sum, E.Y.; Peng, B.; Yu, X.; Chen, J.; Byrne, J.; Lindeman, G.J.; Visvader, J.E. The LIM domain protein LMO4 interacts with the cofactor CtIP and the tumor suppressor BRCA1 and inhibits BRCA1 activity. J. Biol. Chem. 2002, 277, 7849–7856. [Google Scholar]
- Vidarsson, H.; Mikaelsdottir, E.K.; Rafnar, T.; Bertwistle, D.; Ashworth, A.; Eyfjord, J.E.; Valgeirsdottir, S. BRCA1 and BRCA2 bind Stat5a and suppress its transcriptional activity. FEBS Lett. 2002, 532, 247–252. [Google Scholar]
- Cable, P.L.; Wilson, C.A.; Calzone, F.J.; Rauscher, F.J., 3rd.; Scully, R.; Livingston, D.M.; Li, L.; Blackwell, C.B.; Futreal, P.A.; Afshari, C.A. Novel consensus DNA-binding sequence for BRCA1 protein complexes. Mol. Carcinog. 2003, 38, 85–96. [Google Scholar] [CrossRef]
- Kang, H.J.; Hong, Y.B.; Kim, H.J.; Rodriguez, O.C.; Nath, R.G.; Tilli, E.M.; Albanese, C.; Chung, F.L.; Kwon, S.H.; Bae, I. Detoxification: A novel function of BRCA1 in tumor suppression? Toxicol. Sci. 2011, 122, 26–37. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar]
- Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 307–340. [Google Scholar]
- Bae, I.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Haddad, B.; Kirilyuk, A.; Fan, S.; Avantaggiati, M.L.; Rosen, E.M. BRCA1 regulates gene expression for orderly mitotic progress. Cell Cycle 2005, 4, 1641–1666. [Google Scholar] [CrossRef]
- Bae, I.; Fan, S.; Meng, Q.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Xu, J.; Goldberg, I.D.; Jaiswal, A.K.; Rosen, E.M. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004, 64, 7893–7909. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Wang, Q.; Li, J.; Yang, X.; Sun, H.; Gao, S.; Zhu, H.; Wu, J.; Jin, W. Nrf2 is associated with the regulation of basal transcription activity of the BRCA1 gene. Acta Biochim. Biophys. Sin. 2013, 45, 179–187. [Google Scholar]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA1 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef]
- Ouchi, T. BRCA1 phosphorylation: Biological consequence. Cancer Biol. Ther. 2006, 5, 470–475. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef]
- Guo, Z.; Deshpande, R.; Paull, T.T. ATM activation in the presense of oxidative stress. Cell Cycle 2010, 9, 4805–4811. [Google Scholar] [CrossRef]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Nardo, A.D.; Han, J.M.; Kwiatkowski, E.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Chen, B.P.C.; Li, M.; Asaithamby, A. New insights into the roles of ATM and DNA-PKcs in the cellular response to oxidative stress. Cancer Lett. 2012, 327, 103–110. [Google Scholar] [CrossRef]
- Jorgenson, T.C.; Zhong, W.; Oberley, T.D. Redox imbalance and biochemical changes in cancer. Cancer Res. 2013, 73, 6118–6123. [Google Scholar] [CrossRef]
- Miki, H.; Funato, Y. Regulation of intracellular signaling through cysteine oxidation by reactive oxygen species. J. Biochem. 2012, 151, 255–261. [Google Scholar]
- Paulsen, C.E.; Carroll, K. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013. [Google Scholar] [CrossRef]
- Lau, A.T.Y.; Wang, Y.; Chiu, J.F. Reactive oxygen species: Current knowledge and applications in cancer research and therapeutic. J. Cell. Biochem. 2008, 104, 657–667. [Google Scholar]
- Chibber, S.; Farhan, M.; Hassan, I.; Naseem, I. Novel aspect of chemophototherapy in treatment of cancer. Tumor Biol. 2012, 33, 701–706. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathway: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by small molecule targeting the stress responses to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef]
- Tassone, P.; Tagliaferri, P.; Perricelli, A.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Costanzo, F.; Boland, C.R.; et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar] [CrossRef]
- Santarosa, M.; del Col, L.; Tonin, E.; Caragnano, A.; Viel, A.; Maestro, R. Premature senescence is a major response to DNA cross-linking agents in BRCA1-defective cells: Implication for tailored treatments of BRCA1 mutation carriers. Mol. Cancer Ther. 2009, 8, 844–854. [Google Scholar]
- Fasano, J.; Muggia, F. Breast cancer arising in a BRCA-mutated background: Therapeutic implications from an animal model and drug development. Ann. Oncol. 2009, 20, 609–614. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulcation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Parri, M.; Chiarugi, P. Redox molecular machines involved in tumor progression. Antioxid. Redox Signal. 2013, 19, 1828–1845. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yi, Y.W.; Kang, H.J.; Bae, I. BRCA1 and Oxidative Stress. Cancers 2014, 6, 771-795. https://doi.org/10.3390/cancers6020771
Yi YW, Kang HJ, Bae I. BRCA1 and Oxidative Stress. Cancers. 2014; 6(2):771-795. https://doi.org/10.3390/cancers6020771
Chicago/Turabian StyleYi, Yong Weon, Hyo Jin Kang, and Insoo Bae. 2014. "BRCA1 and Oxidative Stress" Cancers 6, no. 2: 771-795. https://doi.org/10.3390/cancers6020771
APA StyleYi, Y. W., Kang, H. J., & Bae, I. (2014). BRCA1 and Oxidative Stress. Cancers, 6(2), 771-795. https://doi.org/10.3390/cancers6020771