Updates and Controversies in the Rapidly Evolving Field of Lung Cancer Screening, Early Detection, and Chemoprevention

Abstract
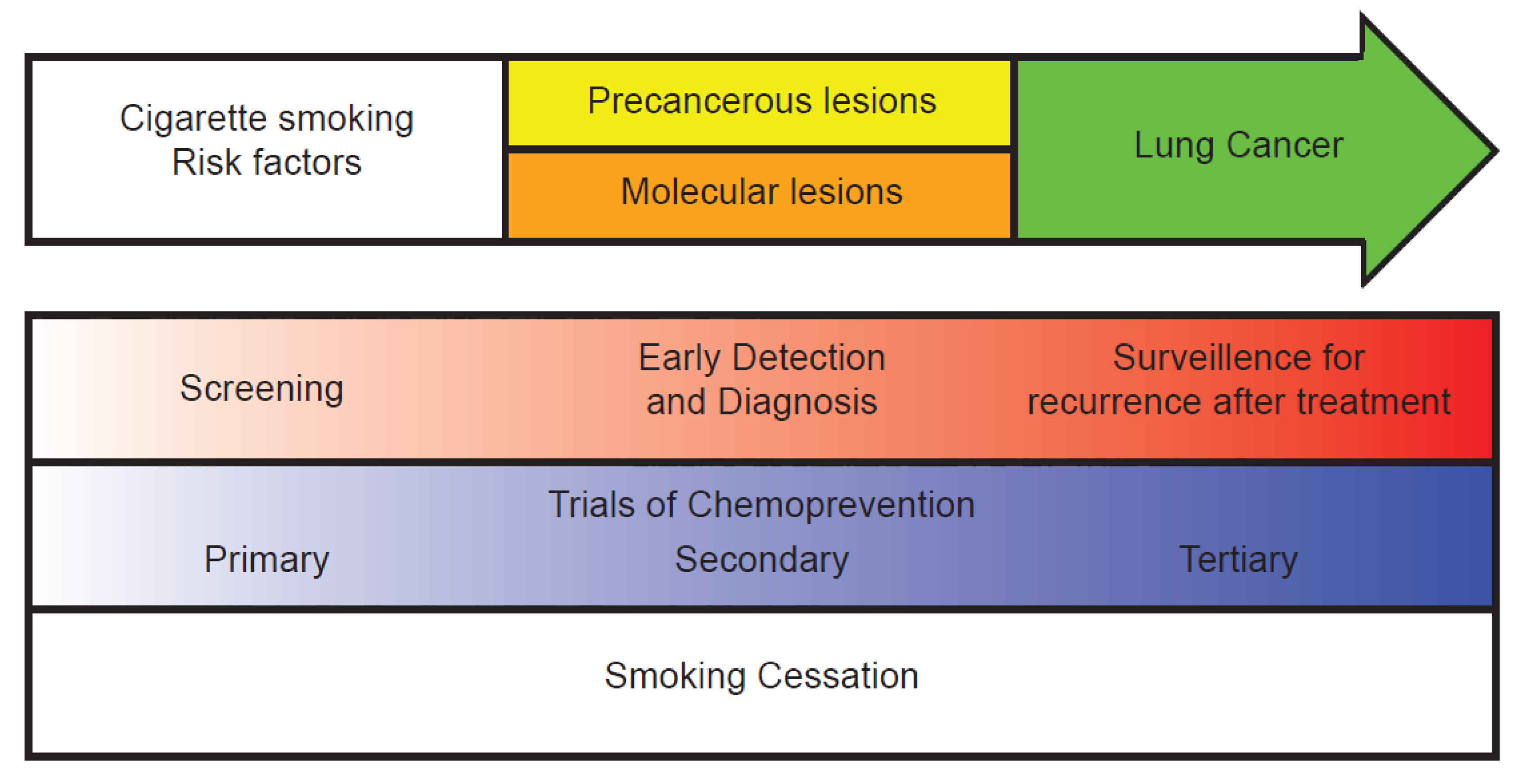
:1. Introduction
2. Quantifying Lung Cancer Risk
3. Imaging-Based Lung Cancer Screening

4. Early Detection Biomarkers for Lung Cancer
4.1. Bronchial Airway Biomarkers
4.2. Sputum Biomarkers
4.3. Blood-Based Biomarkers
5. Risk Reduction and Chemoprevention
5.1. Smoking Cessation
5.2. Chemoprevention
5.3. Clinical Trials

{kind=link}
| Chemoprevention group | Antioxidants | Anti-inflammatories | PI3K/AKT/mTOR Pathway inhibitors |
|---|---|---|---|
| Primary chemoprevention Healthy patients at high risk for lung cancer | α-Tocopherol [82,83,84] β-Carotene [82,83,84,85,86,87] Selenium [88,89,90,91] | NSAIDs [92,93,94,95] PPARγ agonists [96,97,98] | |
| Secondary chemoprevention Patients with pre-cancerous lesions | Retinoids [99,100,101,102] Retinoids + β-carotene [103] ADT [104] | Celecoxib [105,106] Iloprost [107] | Myoinositol [108,109] |
| Tertiary chemoprevention Patients with a previous lung cancer that has been treated | Retinoids [110,111] Selenium [112] |
5.4. Primary Chemoprevention
5.4.1. Antioxidants
5.4.2. Anti-Inflammatories
5.5. Secondary Chemoprevention
5.5.1. Antioxidants
5.5.2. Anti-Inflammatories
5.5.3. PI3K/AKT/mTOR Pathway Inhibitors
5.6. Tertiary Chemoprevention: Antioxidants
6. Conclusions
6.1. The Future of Lung Cancer Prevention, Screening, and Early Detection
6.2. Intermediate Markers of Disease Risk as Potential Targets for Personalized Chemoprevention
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Yabroff, K.R.; Lamont, E.B.; Mariotto, A.; Warren, J.L.; Topor, M.; Meekins, A.; Brown, M.L. Cost of care for elderly cancer patients in the United States. J. Natl. Cancer Inst. 2008, 100, 630–641. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Neyman, N.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. SEER cancer statistics review (CSR), 1975–2010. Available online: http://seer.cancer.gov/csr/1975_2010/sections.html (accessed on 11 December 2013).
- Humphrey, L.L.; Deffebach, M.; Pappas, M.; Baumann, C.; Artis, K.; Mitchell, J.P.; Zakher, B.; Fu, R.; Slatore, C.G. Screening for lung cancer with low-dose computed tomography: A systematic review to update the US Preventive services task force recommendation. Ann. Intern. Med. 2013, 159, 411–420. [Google Scholar] [CrossRef]
- Wakelee, H.A.; Chang, E.T.; Gomez, S.L.; Keegan, T.H.; Feskanich, D.; Clarke, C.A.; Holmberg, L.; Yong, L.C.; Kolonel, L.N.; Gould, M.K.; et al. Lung cancer incidence in never smokers. J. Clin. Oncol. 2007, 25, 472–478. [Google Scholar] [CrossRef]
- Thu, K.L.; Vucic, E.A.; Chari, R.; Zhang, W.; Lockwood, W.W.; English, J.C.; Fu, R.; Wang, P.; Feng, Z.; MacAulay, C.E.; et al. Lung adenocarcinoma of never smokers and smokers harbor differential regions of genetic alteration and exhibit different levels of genomic instability. PLoS One 2012, 7, e33003. [Google Scholar]
- Bach, P.B.; Kattan, M.W.; Thornquist, M.D.; Kris, M.G.; Tate, R.C.; Barnett, M.J.; Hsieh, L.J.; Begg, C.B. Variations in lung cancer risk among smokers. J. Natl. Cancer Inst. 2003, 95, 470–478. [Google Scholar] [CrossRef]
- Cronin, K.A.; Gail, M.H.; Zou, Z.; Bach, P.B.; Virtamo, J.; Albanes, D. Validation of a model of lung cancer risk prediction among smokers. J. Natl. Cancer Inst. 2006, 98, 637–640. [Google Scholar] [CrossRef]
- Spitz, M.R.; Hong, W.K.; Amos, C.I.; Wu, X.; Schabath, M.B.; Dong, Q.; Shete, S.; Etzel, C.J. A risk model for prediction of lung cancer. J. Natl. Cancer Inst. 2007, 99, 715–726. [Google Scholar] [CrossRef]
- Cassidy, A.; Myles, J.P.; van Tongeren, M.; Page, R.D.; Liloglou, T.; Duffy, S.W.; Field, J.K. The LLP risk model: An individual risk prediction model for lung cancer. Br. J. Cancer 2008, 98, 270–276. [Google Scholar] [CrossRef]
- D’Amelio, A.M.; Cassidy, A.; Asomaning, K.; Raji, O.Y.; Duffy, S.W.; Field, J.K.; Spitz, M.R.; Christiani, D.; Etzel, C.J. Comparison of discriminatory power and accuracy of three lung cancer risk models. Cancer Prev. Res. (Phila) 2010, 3. Abstract A127. [Google Scholar]
- Iyen-Omofoman, B.; Tata, L.J.; Baldwin, D.R.; Smith, C.J.P.; Hubbard, R.B. Using socio-demographic and early clinical features in general practice to identify people with lung cancer earlier. Thorax 2013, 68, 451–459. [Google Scholar] [CrossRef]
- Maisonneuve, P.; Bagnardi, V.; Bellomi, M.; Spaggiari, L.; Pelosi, G.; Rampinelli, C.; Bertolotti, R.; Rotmensz, N.; Field, J.K.; Decensi, A.; et al. Lung cancer risk prediction to select smokers for screening CT—A model based on the Italian COSMOS trial. Cancer Prev. Res. (Phila) 2011, 4, 1778–1789. [Google Scholar] [CrossRef]
- Thun, M.J.; Henley, S.J.; Calle, E.E. Tobacco use and cancer: An epidemiologic perspective for geneticists. Oncogene 2002, 21, 7307–7325. [Google Scholar] [CrossRef]
- Cooper, D.N. The Molecular Genetics of Lung Cancer; Springer: Heidelberg, Germany, 2005. [Google Scholar]
- Zienolddiny, S.; Campa, D.; Lind, H.; Ryberg, D.; Skaug, V.; Stangeland, L.; Phillips, D.H.; Canzian, F.; Haugen, A. Polymorphisms of DNA repair genes and risk of non-small cell lung cancer. Carcinogenesis 2006, 27, 560–567. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Hay, B.A.; Epton, M.J.; Mills, G.D.; Black, P.N.; Gardner, H.D.; Sullivan, R.; Gamble, G.D. Lung cancer susceptibility model based on age, family history and genetic variants. PLoS One 2009, 4, e5302. [Google Scholar] [CrossRef]
- Spitz, M.R.; Etzel, C.J.; Dong, Q.; Amos, C.I.; Wei, Q.; Wu, X.; Hong, W.K. An expanded risk prediction model for lung cancer. Cancer Prev. Res. (Phila) 2008, 1, 250–254. [Google Scholar] [CrossRef]
- Patz, E.F., Jr.; Goodman, P.C.; Bepler, G. Screening for lung cancer. N. Engl. J. Med. 2000, 343, 1627–1633. [Google Scholar] [CrossRef]
- Brett, G.Z. Earlier diagnosis and survival in lung cancer. Br. Med. J. 1969, 4, 260–262. [Google Scholar] [CrossRef]
- Oken, M.M.; Hocking, W.G.; Kvale, P.A.; Andriole, G.L.; Buys, S.S.; Church, T.R.; Crawford, E.D.; Fouad, M.N.; Isaacs, C.; Reding, D.J.; et al. Screening by chest radiograph and lung cancer mortality: The Prostate, Lung, Colorectal, and Ovarian (PLCO) randomized trial. JAMA 2011, 306, 1865–1873. [Google Scholar] [CrossRef]
- Marcus, P.M.; Bergstralh, E.J.; Fagerstrom, R.M.; Williams, D.E.; Fontana, R.; Taylor, W.F.; Prorok, P.C. Lung cancer mortality in the Mayo Lung Project: Impact of extended follow-up. J. Natl. Cancer Inst. 2000, 92, 1308–1316. [Google Scholar] [CrossRef]
- Jett, J.R.; Midthun, D.E. Screening for lung cancer: Current status and future directions: Thomas A. Neff lecture. Chest 2004, 125, 158S–162S. [Google Scholar] [CrossRef]
- Manser, R.; Lethaby, A.; Irving, L.B.; Stone, C.; Byrnes, G.; Abramson, M.J.; Campbell, D. Screening for lung cancer. Cochrane Database Syst. Rev. 2013, 6. [Google Scholar] [CrossRef]
- Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; et al. Trial research team reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef]
- Moyer, V.A. Screening for Lung Cancer: U.S. preventive services task force recommendation statement. Ann. Intern. Med. 2013, 160, 330–338. [Google Scholar]
- Van Iersel, C.A.; de Koning, H.J.; Draisma, G.; Mali, W.P.T.M.; Scholten, E.T.; Nackaerts, K.; Prokop, M.; Habbema, J.D.F.; Oudkerk, M.; van Klaveren, R.J. Risk-based selection from the general population in a screening trial: Selection criteria, recruitment and power for the Dutch-Belgian randomised lung cancer multi-slice CT screening trial (NELSON). Int. J. Cancer 2007, 120, 868–874. [Google Scholar] [CrossRef]
- Heuvelmans, M.A.; Oudkerk, M.; de Bock, G.H.; de Koning, H.J.; Xie, X.; van Ooijen, P.M.A.; Greuter, M.J.W.; de Jong, P.A.; Groen, H.J.M.; Vliegenthart, R. Optimisation of volume-doubling time cutoff for fast-growing lung nodules in CT lung cancer screening reduces false-positive referrals. Eur. Radiol. 2013, 23, 1836–1845. [Google Scholar] [CrossRef]
- Saghir, Z.; Dirksen, A.; Ashraf, H.; Bach, K.S.; Brodersen, J.; Clementsen, P.F.; Døssing, M.; Hansen, H.; Kofoed, K.F.; Larsen, K.R.; et al. CT screening for lung cancer brings forward early disease. The randomised Danish Lung Cancer Screening Trial: Status after five annual screening rounds with low-dose CT. Thorax 2012, 67, 296–301. [Google Scholar] [CrossRef]
- Nackaerts, K.; Vansteenkiste, J. Low-dose CT screening for lung cancer: Trial and error? J. Thorac. Oncol. 2009, 4, 563–564. [Google Scholar] [CrossRef]
- Van den Bergh, K.A.M.; Essink-Bot, M.-L.; Bunge, E.M.; Scholten, E.T.; Prokop, M.; van Iersel, C.A.; van Klaveren, R.J.; de Koning, H.J. Impact of computed tomography screening for lung cancer on participants in a randomized controlled trial (NELSON trial). Cancer 2008, 113, 396–404. [Google Scholar] [CrossRef]
- Van den Bergh, K.A.M.; Essink-Bot, M.L.; Borsboom, G.J.J.M.; Scholten, E.T.; van Klaveren, R.J.; de Koning, H.J. Long-term effects of lung cancer computed tomography screening on health-related quality of life: The NELSON trial. Eur. Respir. J. 2011, 38, 154–161. [Google Scholar] [CrossRef]
- Braithwaite, D.; Zhu, W.; Hubbard, R.A.; O’Meara, E.S.; Miglioretti, D.L.; Geller, B.; Dittus, K.; Moore, D.; Wernli, K.J.; Mandelblatt, J.; et al. Screening outcomes in older US women undergoing multiple mammograms in community practice: Does interval, age, or comorbidity score affect tumor characteristics or false positive rates? J. Natl. Cancer Inst. 2013, 105, 334–341. [Google Scholar] [CrossRef]
- Kovalchik, S.A.; Tammemagi, M.; Berg, C.D.; Caporaso, N.E.; Riley, T.L.; Korch, M.; Silvestri, G.A.; Chaturvedi, A.K.; Katki, H.A. Targeting of low-dose CT screening according to the risk of lung-cancer death. N. Engl. J. Med. 2013, 369, 245–254. [Google Scholar] [CrossRef]
- Mettler, F.A., Jr.; Huda, W.; Yoshizumi, T.T.; Mahesh, M. Effective doses in radiology and diagnostic nuclear medicine: A catalog. Radiology 2008, 248, 254–263. [Google Scholar] [CrossRef]
- Bach, P.B.; Mirkin, J.N.; Oliver, T.K.; Azzoli, C.G.; Berry, D.A.; Brawley, O.W.; Byers, T.; Colditz, G.A.; Gould, M.K.; Jett, J.R.; et al. Benefits and harms of CT screening for lung cancer: A systematic review. JAMA 2012, 307, 2418–2429. [Google Scholar] [CrossRef]
- Tokarskaya, Z.B.; Scott, B.R.; Zhuntova, G.V.; Okladnikova, N.D.; Belyaeva, Z.D.; Khokhryakov, V.F.; Schöllnberger, H.; Vasilenko, E.K. Interaction of radiation and smoking in lung cancer induction among workers at the Mayak nuclear enterprise. Health Phys. 2002, 83, 833–846. [Google Scholar] [CrossRef]
- Silvestri, G.A. Screening for lung cancer: It works, but does it really work? Ann. Intern. Med. 2011, 155, 537–539. [Google Scholar] [CrossRef]
- Patz, E.F., Jr.; Pinsky, P.; Gatsonis, C.; Sicks, J.D.; Kramer, B.S.; Tammemägi, M.C.; Chiles, C.; Black, W.C.; Aberle, D.R. Overdiagnosis in low-dose computed tomography screening for lung cancer. JAMA Intern. Med. 2013, 174, 269–274. [Google Scholar] [CrossRef]
- Yu, L.; Todd, N.W.; Xing, L.; Xie, Y.; Zhang, H.; Liu, Z.; Fang, H.; Zhang, J.; Katz, R.L.; Jiang, F. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int. J. Cancer 2010, 127, 2870–2878. [Google Scholar] [CrossRef]
- Franklin, W.A.; Gazdar, A.F.; Haney, J.; Wistuba, I.I.; LaRosa, F.G.; Kennedy, T.; Ritchey, D.M.; Miller, Y.E. Widely dispersed p53 mutation in respiratory epithelium. A novel mechanism for field carcinogenesis. J. Clin. Investig. 1997, 100, 2133–2137. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Lam, S.; Behrens, C.; Virmani, A.K.; Fong, K.M.; LeRiche, J.; Samet, J.M.; Srivastava, S.; Minna, J.D.; Gazdar, A.F. Molcular damage in the bronchial epithelium of current and former smokers. J. Natl. Cancer Inst. 1997, 89, 1366–1373. [Google Scholar] [CrossRef]
- Mao, L.; Lee, J.S.; Kurie, J.M.; Fan, Y.H.; Lippman, S.M.; Lee, J.J.; Ro, J.Y.; Broxson, A.; Yu, R.; Morice, R.C.; et al. Clonal genetic alterations in the lungs of current and former smokers. J. Natl. Cancer Inst. 1997, 89, 857–862. [Google Scholar] [CrossRef]
- Tang, X.; Shigematsu, H.; Bekele, B.N.; Roth, J.A.; Minna, J.D.; Hong, W.K.; Gazdar, A.F.; Wistuba, I.I. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res. 2005, 65, 7568–7572. [Google Scholar]
- Powell, C.A.; Klares, S.; O’Connor, G.; Brody, J.S. Loss of heterozygosity in epithelial cells obtained by bronchial brushing: Clinical utility in lung cancer. Clin. Cancer Res. 1999, 5, 2025–2034. [Google Scholar]
- Spira, A.; Beane, J.E.; Shah, V.; Steiling, K.; Liu, G.; Schembri, F.; Gilman, S.; Dumas, Y.-M.; Calner, P.; Sebastiani, P.; et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med. 2007, 13, 361–366. [Google Scholar] [CrossRef]
- Beane, J.; Sebastiani, P.; Whitfield, T.H.; Steiling, K.; Dumas, Y.-M.; Lenburg, M.E.; Spira, A. A prediction model for lung cancer diagnosis that integrates genomic and clinical features. Cancer Prev. Res. (Phila) 2008, 1, 56–64. [Google Scholar] [CrossRef]
- Li, Q.K.; Shah, P.; Li, Y.; Aiyetan, P.O.; Chen, J.; Yung, R.; Molena, D.; Gabrielson, E.; Askin, F.; Chan, D.W.; et al. Glycoproteomic analysis of bronchoalveolar lavage (BAL) fluid identifies tumor-associated glycoproteins from lung adenocarcinoma. J. Proteome Res. 2013, 12, 3689–3696. [Google Scholar] [CrossRef]
- Hassanein, M.; Callison, J.C.; Callaway-Lane, C.; Aldrich, M.C.; Grogan, E.L.; Massion, P.P. The state of molecular biomarkers for the early detection of lung cancer. Cancer Prev. Res. (Phila) 2012, 5, 992–1006. [Google Scholar] [CrossRef]
- Spira, A.; Beane, J.; Shah, V.; Liu, G.; Schembri, F.; Yang, X.; Palma, J.; Brody, J.S. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc. Natl. Acad. Sci. USA 2004, 101, 10143–10148. [Google Scholar]
- Steiling, K.; Kadar, A.Y.; Bergerat, A.; Flanigon, J.; Sridhar, S.; Shah, V.; Ahman, R.Q.; Brody, J.S.; Lenburg, M.E.; Steffen, M.; et al. Comparison of proteomic and transcriptomic profiles in the bronchial airway of current and never smokers. PLoS One 2009, 4, e5043. [Google Scholar] [CrossRef]
- Sridhar, S.; Schembri, F.; Zeskind, J.; Shah, V.; Gustafson, A.M.; Steiling, K.; Liu, G.; Dumas, Y.-M.; Zhang, X.; Brody, J.S.; et al. Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genomics 9, 2008. [Google Scholar] [CrossRef]
- Zhang, X.; Sebastiani, P.; Liu, G.; Schembri, F.; Zhang, X.; Dumas, Y.M.; Langer, E.M.; Alekseyev, Y.; O’Connor, G.T.; Brooks, D.R.; et al. Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol. Genomics 2010, 41, 1–8. [Google Scholar] [CrossRef]
- Boyle, J.O.; Gümüs, Z.H.; Kacker, A.; Choksi, V.L.; Bocker, J.M.; Zhou, X.K.; Yantiss, R.K.; Hughes, D.B.; Du, B.; Judson, B.L.; et al. Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res. (Phila) 2010, 3, 266–278. [Google Scholar] [CrossRef]
- Flehinger, B.J.; Melamed, M.R.; Zaman, M.B.; Heelan, R.T.; Perchick, W.B.; Martini, N. Early lung cancer detection: Results of the initial (prevalence) radiologic and cytologic screening in the Memorial Sloan-Kettering study. Am. Rev. Respir. Dis. 1984, 130, 555–560. [Google Scholar]
- Mao, L.; Hruban, R.H.; Boyle, J.O.; Tockman, M.; Sidransky, D. Detection of oncogene mutations in sputum precedes diagnosis of lung cancer. Cancer Res. 1994, 54, 1634–1637. [Google Scholar]
- Li, R.; Todd, N.W.; Qiu, Q.; Fan, T.; Zhao, R.Y.; Rodgers, W.H.; Fang, H.-B.; Katz, R.L.; Stass, S.A.; Jiang, F. Genetic deletions in sputum as diagnostic markers for early detection of stage I non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 482–487. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Palmisano, W.A.; Gilliland, F.D.; Crooks, L.A.; Divine, K.K.; Winters, S.A.; Grimes, M.J.; Harms, H.J.; Tellez, C.S.; Smith, T.M.; et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002, 62, 2370–2377. [Google Scholar]
- Palmisano, W.A.; Divine, K.K.; Saccomanno, G.; Gilliland, F.D.; Baylin, S.B.; Herman, J.G.; Belinsky, S.A. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000, 60, 5954–5958. [Google Scholar]
- Diamandis, E.P.; Goodglick, L.; Planque, C.; Thornquist, M.D. Pentraxin-3 is a novel biomarker of lung carcinoma. Clin. Cancer Res. 2011, 17, 2395–2399. [Google Scholar] [CrossRef]
- Ajona, D.; Pajares, M.J.; Corrales, L.; Perez-Gracia, J.L.; Agorreta, J.; Lozano, M.D.; Torre, W.; Massion, P.P.; de-Torres, J.P.; Jantus-Lewintre, E.; et al. Investigation of complement activation product c4d as a diagnostic and prognostic biomarker for lung cancer. J. Natl. Cancer Inst. 2013, 105, 1385–1393. [Google Scholar] [CrossRef]
- Boeri, M.; Verri, C.; Conte, D.; Roz, L.; Modena, P.; Facchinetti, F.; Calabrò, E.; Croce, C.M.; Pastorino, U.; Sozzi, G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3713–3718. [Google Scholar] [CrossRef]
- Bianchi, F.; Nicassio, F.; Marzi, M.; Belloni, E.; Dall’olio, V.; Bernard, L.; Pelosi, G.; Maisonneuve, P.; Veronesi, G.; di Fiore, P.P. A serum circulating miRNA diagnostic test to identify asymptomatic high-risk individuals with early stage lung cancer. EMBO Mol. Med. 2011, 3, 495–503. [Google Scholar] [CrossRef]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef]
- Wilson, I.M.; Davies, J.J.; Weber, M.; Brown, C.J.; Alvarez, C.E.; MacAulay, C.; Schübeler, D.; Lam, W.L. Epigenomics: Mapping the methylome. Cell Cycle 2006, 5, 155–158. [Google Scholar] [CrossRef]
- Ostroff, R.M.; Bigbee, W.L.; Franklin, W.; Gold, L.; Mehan, M.; Miller, Y.E.; Pass, H.I.; Rom, W.N.; Siegfried, J.M.; Stewart, A.; et al. Unlocking biomarker discovery: Large scale application of aptamer proteomic technology for early detection of lung cancer. PLoS One 2010, 5, e15003. [Google Scholar] [CrossRef]
- Tanaka, F.; Yoneda, K.; Kondo, N.; Hashimoto, M.; Takuwa, T.; Matsumoto, S.; Okumura, Y.; Rahman, S.; Tsubota, N.; Tsujimura, T.; et al. Circulating tumor cell as a diagnostic marker in primary lung cancer. Clin. Cancer Res. 2009, 15, 6980–6986. [Google Scholar] [CrossRef]
- Yildiz, P.B.; Shyr, Y.; Rahman, J.S.M.; Wardwell, N.R.; Zimmerman, L.J.; Shakhtour, B.; Gray, W.H.; Chen, S.; Li, M.; Roder, H.; et al. Diagnostic accuracy of MALDI mass spectrometric analysis of unfractionated serum in lung cancer. J. Thorac. Oncol. 2007, 2, 893–901. [Google Scholar] [CrossRef]
- Pecot, C.V.; Li, M.; Zhang, X.J.; Rajanbabu, R.; Calitri, C.; Bungum, A.; Jett, J.R.; Putnam, J.B.; Callaway-Lane, C.; Deppen, S.; et al. Added value of a serum proteomic signature in the diagnostic evaluation of lung nodules. Cancer Epidemiol. Biomark. Prev. 2012, 21, 786–792. [Google Scholar] [CrossRef]
- Li, X.; Hayward, C.; Fong, P.-Y.; Dominguez, M.; Hunsucker, S.W.; Lee, L.W.; McLean, M.; Law, S.; Butler, H.; Schirm, M.; et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Shopland, D.R. Tobacco use and its contribution to early cancer mortality with a special emphasis on cigarette smoking. Environ. Health Perspect. 1995, 103, 131–142. [Google Scholar] [CrossRef]
- King, B.; Dube, S.; Kaufmann, R.; Shaw, L.; Pechacek, T. Vital signs: Current cigarette smoking among adults aged ≥18 Years—United States, 2005–2010. Morb. Mortal. Wkly. Rep. 2011, 60, 1207–1212. [Google Scholar]
- Anthonisen, N.R.; Skeans, M.A.; Wise, R.A.; Manfreda, J.; Kanner, R.E.; Connett, J.E. The effects of a smoking cessation intervention on 14.5-year mortality: A randomized clinical trial. Ann. Intern. Med. 2005, 142, 233–239. [Google Scholar] [CrossRef]
- Villanti, A.C.; Jiang, Y.; Abrams, D.B.; Pyenson, B.S. A cost-utility analysis of lung cancer screening and the additional benefits of incorporating smoking cessation interventions. PLoS One 2013, 8, e71379. [Google Scholar]
- Sporn, M.B. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Res. 1976, 36, 2699–2702. [Google Scholar]
- Hirsch, F.R.; Lippman, S.M. Advances in the biology of lung cancer chemoprevention. J. Clin. Oncol. 2005, 23, 3186–3197. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Lippman, S.M.; Dannenberg, A.J.; Sigman, C.C.; Pearce, H.L.; Reid, B.J.; Szabo, E.; Jordan, V.C.; Spitz, M.R.; Mills, G.B.; et al. Progress in chemoprevention drug development: The promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer—A plan to move forward. Clin. Cancer Res. 2006, 12, 3661–3697. [Google Scholar] [CrossRef]
- Khuri, F.R.; Cohen, V. Molecularly targeted approaches to the chemoprevention of lung cancer. Clin. Cancer Res. 2004, 10, 4249s–4253s. [Google Scholar]
- Keith, R.L. Chemoprevention of lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 187–193. [Google Scholar] [CrossRef]
- Greenberg, A.K.; Tsay, J.-C.; Tchou-Wong, K.-M.; Jorgensen, A.; Rom, W.N. Chemoprevention of lung cancer: Prospects and disappointments in human clinical trials. Cancers 2013, 5, 131–148. [Google Scholar] [CrossRef]
- Keith, R.L.; Miller, Y.E. Lung cancer chemoprevention: Current status and future prospects. Nat. Rev. Clin. Oncol. 2013, 10, 334–343. [Google Scholar] [CrossRef]
- The α-Tocopherol, β carotene cancer prevention study group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Lee, I.M.; Cook, N.R.; Manson, J.E.; Buring, J.E.; Hennekens, C.H. Beta-carotene supplementation and incidence of cancer and cardiovascular disease: The Women’s Health Study. J. Natl. Cancer Inst. 1999, 91, 2102–2106. [Google Scholar] [CrossRef]
- Hennekens, C.H.; Buring, J.E.; Manson, J.E.; Stampfer, M.; Rosner, B.; Cook, N.R.; Belanger, C.; LaMotte, F.; Gaziano, J.M.; Ridker, P.M.; et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1145–1149. [Google Scholar] [CrossRef]
- Albanes, D.; Heinonen, O.P.; Taylor, P.R.; Virtamo, J.; Edwards, B.K.; Rautalahti, M.; Hartman, A.M.; Palmgren, J.; Freedman, L.S.; Haapakoski, J.; et al. α-Tocopherol and beta-carotene supplements and lung cancer incidence in the α-tocopherol, beta-carotene cancer prevention study: Effects of base-line characteristics and study compliance. J. Natl. Cancer Inst. 1996, 88, 1560–1570. [Google Scholar] [CrossRef]
- Virtamo, J.; Pietinen, P.; Huttunen, J.K.; Korhonen, P.; Malila, N.; Virtanen, M.J.; Albanes, D.; Taylor, P.R.; Albert, P.; ATBC Study Group. Incidence of cancer and mortality following alpha-tocopherol and beta-carotene supplementation: A postintervention follow-up. JAMA 2003, 290, 476–485. [Google Scholar]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.; Williams, J.H.; et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155. [Google Scholar] [CrossRef]
- Clark, L.C.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA 1996, 276, 1957–1963. [Google Scholar] [CrossRef]
- Reid, M.E.; Duffield-Lillico, A.J.; Garland, L.; Turnbull, B.W.; Clark, L.C.; Marshall, J.R. Selenium supplementation and lung cancer incidence: An update of the nutritional prevention of cancer trial. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1285–1291. [Google Scholar]
- Fleet, J.C. Dietary selenium repletion may reduce cancer incidence in people at high risk who live in areas with low soil selenium. Nutr. Rev. 1997, 55, 277–279. [Google Scholar] [CrossRef]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar]
- Xu, J.; Yin, Z.; Gao, W.; Liu, L.; Wang, R.; Huang, P.; Yin, Y.; Liu, P.; Yu, R.; Shu, Y. Meta-analysis on the association between nonsteroidal anti-inflammatory drug use and lung cancer risk. Clin. Lung Cancer 2012, 13, 44–51. [Google Scholar] [CrossRef]
- Slatore, C.G.; Au, D.H.; Littman, A.J.; Satia, J.A.; White, E. Association of nonsteroidal anti-inflammatory drugs with lung cancer: Results from a large cohort study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1203–1207. [Google Scholar] [CrossRef]
- Cook, N.R.; Lee, I.-M.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Low-dose aspirin in the primary prevention of cancer: The Women’s Health Study: A randomized controlled trial. JAMA 2005, 294, 47–55. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Govindarajan, R.; Ratnasinghe, L.; Simmons, D.L.; Siegel, E.R.; Midathada, M.V.; Kim, L.; Kim, P.J.; Owens, R.J.; Lang, N.P. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J. Clin. Oncol. 2007, 25, 1476–1481. [Google Scholar] [CrossRef]
- Colmers, I.N.; Bowker, S.L.; Johnson, J.A. Thiazolidinedione use and cancer incidence in type 2 diabetes: A systematic review and meta-analysis. Diabetes Metab. 2012, 38, 475–484. [Google Scholar] [CrossRef]
- Pioglitazone for Lung Cancer Chemoprevention. Available online: http://clinicaltrials.gov/show/NCT00780234 (accessed on 14 December 2013).
- Lee, J.S.; Lippman, S.M.; Benner, S.E.; Lee, J.J.; Ro, J.Y.; Lukeman, J.M.; Morice, R.C.; Peters, E.J.; Pang, A.C.; Fritsche, H.A., Jr. Randomized placebo-controlled trial of isotretinoin in chemoprevention of bronchial squamous metaplasia. J. Clin. Oncol. 1994, 12, 937–945. [Google Scholar]
- Kelly, K.; Kittelson, J.; Franklin, W.A.; Kennedy, T.C.; Klein, C.E.; Keith, R.L.; Dempsey, E.C.; Lewis, M.; Jackson, M.K.; Hirsch, F.R.; et al. A randomized phase II chemoprevention trial of 13-CIS retinoic acid with or without alpha tocopherol or observation in subjects at high risk for lung cancer. Cancer Prev. Res. (Phila) 2009, 2, 440–449. [Google Scholar] [CrossRef]
- Arnold, A.M.; Browman, G.P.; Levine, M.N.; D’Souza, T.; Johnstone, B.; Skingley, P.; Turner-Smith, L.; Cayco, R.; Booker, L.; Newhouse, M. The effect of the synthetic retinoid etretinate on sputum cytology: Results from a randomised trial. Br. J. Cancer 1992, 65, 737–743. [Google Scholar] [CrossRef]
- Kurie, J.M.; Lee, J.S.; Khuri, F.R.; Mao, L.; Morice, R.C.; Lee, J.J.; Walsh, G.L.; Broxson, A.; Lippman, S.M.; Ro, J.Y.; et al. N-(4-hydroxyphenyl)retinamide in the chemoprevention of squamous metaplasia and dysplasia of the bronchial epithelium. Clin. Cancer Res. 2000, 6, 2973–2979. [Google Scholar]
- McLarty, J.W.; Holiday, D.B.; Girard, W.M.; Yanagihara, R.H.; Kummet, T.D.; Greenberg, S.D. Beta-Carotene, vitamin A, and lung cancer chemoprevention: Results of an intermediate endpoint study. Am. J. Clin. Nutr. 1995, 62, 1431S–1438S. [Google Scholar]
- Lam, S.; MacAulay, C.; le Riche, J.C.; Dyachkova, Y.; Coldman, A.; Guillaud, M.; Hawk, E.; Christen, M.-O.; Gazdar, A.F. A randomized phase IIb trial of anethole dithiolethione in smokers with bronchial dysplasia. J. Natl. Cancer Inst. 2002, 94, 1001–1009. [Google Scholar] [CrossRef]
- Mao, J.T.; Fishbein, M.C.; Adams, B.; Roth, M.D.; Goodglick, L.; Hong, L.; Burdick, M.; Strieter, E.R.M.; Holmes, C.; Tashkin, D.P.; et al. Celecoxib decreases Ki-67 proliferative index in active smokers. Clin. Cancer Res. 2006, 12, 314–320. [Google Scholar] [CrossRef]
- Kim, E.S.; Hong, W.K.; Lee, J.J.; Mao, L.; Morice, R.C.; Liu, D.D.; Jimenez, C.A.; Eapen, G.A.; Lotan, R.; Tang, X.; et al. Biological activity of celecoxib in the bronchial epithelium of current and former smokers. Cancer Prev. Res. (Phila) 2010, 3, 148–159. [Google Scholar] [CrossRef]
- Keith, R.L.; Blatchford, P.J.; Kittelson, J.; Minna, J.D.; Kelly, K.; Massion, P.P.; Franklin, W.A.; Mao, J.; Wilson, D.O.; Merrick, D.T.; et al. Oral iloprost improves endobronchial dysplasia in former smokers. Cancer Prev. Res. (Phila) 2011, 4, 793–802. [Google Scholar] [CrossRef]
- Lam, S.; McWilliams, A.; LeRiche, J.; MacAulay, C.; Wattenberg, L.; Szabo, E. A phase I study of myo-inositol for lung cancer chemoprevention. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1526–1531. [Google Scholar] [CrossRef]
- Gustafson, A.M.; Soldi, R.; Anderlind, C.; Scholand, M.B.; Qian, J.; Zhang, X.; Cooper, K.; Walker, D.; McWilliams, A.; Liu, G.; et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef]
- Lippman, S.M.; Lee, J.J.; Karp, D.D.; Vokes, E.E.; Benner, S.E.; Goodman, G.E.; Khuri, F.R.; Marks, R.; Winn, R.J.; Fry, W.; et al. Randomized phase III intergroup trial of isotretinoin to prevent second primary tumors in stage I non-small-cell lung cancer. J. Natl. Cancer Inst. 2001, 93, 605–618. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Dalesio, O.; Pastorino, U.; de Vries, N.; van Tinteren, H. EUROSCAN, a randomized trial of vitamin A and N-acetylcysteine in patients with head and neck cancer or lung cancer. J. Natl. Cancer Inst. 2000, 92, 977–986. [Google Scholar] [CrossRef]
- Karp, D.D.; Lee, S.J.; Keller, S.M.; Wright, G.S.; Aisner, S.; Belinsky, S.A.; Johnson, D.H.; Johnston, M.R.; Goodman, G.; Clamon, G.; et al. Randomized, double-blind, placebo-controlled, phase III chemoprevention trial of selenium supplementation in patients with resected stage I non-small-cell lung cancer: ECOG 5597. J. Clin. Oncol. 2013, 31, 4179–4187. [Google Scholar] [CrossRef]
- Wick, M.; Hurteau, G.; Dessev, C.; Chan, D.; Geraci, M.W.; Winn, R.A.; Heasley, L.E.; Nemenoff, R.A. Peroxisome proliferator-activated receptor-gamma is a target of nonsteroidal anti-inflammatory drugs mediating cyclooxygenase-independent inhibition of lung cancer cell growth. Mol. Pharmacol. 2002, 62, 1207–1214. [Google Scholar] [CrossRef]
- Bren-Mattison, Y.; van Putten, V.; Chan, D.; Winn, R.; Geraci, M.W.; Nemenoff, R.A. Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene 2005, 24, 1412–1422. [Google Scholar]
- Nemenoff, R.; Meyer, A.M.; Hudish, T.M.; Mozer, A.B.; Snee, A.; Narumiya, S.; Stearman, R.S.; Winn, R.A.; Weiser-Evans, M.; Geraci, M.W.; et al. Prostacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator—Activated receptor gamma. Cancer Prev. Res. (Phila) 2008, 1, 349–356. [Google Scholar] [CrossRef]
- Lam, S.; leRiche, J.C.; McWilliams, A.; Macaulay, C.; Dyachkova, Y.; Szabo, E.; Mayo, J.; Schellenberg, R.; Coldman, A.; Hawk, E.; et al. A randomized phase IIb trial of pulmicort turbuhaler (budesonide) in people with dysplasia of the bronchial epithelium. Clin. Cancer Res. 2004, 10, 6502–6511. [Google Scholar] [CrossRef]
- Veronesi, G.; Szabo, E.; Decensi, A.; Guerrieri-Gonzaga, A.; Bellomi, M.; Radice, D.; Ferretti, S.; Pelosi, G.; Lazzeroni, M.; Serrano, D.; et al. Randomized phase II trial of inhaled budesonide versus placebo in high-risk individuals with CT screen-detected lung nodules. Cancer Prev. Res. (Phila) 2011, 4, 34–42. [Google Scholar] [CrossRef]
- Crowell, J.A. The chemopreventive agent development research program in the Division of Cancer Prevention of the US National Cancer Institute: An overview. Eur. J. Cancer 2005, 41, 1889–1910. [Google Scholar] [CrossRef]
- Gerhauser, C.; Bartsch, H.; Crowell, J.; de Flora, S.; D’Incalci, M.; Dittrich, C.; Frank, N.; Mihich, E.; Steffen, C.; Tortora, G.; et al. Development of novel cancer chemopreventive agents in Europe—neglected Cinderella or rising phoenix? A critical commentary. ESF Workshop on Cancer Chemoprevention, DKFZ, Heidelberg, September 18–20, 2005. Eur. J. Cancer 2006, 42, 1338–1343. [Google Scholar] [CrossRef]
- Mascaux, C.; Feser, W.J.; Lewis, M.T.; Barón, A.E.; Coldren, C.D.; Merrick, D.T.; Kennedy, T.C.; Eckelberger, J.I.; Rozeboom, L.M.; Franklin, W.A.; et al. Endobronchial miRNAs as biomarkers in lung cancer chemoprevention. Cancer Prev. Res. (Phila) 2013, 6, 100–108. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kathuria, H.; Gesthalter, Y.; Spira, A.; Brody, J.S.; Steiling, K. Updates and Controversies in the Rapidly Evolving Field of Lung Cancer Screening, Early Detection, and Chemoprevention. Cancers 2014, 6, 1157-1179. https://doi.org/10.3390/cancers6021157
Kathuria H, Gesthalter Y, Spira A, Brody JS, Steiling K. Updates and Controversies in the Rapidly Evolving Field of Lung Cancer Screening, Early Detection, and Chemoprevention. Cancers. 2014; 6(2):1157-1179. https://doi.org/10.3390/cancers6021157
Chicago/Turabian StyleKathuria, Hasmeena, Yaron Gesthalter, Avrum Spira, Jerome S. Brody, and Katrina Steiling. 2014. "Updates and Controversies in the Rapidly Evolving Field of Lung Cancer Screening, Early Detection, and Chemoprevention" Cancers 6, no. 2: 1157-1179. https://doi.org/10.3390/cancers6021157
APA StyleKathuria, H., Gesthalter, Y., Spira, A., Brody, J. S., & Steiling, K. (2014). Updates and Controversies in the Rapidly Evolving Field of Lung Cancer Screening, Early Detection, and Chemoprevention. Cancers, 6(2), 1157-1179. https://doi.org/10.3390/cancers6021157