Genetic Interactions of STAT3 and Anticancer Drug Development

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. STAT3-Associated Single-Gene Diseases
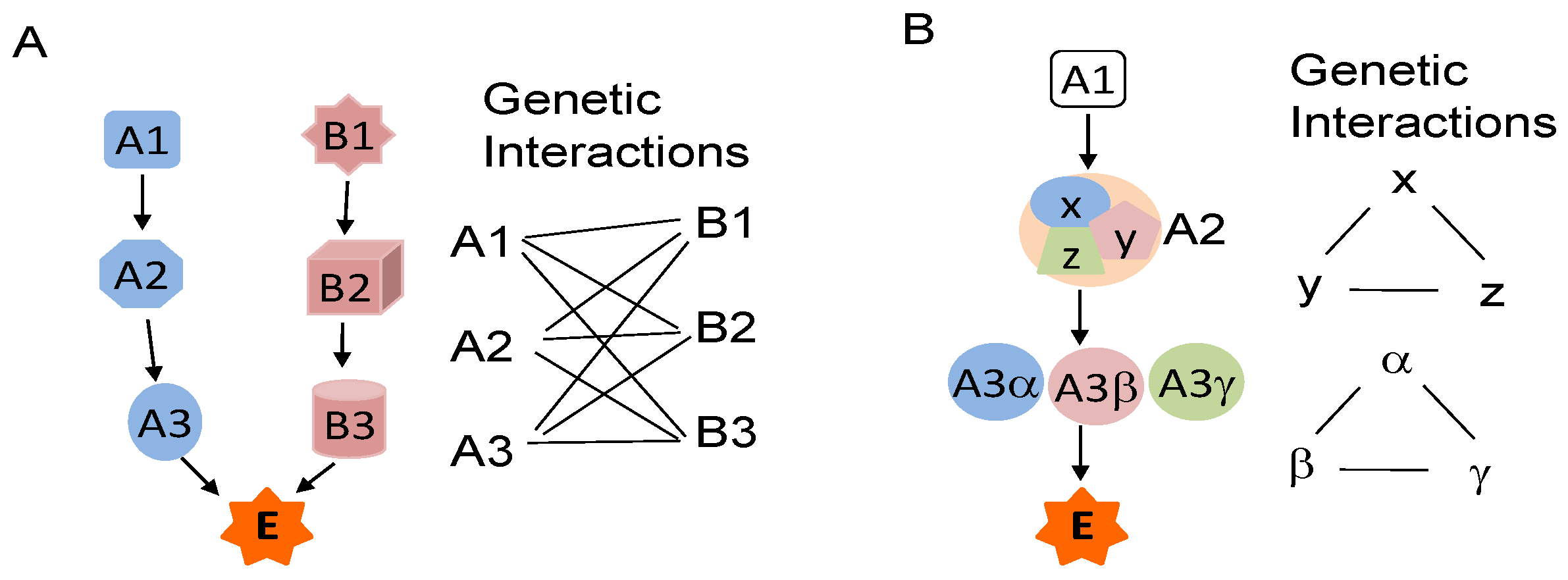
3. Genetic Interactions and STAT3 Activation
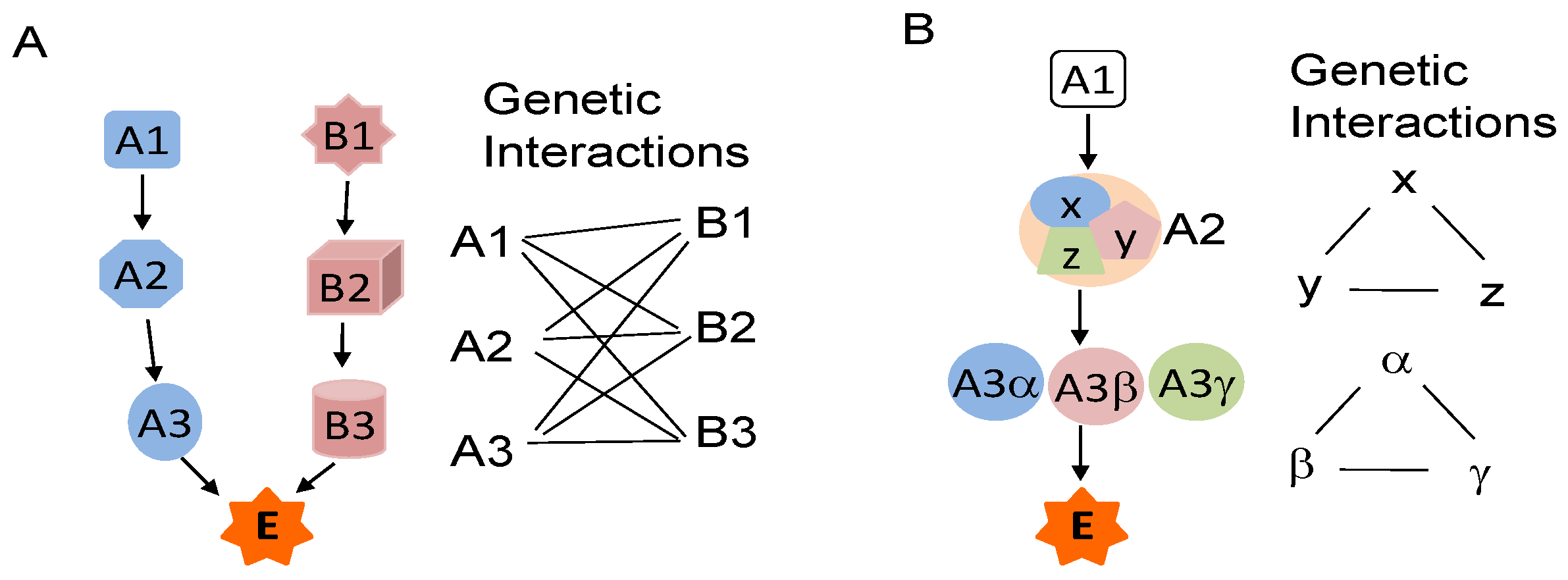
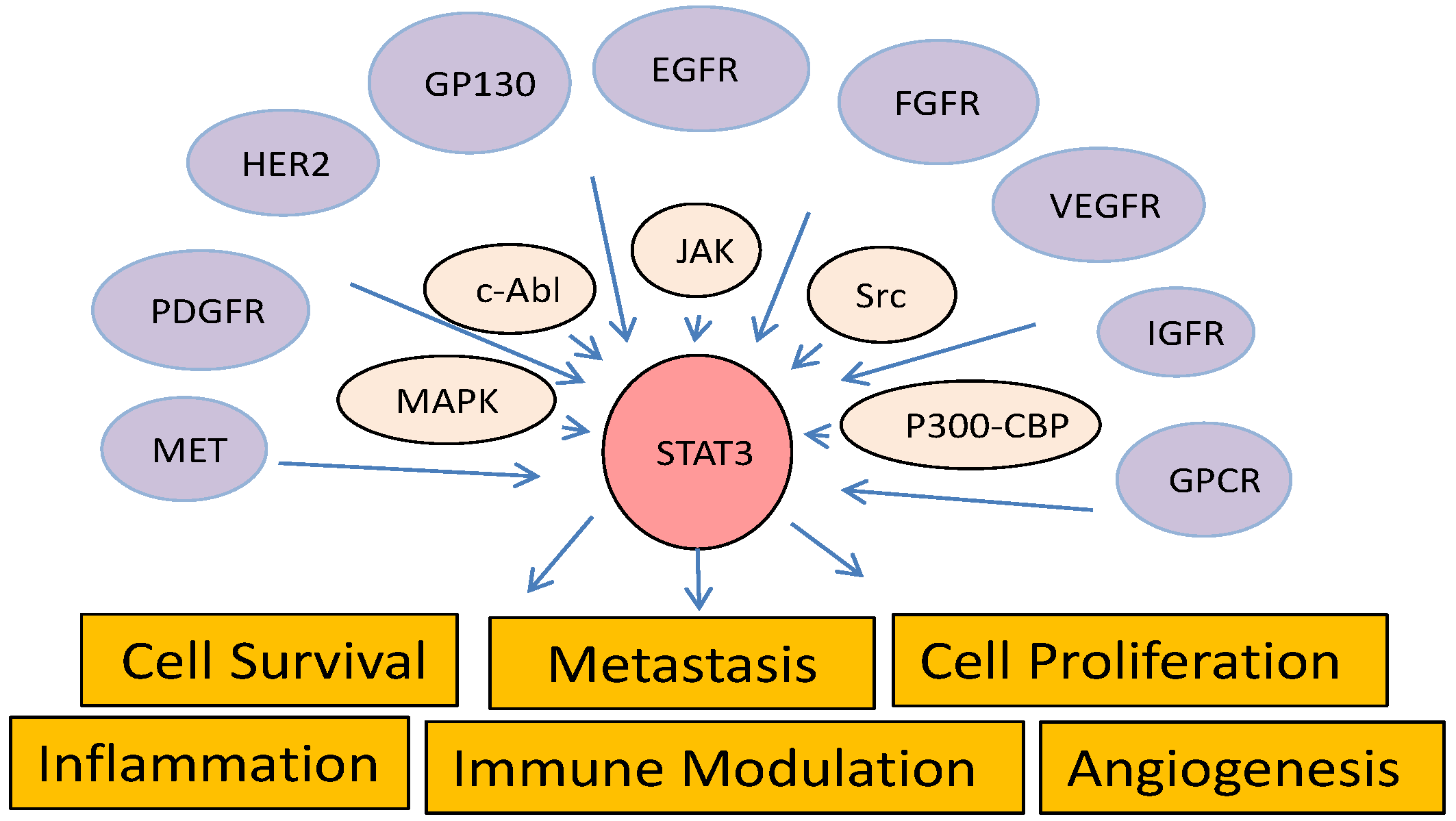
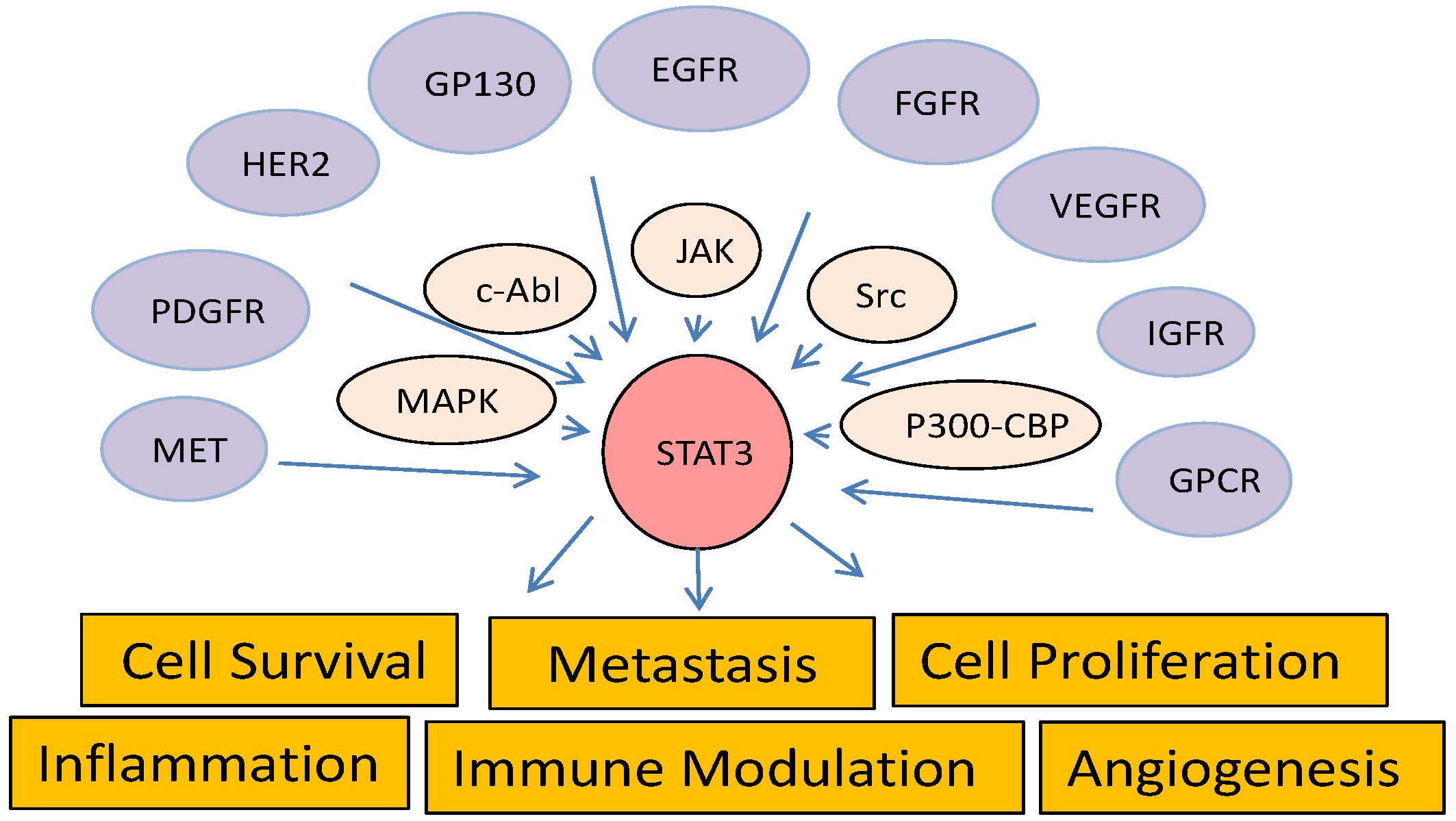
4. Cross-Talk of STAT3 with Other Cancer-Associated Pathways
4.1. RAS Pathway
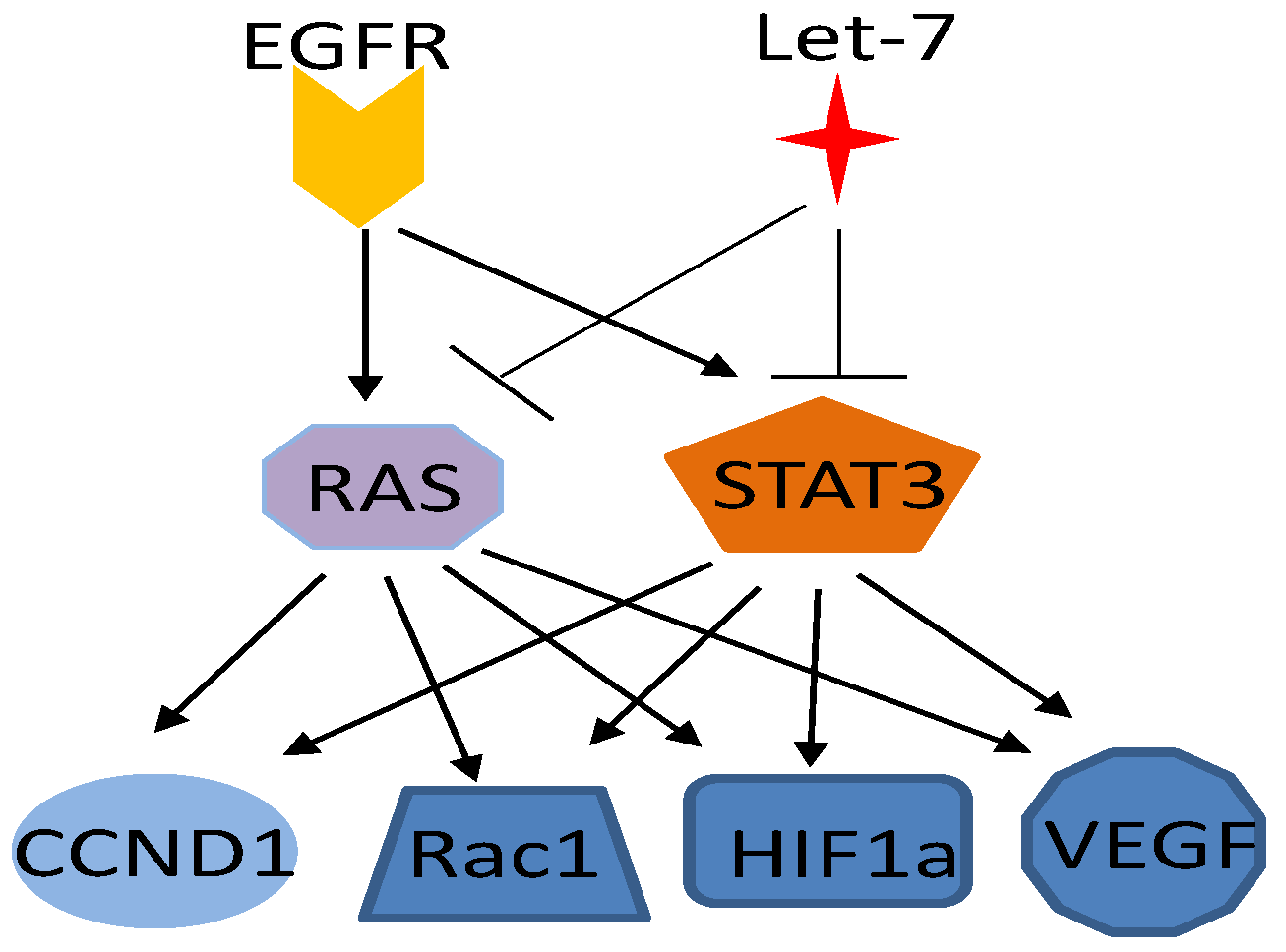
4.2. EGFR Pathway
4.3. Reduction-Oxidation Pathways
5. Conclusions and Perspectives
Acknowledgment
Conflicts of Interest
References
- Barbieri, I.; Pensa, S.; Pannellini, T.; Quaglino, E.; Maritano, D.; Demaria, M.; Voster, A.; Turkson, J.; Cavallo, F.; Watson, C.J.; et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010, 70, 2558–2567. [Google Scholar] [CrossRef]
- Ranger, J.J.; Levy, D.E.; Shahalizadeh, S.; Hallett, M.; Muller, W.J. Identification of a Stat3-dependent transcription regulatory network involved in metastatic progression. Cancer Res. 2009, 69, 6823–6830. [Google Scholar] [CrossRef]
- Li, Y.; Du, H.; Qin, Y.; Roberts, J.; Cummings, O.W.; Yan, C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007, 67, 8494–8503. [Google Scholar] [CrossRef]
- Okamoto, N.; Yasukawa, M.; Nguyen, C.; Kasim, V.; Maida, Y.; Possemato, R.; Shibata, T.; Ligon, K.L.; Fukami, K.; Hahn, W.C.; et al. Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc. Natl. Acad. Sci. USA 2011, 108, 20388–20393. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Zhang, W.Z.; Sun, M.; Wang, Q.; Coppola, D.; Mansour, M.; Xu, L.M.; Costanzo, C.; Cheng, J.Q.; Wang, L.H. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J. Biol. Chem. 2008, 283, 14665–14673. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. STAT3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef]
- Lin, L.; Amin, R.; Gallicano, G.I.; Glasgow, E.; Jogunoori, W.; Jessup, J.M.; Zasloff, M.; Marshall, J.L.; Shetty, K.; Johnson, L.; et al. The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene 2009, 28, 961–972. [Google Scholar] [CrossRef]
- Zhang, X.; Yue, P.; Fletcher, S.; Zhao, W.; Gunning, P.T.; Turkson, J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem. Pharmacol. 2010, 79, 1398–1409. [Google Scholar] [CrossRef]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef]
- Zhao, W.; Jaganathan, S.; Turkson, J. A cell-permeable Stat3 SH2 domain mimetic inhibits Stat3 activation and induces antitumor cell effects in vitro. J. Biol. Chem. 2010, 285, 35855–35865. [Google Scholar] [CrossRef]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef]
- Bhasin, D.; Cisek, K.; Pandharkar, T.; Regan, N.; Li, C.; Pandit, B.; Lin, J.; Li, P.K. Design, synthesis, and studies of small molecule STAT3 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 391–395. [Google Scholar] [CrossRef]
- Deng, J.; Grande, F.; Neamati, N. Small molecule inhibitors of Stat3 signaling pathway. Curr. Cancer Drug Targets 2007, 7, 91–107. [Google Scholar] [CrossRef]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Exp. Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef]
- Sansone, P.; Bromberg, J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J. Clin. Oncol. 2012, 30, 1005–1014. [Google Scholar] [CrossRef]
- Kayentao, K.; Garner, P.; van Eijk, A.M.; Naidoo, I.; Roper, C.; Mulokozi, A.; MacArthur, J.R.; Luntamo, M.; Ashorn, P.; Doumbo, O.K.; et al. Intermittent preventive therapy for malaria during pregnancy using 2 vs. 3 or more doses of sulfadoxine-pyrimethamine and risk of low birth weight in Africa: Systematic review and meta-analysis. JAMA 2013, 309, 594–604. [Google Scholar] [CrossRef]
- Aponte, J.J.; Schellenberg, D.; Egan, A.; Breckenridge, A.; Carneiro, I.; Critchley, J.; Danquah, I.; Dodoo, A.; Kobbe, R.; Lell, B.; et al. Efficacy and safety of intermittent preventive treatment with sulfadoxine-pyrimethamine for malaria in African infants: A pooled analysis of six randomised, placebo-controlled trials. Lancet 2009, 374, 1533–1542. [Google Scholar] [CrossRef]
- Takakura, A.; Nelson, E.A.; Haque, N.; Humphreys, B.D.; Zandi-Nejad, K.; Frank, D.A.; Zhou, J. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum. Mol. Genet. 2011, 20, 4143–4154. [Google Scholar] [CrossRef]
- Lowinger, T.B.; Riedl, B.; Dumas, J.; Smith, R.A. Design and discovery of small molecules targeting raf-1 kinase. Curr. Pharm. Design 2002, 8, 2269–2278. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Blechacz, B.R.; Smoot, R.L.; Bronk, S.F.; Werneburg, N.W.; Sirica, A.E.; Gores, G.J. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology 2009, 50, 1861–1870. [Google Scholar] [CrossRef]
- Yang, F.; Brown, C.; Buettner, R.; Hedvat, M.; Starr, R.; Scuto, A.; Schroeder, A.; Jensen, M.; Jove, R. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol. Cancer Ther. 2010, 9, 953–962. [Google Scholar] [CrossRef]
- Wetzler, M.; Brady, M.T.; Tracy, E.; Li, Z.R.; Donohue, K.A.; O’Loughlin, K.L.; Cheng, Y.; Mortazavi, A.; McDonald, A.A.; Kunapuli, P.; et al. Arsenic trioxide affects signal transducer and activator of transcription proteins through alteration of protein tyrosine kinase phosphorylation. Clin. Cancer Res. 2006, 12, 6817–6825. [Google Scholar] [CrossRef]
- Jia, Y.; Liu, D.; Xiao, D.; Ma, X.; Han, S.; Zheng, Y.; Sun, S.; Zhang, M.; Gao, H.; Cui, X.; et al. Expression of AFP and STAT3 is involved in arsenic trioxide-induced apoptosis and inhibition of proliferation in AFP-producing gastric cancer cells. PLoS One 2013, 8, e54774. [Google Scholar]
- Bernhard, G.C. Auranofin therapy in rheumatoid arthritis. J. Lab. Clin. Med. 1982, 100, 167–177. [Google Scholar]
- Kim, N.H.; Lee, M.Y.; Park, S.J.; Choi, J.S.; Oh, M.K.; Kim, I.S. Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology 2007, 122, 607–614. [Google Scholar] [CrossRef]
- Schuh, E.; Pfluger, C.; Citta, A.; Folda, A.; Rigobello, M.P.; Bindoli, A.; Casini, A.; Mohr, F. Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 2012, 55, 5518–5528. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef]
- Stephens, P.; Edkins, S.; Davies, H.; Greenman, C.; Cox, C.; Hunter, C.; Bignell, G.; Teague, J.; Smith, R.; Stevens, C.; et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat. Genet. 2005, 37, 590–592. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordonez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar]
- Grant, S. Is the focus moving toward a combination of targeted drugs? Bailliere’s Best Pract. Clin. Haematol. 2008, 21, 629–637. [Google Scholar] [CrossRef]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Disc. 2007, 6, 734–745. [Google Scholar]
- Schaefer, T.S.; Sanders, L.K.; Nathans, D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc. Natl. Acad. Sci. USA 1995, 92, 9097–9101. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar]
- Huang, Y.; Qiu, J.; Dong, S.; Redell, M.S.; Poli, V.; Mancini, M.A.; Tweardy, D.J. Stat3 isoforms, alpha and beta, demonstrate distinct intracellular dynamics with prolonged nuclear retention of Stat3beta mapping to its unique C-terminal end. J. Biol. Chem. 2007, 282, 34958–34967. [Google Scholar]
- Minegishi, Y.; Saito, M.; Tsuchiya, S.; Tsuge, I.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; Pasic, S.; Stojkovic, O.; et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007, 448, 1058–1062. [Google Scholar] [CrossRef]
- Liu, J.Y.; Li, Q.; Chen, T.T.; Guo, X.; Ge, J.; Yuan, L.X. Destructive pulmonary staphylococcal infection in a boy with hyper-IgE syndrome: A novel mutation in the signal transducer and activator of transcription 3 (STAT3) gene (p.Y657S). Eur. J. Pediat. 2011, 170, 661–666. [Google Scholar] [CrossRef]
- Renner, E.D.; Rylaarsdam, S.; Anover-Sombke, S.; Rack, A.L.; Reichenbach, J.; Carey, J.C.; Zhu, Q.; Jansson, A.F.; Barboza, J.; Schimke, L.F.; et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol. 2008, 122, 181–187. [Google Scholar] [CrossRef]
- Miyazaki, K.; Miyazawa, T.; Sugimoto, K.; Fujita, S.; Yanagida, H.; Okada, M.; Takemura, T. An adolescent with marked hyperimmuno-globulinemia E showing minimal change nephrotic syndrome and a STAT3 gene mutation. Clin. Nephrol. 2011, 75, 369–373. [Google Scholar] [CrossRef]
- Jiao, H.; Toth, B.; Erdos, M.; Fransson, I.; Rakoczi, E.; Balogh, I.; Magyarics, Z.; Derfalvi, B.; Csorba, G.; Szaflarska, A.; et al. Novel and recurrent STAT3 mutations in hyper-IgE syndrome patients from different ethnic groups. Mol. Immunol. 2008, 46, 202–206. [Google Scholar]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef]
- De Beaucoudrey, L.; Puel, A.; Filipe-Santos, O.; Cobat, A.; Ghandil, P.; Chrabieh, M.; Feinberg, J.; von, B.H.; Samarina, A.; Janniere, L.; et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J. Exp. Med. 2008, 205, 1543–1550. [Google Scholar] [CrossRef]
- Kumanovics, A.; Perkins, S.L.; Gilbert, H.; Cessna, M.H.; Augustine, N.H.; Hill, H.R. Diffuse large B cell lymphoma in hyper-IgE syndrome due to STAT3 mutation. J. Clin. Immunol. 2010, 30, 886–893. [Google Scholar] [CrossRef]
- Leonard, G.D.; Posadas, E.; Herrmann, P.C.; Anderson, V.L.; Jaffe, E.S.; Holland, S.M.; Wilson, W.H. Non-Hodgkin’s lymphoma in Job’s syndrome: A case report and literature review. Leuk. Lymphoma 2004, 45, 2521–2525. [Google Scholar] [CrossRef]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef]
- Sanchez-Ceja, S.G.; Reyes-Maldonado, E.; Vazquez-Manriquez, M.E.; Lopez-Luna, J.J.; Belmont, A.; Gutierrez-Castellanos, S. Differential expression of STAT5 and Bcl-xL, and high expression of Neu and STAT3 in non-small-cell lung carcinoma. Lung Cancer 2006, 54, 163–168. [Google Scholar] [CrossRef]
- Abou-Ghazal, M.; Yang, D.S.; Qiao, W.; Reina-Ortiz, C.; Wei, J.; Kong, L.Y.; Fuller, G.N.; Hiraoka, N.; Priebe, W.; Sawaya, R.; et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin. Cancer Res. 2008, 14, 8228–8235. [Google Scholar] [CrossRef]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in kras-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef]
- Hsiao, J.R.; Jin, Y.T.; Tsai, S.T.; Shiau, A.L.; Wu, C.L.; Su, W.C. Constitutive activation of STAT3 and STAT5 is present in the majority of nasopharyngeal carcinoma and correlates with better prognosis. Br. J. Cancer 2003, 89, 344–349. [Google Scholar] [CrossRef]
- Abdulghani, J.; Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Zellweger, T.; Alanen, K.; Mirtti, T.; et al. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 2008, 172, 1717–1728. [Google Scholar] [CrossRef]
- Migone, T.S.; Lin, J.X.; Cereseto, A.; Mulloy, J.C.; O’Shea, J.J.; Franchini, G.; Leonard, W.J. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 1995, 269, 79–81. [Google Scholar]
- Benekli, M.; Xia, Z.; Donohue, K.A.; Ford, L.A.; Pixley, L.A.; Baer, M.R.; Baumann, H.; Wetzler, M. Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood 2002, 99, 252–257. [Google Scholar] [CrossRef]
- Stewart, D.A.; Bahlis, N.; Mansoor, A. pY-STAT3 and p53 expression predict outcome for poor prognosis diffuse large B-cell lymphoma treated with high dose chemotherapy and autologous stem cell transplantation. Leuk. Lymphoma 2009, 50, 1276–1282. [Google Scholar] [CrossRef]
- Wang, Y.C.; Zheng, L.H.; Ma, B.A.; Zhou, Y.; Zhang, M.H.; Zhang, D.Z.; Fan, Q.Y. Clinical value of signal transducers and activators of transcription 3 (STAT3) gene expression in human osteosarcoma. Acta Histochem. 2011, 113, 402–408. [Google Scholar] [CrossRef]
- Ryu, K.; Choy, E.; Yang, C.; Susa, M.; Hornicek, F.J.; Mankin, H.; Duan, Z. Activation of signal transducer and activator of transcription 3 (Stat3) pathway in osteosarcoma cells and overexpression of phosphorylated-Stat3 correlates with poor prognosis. J. Orthopaed. Res. 2010, 28, 971–978. [Google Scholar]
- Chatterjee, D.; Sabo, E.; Tavares, R.; Resnick, M.B. Inverse association between Raf Kinase Inhibitory Protein and signal transducers and activators of transcription 3 expression in gastric adenocarcinoma patients: Implications for clinical outcome. Clin. Cancer Res. 2008, 14, 2994–3001. [Google Scholar] [CrossRef]
- Yakata, Y.; Nakayama, T.; Yoshizaki, A.; Kusaba, T.; Inoue, K.; Sekine, I. Expression of p-STAT3 in human gastric carcinoma: Significant correlation in tumour invasion and prognosis. Int. J. Oncol. 2007, 30, 437–442. [Google Scholar]
- Morikawa, T.; Baba, Y.; Yamauchi, M.; Kuchiba, A.; Nosho, K.; Shima, K.; Tanaka, N.; Huttenhower, C.; Frank, D.A.; Fuchs, C.S.; et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin. Cancer Res. 2011, 17, 1452–1462. [Google Scholar] [CrossRef]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Inoue, K.; Nagayasu, T.; Sekine, I. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol. Rep. 2006, 15, 1445–1451. [Google Scholar]
- Guo, S.; Sun, F.; Guo, Z.; Li, W.; Alfano, A.; Chen, H.; Magyar, C.E.; Huang, J.; Chai, T.C.; Qiu, S.; et al. Tyrosine kinase ETK/BMX is up-regulated in bladder cancer and predicts poor prognosis in patients with cystectomy. PLoS One 2011, 6, e17778. [Google Scholar] [CrossRef]
- Takemoto, S.; Ushijima, K.; Kawano, K.; Yamaguchi, T.; Terada, A.; Fujiyoshi, N.; Nishio, S.; Tsuda, N.; Ijichi, M.; Kakuma, T.; et al. Expression of activated signal transducer and activator of transcription-3 predicts poor prognosis in cervical squamous-cell carcinoma. Br. J. Cancer 2009, 101, 967–972. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Licciulli, S.; Kissil, J.L. WT1: A weak spot in KRAS-induced transformation. J. Clin. Investig. 2010, 120, 3804–3807. [Google Scholar] [CrossRef]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef]
- Scholl, C.; Frohling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef]
- Whitehurst, A.W.; Bodemann, B.O.; Cardenas, J.; Ferguson, D.; Girard, L.; Peyton, M.; Minna, J.D.; Michnoff, C.; Hao, W.; Roth, M.G.; et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar] [CrossRef]
- Astsaturov, I.; Ratushny, V.; Sukhanova, A.; Einarson, M.B.; Bagnyukova, T.; Zhou, Y.; Devarajan, K.; Silverman, J.S.; Tikhmyanova, N.; Skobeleva, N.; et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signal 2010, 3. [Google Scholar] [CrossRef]
- Torrance, C.J.; Agrawal, V.; Vogelstein, B.; Kinzler, K.W. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat. Biotechnol. 2001, 19, 940–945. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Guo, W.; Wu, S.; Liu, J.; Fang, B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008, 68, 7403–7408. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Le Meur, N.; Gentleman, R. Modeling synthetic lethality. Genome Biol. 2008, 9. [Google Scholar] [CrossRef]
- Ooi, S.L.; Pan, X.; Peyser, B.D.; Ye, P.; Meluh, P.B.; Yuan, D.S.; Irizarry, R.A.; Bader, J.S.; Spencer, F.A.; Boeke, J.D. Global synthetic-lethality analysis and yeast functional profiling. Trends Genet. 2006, 22, 56–63. [Google Scholar] [CrossRef]
- Fang, B. Genetic interactions in translational research on cancer. World J. Med. Genet. 2011, 1, 14–22. [Google Scholar] [CrossRef]
- Argetsinger, L.S.; Campbell, G.S.; Yang, X.; Witthuhn, B.A.; Silvennoinen, O.; Ihle, J.N.; Carter-Su, C. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 1993, 74, 237–244. [Google Scholar] [CrossRef]
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998, 93, 385–395. [Google Scholar] [CrossRef]
- Bromberg, J.F. Activation of STAT proteins and growth control. Bioessays 2001, 23, 161–169. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar]
- Xu, F.; Mukhopadhyay, S.; Sehgal, P.B. Live cell imaging of interleukin-6-induced targeting of “transcription factor” STAT3 to sequestering endosomes in the cytoplasm. Am. J. Physiol. 2007, 293, C1374–C1382. [Google Scholar]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef]
- Zhang, X.; Blenis, J.; Li, H.C.; Schindler, C.; Chen-Kiang, S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science 1995, 267, 1990–1994. [Google Scholar]
- Qin, H.R.; Kim, H.J.; Kim, J.Y.; Hurt, E.M.; Klarmann, G.J.; Kawasaki, B.T.; Duhagon Serrat, M.A.; Farrar, W.L. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008, 68, 7736–7741. [Google Scholar] [CrossRef]
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D.E.; Darnell, J.E., Jr. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol. Cell. Biol. 2004, 24, 407–419. [Google Scholar] [CrossRef]
- Chung, J.; Uchida, E.; Grammer, T.C.; Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 1997, 17, 6508–6516. [Google Scholar]
- Lim, C.P.; Cao, X. Serine phosphorylation and negative regulation of Stat3 by JNK. J. Biol. Chem. 1999, 274, 31055–31061. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar]
- Nadiminty, N.; Lou, W.; Lee, S.O.; Lin, X.; Trump, D.L.; Gao, A.C. Stat3 activation of NFkappaB p100 processing involves CBP/p300-mediated acetylation. Proc. Natl. Acad. Sci. USA 2006, 103, 7264–7269. [Google Scholar]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell. Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef]
- Klingmuller, U.; Lorenz, U.; Cantley, L.C.; Neel, B.G.; Lodish, H.F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 1995, 80, 729–738. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B.; et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Lee, C.K.; Bluyssen, H.A.; Levy, D.E. Regulation of interferon-alpha responsiveness by the duration of Janus kinase activity. J. Biol. Chem. 1997, 272, 21872–21877. [Google Scholar]
- Ray, S.; Lee, C.; Hou, T.; Boldogh, I.; Brasier, A.R. Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res. 2008, 36, 4510–4520. [Google Scholar] [CrossRef]
- Endo, T.A.; Masuhara, M.; Yokouchi, M.; Suzuki, R.; Sakamoto, H.; Mitsui, K.; Matsumoto, A.; Tanimura, S.; Ohtsubo, M.; Misawa, H.; et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997, 387, 921–924. [Google Scholar] [CrossRef]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ohkubo, T.; Kiguchi, T.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Hara, T.; Miyajima, A. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995, 14, 2816–2826. [Google Scholar]
- Haspel, R.L.; Darnell, J.E., Jr. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc. Natl. Acad. Sci. USA 1999, 96, 10188–10193. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Betz, A.; Lampen, N.; Martinek, S.; Young, M.W.; Darnell, J.E., Jr. A Drosophila PIAS homologue negatively regulates stat92E. Proc. Natl. Acad. Sci. USA 2001, 98, 9563–9568. [Google Scholar] [CrossRef]
- Nishimoto, A.; Yu, Y.; Lu, Z.; Mao, X.; Ren, Z.; Watowich, S.S.; Mills, G.B.; Liao, W.S.; Chen, X.; Bast, R.C., Jr.; et al. A Ras homologue member I directly inhibits signal transducers and activators of transcription 3 translocation and activity in human breast and ovarian cancer cells. Cancer Res. 2005, 65, 6701–6710. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef]
- Li, J.; Favata, M.; Kelley, J.A.; Caulder, E.; Thomas, B.; Wen, X.; Sparks, R.B.; Arvanitis, A.; Rogers, J.D.; Combs, A.P.; et al. INCB16562, a JAK1/2 selective inhibitor, is efficacious against multiple myeloma cells and reverses the protective effects of cytokine and stromal cell support. Neoplasia 2010, 12, 28–38. [Google Scholar]
- Manshouri, T.; Quintas-Cardama, A.; Nussenzveig, R.H.; Gaikwad, A.; Estrov, Z.; Prchal, J.; Cortes, J.E.; Kantarjian, H.M.; Verstovsek, S. The JAK kinase inhibitor CP-690,550 suppresses the growth of human polycythemia vera cells carrying the JAK2V617F mutation. Cancer Sci. 2008, 99, 1265–1273. [Google Scholar]
- Deisseroth, A.; Kaminskas, E.; Grillo, J.; Chen, W.; Saber, H.; Lu, H.L.; Rothmann, M.D.; Brar, S.; Wang, J.; Garnett, C.; et al. Food and Drug Administration approval: Ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin. Cancer Res. 2012, 18, 3212–3217. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R. Ruxolitinib: The first FDA approved therapy for the treatment of myelofibrosis. Clin. Cancer Res. 2012, 18, 3008–3014. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Burmester, G.R.; Blanco, R.; Charles-Schoeman, C.; Wollenhaupt, J.; Zerbini, C.; Benda, B.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; Zwillich, S.H.; et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: A randomised phase 3 trial. Lancet 2013, 381, 451–460. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W.; Study, A. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef]
- Byers, L.A.; Sen, B.; Saigal, B.; Diao, L.; Wang, J.; Nanjundan, M.; Cascone, T.; Mills, G.B.; Heymach, J.V.; Johnson, F.M. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin. Cancer Res. 2009, 15, 6852–6861. [Google Scholar] [CrossRef]
- Sen, B.; Saigal, B.; Parikh, N.; Gallick, G.; Johnson, F.M. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009, 69, 1958–1965. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004. [Google Scholar] [CrossRef]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell. Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef]
- Bernards, A.; Settleman, J. GAP control: Regulating the regulators of small GTPases. Trends Cell Biol. 2004, 14, 377–385. [Google Scholar] [CrossRef]
- Hoa, M.; Davis, S.L.; Ames, S.J.; Spanjaard, R.A. Amplification of wild-type K-ras promotes growth of head and neck squamous cell carcinoma. Cancer Res. 2002, 62, 7154–7156. [Google Scholar]
- Filmus, J.E.; Buick, R.N. Stability of c-K-ras amplification during progression in a patient with adenocarcinoma of the ovary. Cancer Res. 1985, 45, 4468–4472. [Google Scholar]
- Coleman, W.B.; Throneburg, D.B.; Grisham, J.W.; Smith, G.J. Overexpression of c-K-ras, c-N-ras and transforming growth factor beta co-segregate with tumorigenicity in morphologically transformed C3H 10T1/2 cell lines. Carcinogenesis 1994, 15, 1005–1012. [Google Scholar] [CrossRef]
- Ehrhardt, A.; David, M.D.; Ehrhardt, G.R.; Schrader, J.W. Distinct mechanisms determine the patterns of differential activation of H-Ras, N-Ras, K-Ras 4B, and M-Ras by receptors for growth factors or antigen. Mol. Cell Biol. 2004, 24, 6311–6323. [Google Scholar]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Egan, S.E.; Giddings, B.W.; Brooks, M.W.; Buday, L.; Sizeland, A.M.; Weinberg, R.A. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 1993, 363, 45–51. [Google Scholar] [CrossRef]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar]
- Vojtek, A.B.; Hollenberg, S.M.; Cooper, J.A. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 1993, 74, 205–214. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 2000, 103, 931–943. [Google Scholar] [CrossRef]
- Lowenstein, E.J.; Daly, R.J.; Batzer, A.G.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.Y.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Kumar, M.S.; Erkeland, S.J.; Pester, R.E.; Chen, C.Y.; Ebert, M.S.; Sharp, P.A.; Jacks, T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl. Acad. Sci. USA 2008, 105, 3903–3908. [Google Scholar]
- Sugimura, K.; Miyata, H.; Tanaka, K.; Hamano, R.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; Nakajima, K.; Takiguchi, S.; Mori, M.; et al. Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin. Cancer Res. 2012, 18, 5144–5153. [Google Scholar] [CrossRef]
- Filmus, J.; Robles, A.I.; Shi, W.; Wong, M.J.; Colombo, L.L.; Conti, C.J. Induction of cyclin D1 overexpression by activated ras. Oncogene 1994, 9, 3627–3633. [Google Scholar]
- Liu, J.J.; Chao, J.R.; Jiang, M.C.; Ng, S.Y.; Yen, J.J.; Yang-Yen, H.F. Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3T3 cells. Mol. Cell Biol. 1995, 15, 3654–3663. [Google Scholar]
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552. [Google Scholar] [CrossRef]
- Rosen, K.; Rak, J.; Leung, T.; Dean, N.M.; Kerbel, R.S.; Filmus, J. Activated Ras prevents downregulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism of Ras-induced resistance to anoikis in intestinal epithelial cells. J. Cell Biol. 2000, 149, 447–456. [Google Scholar] [CrossRef]
- Zushi, S.; Shinomura, Y.; Kiyohara, T.; Miyazaki, Y.; Kondo, S.; Sugimachi, M.; Higashimoto, Y.; Kanayama, S.; Matsuzawa, Y. STAT3 mediates the survival signal in oncogenic ras-transfected intestinal epithelial cells. Int. J. Cancer 1998, 78, 326–330. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.M.; Yang-Yen, H.F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Nagaoka, H.; Takahashi, Y.; Hayashi, R.; Nakamura, T.; Ishii, K.; Matsuda, J.; Ogura, A.; Shirakata, Y.; Karasuyama, H.; Sudo, T.; et al. Ras mediates effector pathways responsible for pre-B cell survival, which is essential for the developmental progression to the late pre-B cell stage. J. Exp. Med. 2000, 192, 171–182. [Google Scholar] [CrossRef]
- Teng, T.S.; Lin, B.; Manser, E.; Ng, D.C.; Cao, X. Stat3 promotes directional cell migration by regulating Rac1 activity via its activator betaPIX. J. Cell Sci. 2009, 122, 4150–4159. [Google Scholar] [CrossRef]
- Khosravi-Far, R.; Solski, P.A.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell Biol. 1995, 15, 6443–6453. [Google Scholar]
- Scita, G.; Tenca, P.; Frittoli, E.; Tocchetti, A.; Innocenti, M.; Giardina, G.; di Fiore, P.P. Signaling from Ras to Rac and beyond: Not just a matter of GEFs. EMBO J. 2000, 19, 2393–2398. [Google Scholar] [CrossRef]
- Blancher, C.; Moore, J.W.; Robertson, N.; Harris, A.L. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3'-kinase/Akt signaling pathway. Cancer Res. 2001, 61, 7349–7355. [Google Scholar]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef]
- Rak, J.; Mitsuhashi, Y.; Bayko, L.; Filmus, J.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995, 55, 4575–4580. [Google Scholar]
- Ancrile, B.; Lim, K.H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Gene Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Turkson, J.; Bowman, T.; Adnane, J.; Zhang, Y.; Djeu, J.Y.; Sekharam, M.; Frank, D.A.; Holzman, L.B.; Wu, J.; Sebti, S.; et al. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol. Cell Biol. 1999, 19, 7519–7528. [Google Scholar]
- Raptis, L.; Arulanandam, R.; Geletu, M.; Turkson, J. The R(h)oads to Stat3: Stat3 activation by the Rho GTPases. Exp. Cell Res. 2011, 317, 1787–1795. [Google Scholar] [CrossRef]
- Pelletier, S.; Duhamel, F.; Coulombe, P.; Popoff, M.R.; Meloche, S. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol. Cell Biol. 2003, 23, 1316–1333. [Google Scholar] [CrossRef]
- Simon, A.R.; Vikis, H.G.; Stewart, S.; Fanburg, B.L.; Cochran, B.H.; Guan, K.L. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science 2000, 290, 144–147. [Google Scholar] [CrossRef]
- Faruqi, T.R.; Gomez, D.; Bustelo, X.R.; Bar-Sagi, D.; Reich, N.C. Rac1 mediates STAT3 activation by autocrine IL-6. Proc. Natl. Acad. Sci. USA 2001, 98, 9014–9019. [Google Scholar] [CrossRef]
- Arulanandam, R.; Geletu, M.; Feracci, H.; Raptis, L. Activated Rac1 requires gp130 for Stat3 activation, cell proliferation and migration. Exp. Cell Res. 2010, 316, 875–886. [Google Scholar] [CrossRef]
- Debidda, M.; Wang, L.; Zang, H.; Poli, V.; Zheng, Y. A role of STAT3 in Rho GTPase-regulated cell migration and proliferation. J. Biol. Chem. 2005, 280, 17275–17285. [Google Scholar] [CrossRef]
- Schuringa, J.J.; Jonk, L.J.; Dokter, W.H.; Vellenga, E.; Kruijer, W. Interleukin-6-induced STAT3 transactivation and Ser727 phosphorylation involves Vav, Rac-1 and the kinase SEK-1/MKK-4 as signal transduction components. Biochem. J. 2000, 347, 89–96. [Google Scholar] [CrossRef]
- Dai, B.; Meng, J.; Peyton, M.; Girard, L.; Bornmann, W.G.; Ji, L.; Minna, J.D.; Fang, B.; Roth, J.A. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011, 71, 3658–3668. [Google Scholar] [CrossRef]
- Park, E.; Park, J.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. NVP-BKM120, a novel PI3K inhibitor, shows synergism with a STAT3 inhibitor in human gastric cancer cells harboring KRAS mutations. Int. J. Oncol. 2012, 40, 1259–1266. [Google Scholar]
- Guo, W.; Wu, S.; Wang, L.; Wang, R.; Wei, L.; Liu, J.; Fang, B. Interruption of RNA processing machinery by a small compound 1-[(4-chlorophenyl) methyl]-1H-indole-3-carboxaldehyde (oncrasin-1). Mol. Cancer Ther. 2009, 8, 441–448. [Google Scholar] [CrossRef]
- Guo, W.; Wu, S.; Wang, L.; Wei, X.; Liu, X.; Wang, J.; Lu, Z.; Hollingshead, M.; Fang, B. Antitumor activity of a novel oncrasin analogue is mediated by JNK activation and STAT3 inhibition. PLoS One 2011, 6, e28487. [Google Scholar]
- Liu, X.; Guo, W.; Wu, S.; Wang, L.; Wang, J.; Dai, B.; Kim, E.S.; Heymach, J.V.; Wang, M.; Girard, L.; et al. Antitumor activity of a novel STAT3 inhibitor and redox modulator in non-small cell lung cancer cells. Biochem. Pharmacol. 2012, 83, 1456–1464. [Google Scholar] [CrossRef]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Marchetti, A.; Martella, C.; Felicioni, L.; Barassi, F.; Salvatore, S.; Chella, A.; Camplese, P.P.; Iarussi, T.; Mucilli, F.; Mezzetti, A.; et al. EGFR mutations in non-small-cell lung cancer: Analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J. Clin. Oncol. 2005, 23, 857–865. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; di Maria, M.V.; Veve, R.; Bremmes, R.M.; Baron, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Hirsch, F.R.; Rossi, E.; Bartolini, S.; Ceresoli, G.L.; Bemis, L.; Haney, J.; Witta, S.; Danenberg, K.; Domenichini, I.; et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J. Natl. Cancer Inst. 2005, 97, 643–655. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef]
- Shia, J.; Klimstra, D.S.; Li, A.R.; Qin, J.; Saltz, L.; Teruya-Feldstein, J.; Akram, M.; Chung, K.Y.; Yao, D.; Paty, P.B.; et al. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: An immunohistochemical and chromogenic in situ hybridization study. Mod. Pathol. 2005, 18, 1350–1356. [Google Scholar] [CrossRef]
- Boeck, S.; Jung, A.; Laubender, R.P.; Neumann, J.; Egg, R.; Goritschan, C.; Vehling-Kaiser, U.; Winkelmann, C.; Fischer von, W.L.; Clemens, M.R.; et al. EGFR pathway biomarkers in erlotinib-treated patients with advanced pancreatic cancer: Translational results from the randomised, crossover phase 3 trial AIO-PK0104. Br. J. Cancer 2013, 108, 469–476. [Google Scholar] [CrossRef]
- Lee, J.; Jang, K.T.; Ki, C.S.; Lim, T.; Park, Y.S.; Lim, H.Y.; Choi, D.W.; Kang, W.K.; Park, K.; Park, J.O. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer 2007, 109, 1561–1569. [Google Scholar] [CrossRef]
- Chung, C.H.; Ely, K.; McGavran, L.; Varella-Garcia, M.; Parker, J.; Parker, N.; Jarrett, C.; Carter, J.; Murphy, B.A.; Netterville, J.; et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J. Clin. Oncol. 2006, 24, 4170–4176. [Google Scholar] [CrossRef]
- Yang, L.; Luquette, L.J.; Gehlenborg, N.; Xi, R.; Haseley, P.S.; Hsieh, C.H.; Zhang, C.; Ren, X.; Protopopov, A.; Chin, L.; et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 2013, 153, 919–929. [Google Scholar] [CrossRef]
- Ji, H.; Li, D.; Chen, L.; Shimamura, T.; Kobayashi, S.; McNamara, K.; Mahmood, U.; Mitchell, A.; Sun, Y.; Al-Hashem, R.; et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 2006, 9, 485–495. [Google Scholar] [CrossRef]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Gene Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Greulich, H.; Sellers, W.R.; Meyerson, M.; Frank, D.A. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006, 66, 3162–3168. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Shao, H.; Cheng, H.Y.; Cook, R.G.; Tweardy, D.J. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003, 63, 3923–3930. [Google Scholar]
- Shi, C.S.; Kehrl, J.H. Pyk2 amplifies epidermal growth factor and c-Src-induced Stat3 activation. J. Biol. Chem. 2004, 279, 17224–17231. [Google Scholar] [CrossRef]
- Goi, T.; Shipitsin, M.; Lu, Z.; Foster, D.A.; Klinz, S.G.; Feig, L.A. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. EMBO J. 2000, 19, 623–630. [Google Scholar] [CrossRef]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef]
- Batzer, A.G.; Rotin, D.; Urena, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar]
- Okutani, T.; Okabayashi, Y.; Kido, Y.; Sugimoto, Y.; Sakaguchi, K.; Matuoka, K.; Takenawa, T.; Kasuga, M. Grb2/Ash binds directly to tyrosines 1068 and 1086 and indirectly to tyrosine 1148 of activated human epidermal growth factor receptors in intact cells. J. Biol. Chem. 1994, 269, 31310–31314. [Google Scholar]
- Okabayashi, Y.; Kido, Y.; Okutani, T.; Sugimoto, Y.; Sakaguchi, K.; Kasuga, M. Tyrosines 1148 and 1173 of activated human epidermal growth factor receptors are binding sites of Shc in intact cells. J. Biol. Chem. 1994, 269, 18674–18678. [Google Scholar]
- Rotin, D.; Margolis, B.; Mohammadi, M.; Daly, R.J.; Daum, G.; Li, N.; Fischer, E.H.; Burgess, W.H.; Ullrich, A.; Schlessinger, J. SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: Identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase C gamma. EMBO J. 1992, 11, 559–567. [Google Scholar]
- Keilhack, H.; Tenev, T.; Nyakatura, E.; Godovac-Zimmermann, J.; Nielsen, L.; Seedorf, K.; Bohmer, F.D. Phosphotyrosine 1173 mediates binding of the protein-tyrosine phosphatase SHP-1 to the epidermal growth factor receptor and attenuation of receptor signaling. J. Biol. Chem. 1998, 273, 24839–24846. [Google Scholar] [CrossRef]
- Grandis, J.R.; Drenning, S.D.; Chakraborty, A.; Zhou, M.Y.; Zeng, Q.; Pitt, A.S.; Tweardy, D.J. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. J. Clin. Investig. 1998, 102, 1385–1392. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Suzuki, S.; Thomas, S.M.; Sen, M.; Leeman-Neill, R.J.; Chiosea, S.I.; Kuan, C.T.; Bigner, D.D.; Gooding, W.E.; Lai, S.Y.; et al. Epidermal growth factor receptor variant III mediates head and neck cancer cell invasion via STAT3 activation. Oncogene 2010, 29, 5135–5145. [Google Scholar] [CrossRef]
- Zhou, W.; Grandis, J.R.; Wells, A. STAT3 is required but not sufficient for EGF receptor-mediated migration and invasion of human prostate carcinoma cell lines. Br. J. Cancer 2006, 95, 164–171. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, E.; Wu, Q.; Guryanova, O.; Hitomi, M.; Lathia, J.D.; Serwanski, D.; Sloan, A.E.; Weil, R.J.; Lee, J.; et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Gene Dev. 2012, 26, 1247–1262. [Google Scholar] [CrossRef]
- Vignais, M.L.; Gilman, M. Distinct mechanisms of activation of Stat1 and Stat3 by platelet-derived growth factor receptor in a cell-free system. Mol. Cell. Biol. 1999, 19, 3727–3735. [Google Scholar]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; Vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef]
- Kitade, M.; Factor, V.M.; Andersen, J.B.; Tomokuni, A.; Kaji, K.; Akita, H.; Holczbauer, A.; Seo, D.; Marquardt, J.U.; Conner, E.A.; et al. Specific fate decisions in adult hepatic progenitor cells driven by MET and EGFR signaling. Gene Dev. 2013, 27, 1706–1717. [Google Scholar] [CrossRef]
- Chiu, H.C.; Chou, D.L.; Huang, C.T.; Lin, W.H.; Lien, T.W.; Yen, K.J.; Hsu, J.T. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem. Pharmacol. 2011, 81, 1263–1270. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Ochi, N.; Ninomiya, T.; Yasugi, M.; Kubo, T.; Takeda, H.; Ichihara, E.; Ohashi, K.; Takata, S.; et al. JAK2-related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor-activating mutation. Cancer Sci. 2012, 103, 1795–1802. [Google Scholar] [CrossRef]
- Sen, M.; Joyce, S.; Panahandeh, M.; Li, C.; Thomas, S.M.; Maxwell, J.; Wang, L.; Gooding, W.E.; Johnson, D.E.; Grandis, J.R. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin. Cancer Res. 2012, 18, 4986–4996. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Washington, M.K.; Merchant, N.B. Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes STAT3-mediated resistance of inhibition of pancreatic tumor growth. Clin. Cancer Res. 2011, 17, 483–493. [Google Scholar] [CrossRef]
- Lo, H.W.; Cao, X.; Zhu, H.; Ali-Osman, F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin. Cancer Res. 2008, 14, 6042–6054. [Google Scholar] [CrossRef]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 2012, 11, 2254–2264. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE 2000, 2000, E1. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109–2117. [Google Scholar]
- Chiarugi, P.; Pani, G.; Giannoni, E.; Taddei, L.; Colavitti, R.; Raugei, G.; Symons, M.; Borrello, S.; Galeotti, T.; Ramponi, G. Reactive oxygen species as essential mediators of cell adhesion: The oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol. 2003, 161, 933–944. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar]
- Jagadeeswaran, R.; Jagadeeswaran, S.; Bindokas, V.P.; Salgia, R. Activation of HGF/c-Met pathway contributes to the reactive oxygen species generation and motility of small cell lung cancer cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1488–L1494. [Google Scholar] [CrossRef]
- Smith, J.; Ladi, E.; Mayer-Proschel, M.; Noble, M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc. Natl. Acad. Sci. USA 2000, 97, 10032–10037. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-Nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002, 297, 1186–1190. [Google Scholar] [CrossRef]
- Johnson, T.M.; Yu, Z.X.; Ferrans, V.J.; Lowenstein, R.A.; Finkel, T. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 11848–11852. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer. 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell. 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Zhang, K.H.; Tian, H.Y.; Gao, X.; Lei, W.W.; Hu, Y.; Wang, D.M.; Pan, X.C.; Yu, M.L.; Xu, G.J.; Zhao, F.K.; et al. Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res. 2009, 69, 5340–5348. [Google Scholar] [CrossRef]
- Ferraro, D.; Corso, S.; Fasano, E.; Panieri, E.; Santangelo, R.; Borrello, S.; Giordano, S.; Pani, G.; Galeotti, T. Pro-metastatic signaling by c-Met through RAC-1 and reactive oxygen species (ROS). Oncogene 2006, 25, 3689–3698. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, J.; Ray, P.S.; Guo, H.; Huang, J.; Shin-Sim, M.; Bukoye, B.A.; Liu, B.; Lee, A.V.; Lin, X.; et al. Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-B signaling. J. Clin. Investig. 2011, 121, 212–225. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Zuo, L.; Ikeda, S.; Tojo, T.; Patrushev, N.A.; Alexander, R.W. cAbl tyrosine kinase mediates reactive oxygen species- and caveolin-dependent AT1 receptor signaling in vascular smooth muscle: Role in vascular hypertrophy. Circul. Res. 2005, 97, 829–836. [Google Scholar] [CrossRef]
- Chowdhury, A.K.; Watkins, T.; Parinandi, N.L.; Saatian, B.; Kleinberg, M.E.; Usatyuk, P.V.; Natarajan, V. Src-mediated tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of reactive oxygen species in lung endothelial cells. J. Biol. Chem. 2005, 280, 20700–20711. [Google Scholar] [CrossRef]
- Meng, D.; Shi, X.; Jiang, B.H.; Fang, J. Insulin-like growth factor-I (IGF-I) induces epidermal growth factor receptor transactivation and cell proliferation through reactive oxygen species. Free Rad. Biol. Med. 2007, 42, 1651–1660. [Google Scholar] [CrossRef]
- Chen, C.H.; Cheng, T.H.; Lin, H.; Shih, N.L.; Chen, Y.L.; Chen, Y.S.; Cheng, C.F.; Lian, W.S.; Meng, T.C.; Chiu, W.T.; et al. Reactive oxygen species generation is involved in epidermal growth factor receptor transactivation through the transient oxidization of Src homology 2-containing tyrosine phosphatase in endothelin-1 signaling pathway in rat cardiac fibroblasts. Mol. Pharmacol. 2006, 69, 1347–1355. [Google Scholar] [CrossRef]
- Colavitti, R.; Pani, G.; Bedogni, B.; Anzevino, R.; Borrello, S.; Waltenberger, J.; Galeotti, T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J. Biol. Chem. 2002, 277, 3101–3108. [Google Scholar]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef]
- Mishra, K.P. Cell membrane oxidative damage induced by gamma-radiation and apoptotic sensitivity. J. Environ. Pathol. Toxicol. Oncol. 2004, 23, 61–66. [Google Scholar] [CrossRef]
- Bhosle, S.M.; Pandey, B.N.; Huilgol, N.G.; Mishra, K.P.; Bhosle, S.M.; Pandey, B.N.; Huilgol, N.G.; Mishra, K.P. Membrane oxidative damage and apoptosis in cervical carcinoma cells of patients after radiation therapy. Methods Cell Sci. 2002, 24, 65–68. [Google Scholar] [CrossRef]
- Davis, W., Jr.; Ronai, Z.; Tew, K.D. Cellular thiols and reactive oxygen species in drug-induced apoptosis. J. Pharmacol. Exp. Ther. 2001, 296, 1–6. [Google Scholar]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar]
- Shaw, S.; Wang, X.; Redd, H.; Alexander, G.D.; Isales, C.M.; Marrero, M.B. High glucose augments the angiotensin II-induced activation of JAK2 in vascular smooth muscle cells via the polyol pathway. J. Biol. Chem. 2003, 278, 30634–30641. [Google Scholar]
- Carballo, M.; Conde, M.; El Bekay, R.; Martin-Nieto, J.; Camacho, M.J.; Monteseirin, J.; Conde, J.; Bedoya, F.J.; Sobrino, F. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J. Biol. Chem. 1999, 274, 17580–17586. [Google Scholar] [CrossRef]
- Schieffer, B.; Luchtefeld, M.; Braun, S.; Hilfiker, A.; Hilfiker-Kleiner, D.; Drexler, H. Role of NAD(P)H oxidase in angiotensin II-induced JAK/STAT signaling and cytokine induction. Circul. Res. 2000, 87, 1195–1201. [Google Scholar] [CrossRef]
- Van Montfort, R.L.; Congreve, M.; Tisi, D.; Carr, R.; Jhoti, H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 2003, 423, 773–777. [Google Scholar] [CrossRef]
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef]
- Bingisser, R.M.; Tilbrook, P.A.; Holt, P.G.; Kees, U.R. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J. Immunol. 1998, 160, 5729–5734. [Google Scholar]
- Nishina, T.; Komazawa-Sakon, S.; Yanaka, S.; Piao, X.; Zheng, D.M.; Piao, J.H.; Kojima, Y.; Yamashina, S.; Sano, E.; Putoczki, T.; et al. Interleukin-11 links oxidative stress and compensatory proliferation. Sci. Signal. 2012, 5, ra5. [Google Scholar]
- Uckun, F.M.; Qazi, S.; Ma, H.; Tuel-Ahlgren, L.; Ozer, Z. STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress. Proc. Natl. Acad. Sci. USA 2010, 107, 2902–2907. [Google Scholar] [CrossRef]
- Kurdi, M.; Booz, G.W. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: Parthenolide targets JAK1 activation by generating ROS. J. Cell Physiol. 2007, 212, 424–431. [Google Scholar] [CrossRef]
- Jang, E.H.; Park, C.S.; Lee, S.K.; Pie, J.E.; Kang, J.H. Excessive nitric oxide attenuates leptin-mediated signal transducer and activator of transcription 3 activation. Life Sci. 2007, 80, 609–617. [Google Scholar] [CrossRef]
- Duhe, R.J.; Evans, G.A.; Erwin, R.A.; Kirken, R.A.; Cox, G.W.; Farrar, W.L. Nitric oxide and thiol redox regulation of Janus kinase activity. Proc. Natl. Acad. Sci. USA 1998, 95, 126–131. [Google Scholar]
- Mamoon, N.M.; Smith, J.K.; Chatti, K.; Lee, S.; Kundrapu, K.; Duhe, R.J. Multiple cysteine residues are implicated in Janus kinase 2-mediated catalysis. Biochemistry 2007, 46, 14810–14818. [Google Scholar] [CrossRef]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S-Glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 2009, 150, 1122–1131. [Google Scholar] [CrossRef]
- Negoro, S.; Kunisada, K.; Fujio, Y.; Funamoto, M.; Darville, M.I.; Eizirik, D.L.; Osugi, T.; Izumi, M.; Oshima, Y.; Nakaoka, Y.; et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 2001, 104, 979–981. [Google Scholar] [CrossRef]
- Oshima, Y.; Fujio, Y.; Nakanishi, T.; Itoh, N.; Yamamoto, Y.; Negoro, S.; Tanaka, K.; Kishimoto, T.; Kawase, I.; Azuma, J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc. Res. 2005, 65, 428–435. [Google Scholar] [CrossRef]
- Sarafian, T.A.; Montes, C.; Imura, T.; Qi, J.; Coppola, G.; Geschwind, D.H.; Sofroniew, M.V. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS One 2010, 5, e9532. [Google Scholar]
- Szczepanek, K.; Chen, Q.; Derecka, M.; Salloum, F.N.; Zhang, Q.; Szelag, M.; Cichy, J.; Kukreja, R.C.; Dulak, J.; Lesnefsky, E.J.; et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011, 286, 29610–29620. [Google Scholar] [CrossRef]
- Guo, W.; Wei, X.; Wu, S.; Wang, L.; Peng, H.; Wang, J.; Fang, B. Antagonistic effect of flavonoids on NSC-741909-mediated antitumor activity via scavenging of reactive oxygen species. Eur. J. Pharmacol. 2010, 649, 51–58. [Google Scholar] [CrossRef]
- Wei, X.; Guo, W.; Wu, S.; Wang, L.; Huang, P.; Liu, J.; Fang, B. Oxidative stress in NSC-741909-induced apoptosis of cancer cells. J. Transl. Med. 2010, 8. [Google Scholar] [CrossRef]
- Kola, I. The state of innovation in drug development. Clin. Pharmacol. Ther. 2008, 83, 227–230. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Disc. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Arrowsmith, J. Trial watch: Phase III and submission failures: 2007–2010. Nat. Rev. Drug Disc. 2011, 10. [Google Scholar] [CrossRef]
- Arrowsmith, J. Trial watch: Phase II failures: 2008–2010. Nat. Rev. Drug Disc. 2011, 10, 328–329. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues, P.J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: Results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005, 366, 1527–1537. [Google Scholar] [CrossRef]
- Gatzemeier, U.; Pluzanska, A.; Szczesna, A.; Kaukel, E.; Roubec, J.; De, R.F.; Milanowski, J.; Karnicka-Mlodkowski, H.; Pesek, M.; Serwatowski, P.; et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: The Tarceva Lung Cancer Investigation Trial. J. Clin. Oncol. 2007, 25, 1545–1552. [Google Scholar] [CrossRef]
- Herbst, R.S.; Prager, D.; Hermann, R.; Fehrenbacher, L.; Johnson, B.E.; Sandler, A.; Kris, M.G.; Tran, H.T.; Klein, P.; Li, X.; et al. TRIBUTE: A phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 5892–5899. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fang, B. Genetic Interactions of STAT3 and Anticancer Drug Development. Cancers 2014, 6, 494-525. https://doi.org/10.3390/cancers6010494
Fang B. Genetic Interactions of STAT3 and Anticancer Drug Development. Cancers. 2014; 6(1):494-525. https://doi.org/10.3390/cancers6010494
Chicago/Turabian StyleFang, Bingliang. 2014. "Genetic Interactions of STAT3 and Anticancer Drug Development" Cancers 6, no. 1: 494-525. https://doi.org/10.3390/cancers6010494
APA StyleFang, B. (2014). Genetic Interactions of STAT3 and Anticancer Drug Development. Cancers, 6(1), 494-525. https://doi.org/10.3390/cancers6010494