Induction of Chromosomal Instability via Telomere Dysfunction and Epigenetic Alterations in Myeloid Neoplasia

{kind=link}
Abstract
:1. Introduction
1.1. Chromosomal Instability in Cancer
1.2. Role of Chromosomal Aberrations in Human Leukemia
1.3. Mechanisms to Induce CIN in Myeloid Neoplasia
2. Telomeres
2.1. Telomeres and CIN
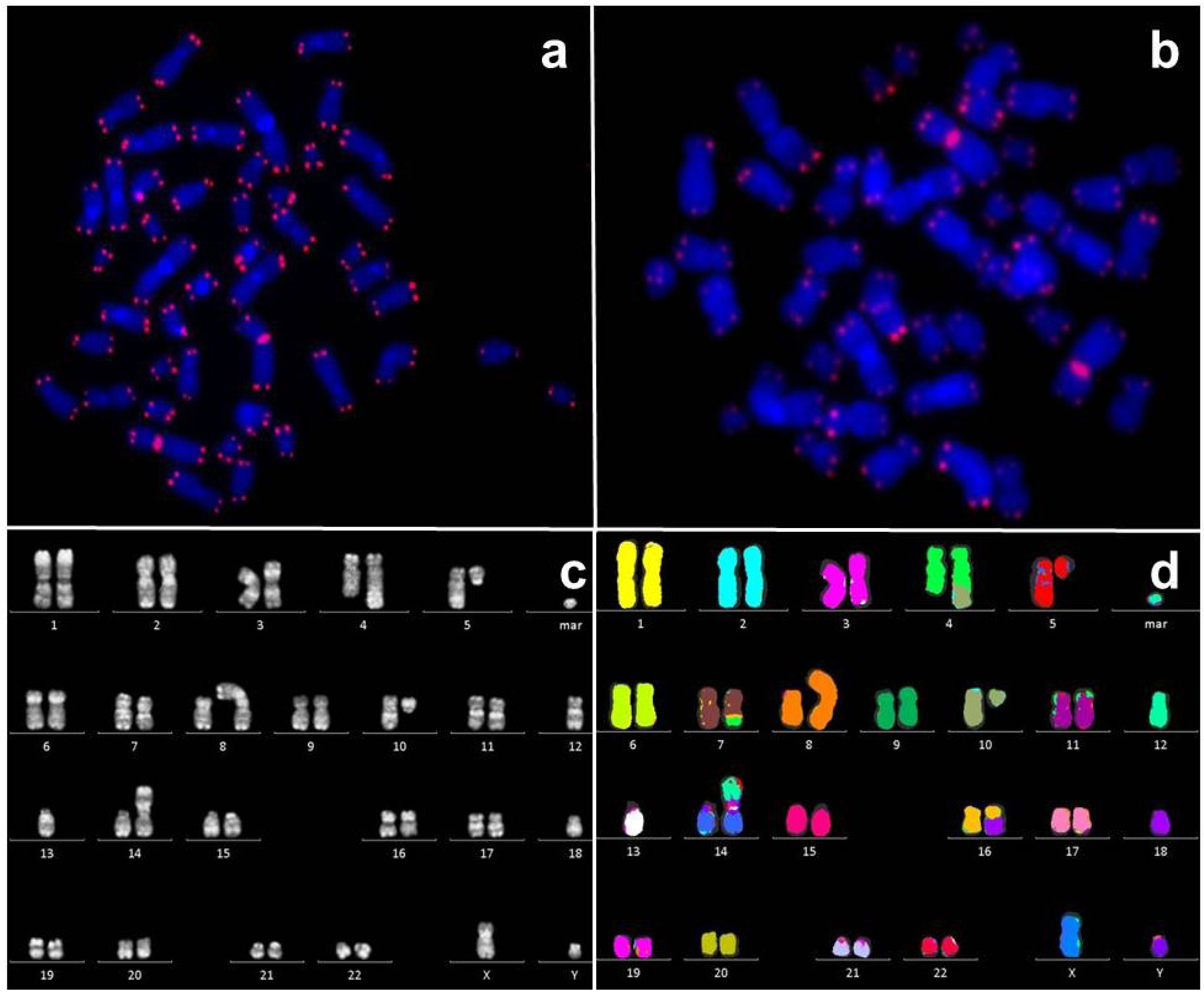
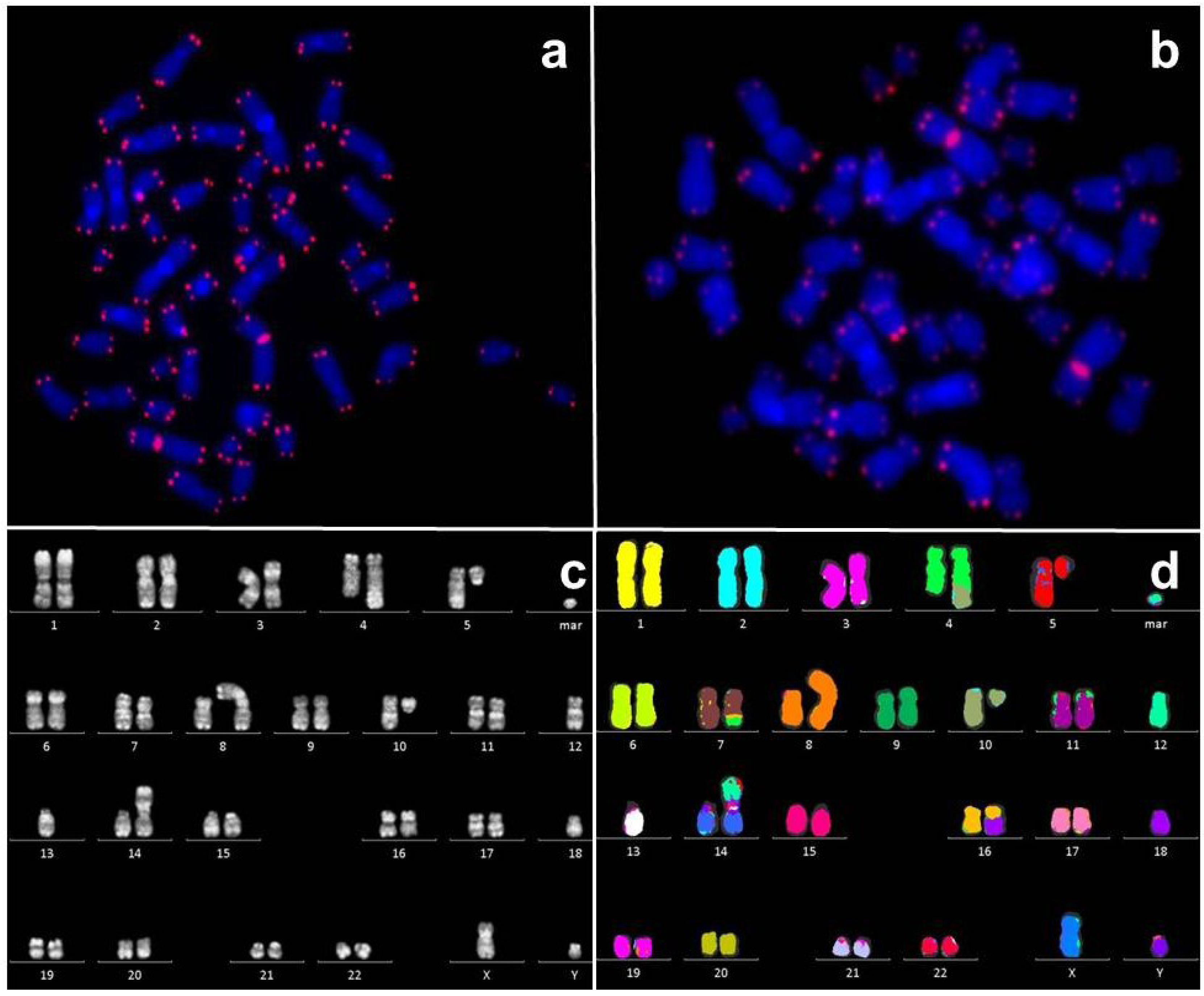
2.2. Telomeres in Leukemia
2.3. Telomere Length Measurement in Primary Cells of Patients and Our Data
3. Epigenetics
3.1. Mutations of Epigenetic Regulators
3.2. Role of Global DNA and Histone Methylation
4. Conclusions
Conflict of Interest
Acknowledgments
References
- Cahill, D.P.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Genetic instability and darwinian selection in tumours. Trends Cell. Biol. 1999, 9, M57–M60. [Google Scholar] [CrossRef]
- Nowak, M.A.; Komarova, N.L.; Sengupta, A.; Jallepalli, P.V.; Shih Ie, M.; Vogelstein, B.; Lengauer, C. The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. USA 2002, 99, 16226–16231. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining “chromosomal instability”. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef]
- Janssen, A.; Medema, R.H. Genetic instability: Tipping the balance. Oncogene 2012. [Google Scholar] [CrossRef]
- Bayani, J.; Selvarajah, S.; Maire, G.; Vukovic, B.; Al-Romaih, K.; Zielenska, M.; Squire, J.A. Genomic mechanisms and measurement of structural and numerical instability in cancer cells. Semin. Cancer Biol. 2007, 17, 5–18. [Google Scholar] [CrossRef]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Wilkens, L.; Flemming, P.; Gebel, M.; Bleck, J.; Terkamp, C.; Wingen, L.; Kreipe, H.; Schlegelberger, B. Induction of aneuploidy by increasing chromosomal instability during dedifferentiation of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2004, 101, 1309–1314. [Google Scholar]
- Heilig, C.E.; Loffler, H.; Mahlknecht, U.; Janssen, J.W.; Ho, A.D.; Jauch, A.; Kramer, A. Chromosomal instability correlates with poor outcome in patients with myelodysplastic syndromes irrespectively of the cytogenetic risk group. J. Cell Mol. Med. 2010, 14, 895–902. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Jadersten, M.; Hellstrom-Lindberg, E. New clues to the molecular pathogenesis of myelodysplastic syndromes. Exp. Cell. Res. 2010, 316, 1390–1396. [Google Scholar] [CrossRef]
- Swerdlow, S.; Campo, E.; Harris, N.; Jaffe, E.; Pileri, S.; Stein, H.; Thiele, J.; Vardiman, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed; IARC: Lyon, France, 2008. [Google Scholar]
- Malcovati, L.; Germing, U.; Kuendgen, A.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Giagounidis, A.; Hildebrandt, B.; Bernasconi, P.; Knipp, S.; et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J. Clin. Oncol. 2007, 25, 3503–3510. [Google Scholar] [CrossRef]
- Germing, U.; Strupp, C.; Giagounidis, A. Clinical features and prognosis of patients with myelodysplastic syndromes. Cancer Treat. Rev. 2007, 33, S15–S18. [Google Scholar] [CrossRef]
- Gohring, G.; Michalova, K.; Beverloo, H.B.; Betts, D.; Harbott, J.; Haas, O.A.; Kerndrup, G.; Sainati, L.; Bergstraesser, E.; Hasle, H.; et al. Complex karyotype newly defined: The strongest prognostic factor in advanced childhood myelodysplastic syndrome. Blood 2010, 116, 3766–3769. [Google Scholar] [CrossRef]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstocker, M.; Nosslinger, T.; Hildebrandt, B.; Kundgen, A.; Lubbert, M.; Kunzmann, R.; Giagounidis, A.A.; et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Andersen, M.T.; Andersen, M.K. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematology Am. Soc. Hematol. Educ. Program 2007, 1, 392–397. [Google Scholar] [CrossRef]
- Corey, S.J.; Minden, M.D.; Barber, D.L.; Kantarjian, H.; Wang, J.C.; Schimmer, A.D. Myelodysplastic syndromes: The complexity of stem-cell diseases. Nat. Rev. Cancer 2007, 7, 118–129. [Google Scholar]
- Huret, J.; Chomienne, C. t(15;17)(q24;q21). Available online: http://atlasgeneticsoncology.org/Anomalies/t1517ID1035.html/ (accessed on 17 June 2013).
- Dohner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- Lugthart, S.; Groschel, S.; Beverloo, H.B.; Kayser, S.; Valk, P.J.; van Zelderen-Bhola, S.L.; Jan Ossenkoppele, G.; Vellenga, E.; van den Berg-de Ruiter, E.; Schanz, U.; et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 3890–3898. [Google Scholar] [CrossRef]
- Dohner, H.; Gaidzik, V.I. Impact of genetic features on treatment decisions in AML. Hematology Am. Soc. Hematol. Educ. Program 2011, 2011, 36–42. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Swiggers, S.J.; Kuijpers, M.A.; de Cort, M.J.; Beverloo, H.B.; Zijlmans, J.M. Critically short telomeres in acute myeloid leukemia with loss or gain of parts of chromosomes. Genes Chromosomes Cancer 2006, 45, 247–256. [Google Scholar] [CrossRef]
- Sieglova, Z.; Zilovcova, S.; Cermak, J.; Rihova, H.; Brezinova, D.; Dvorakova, R.; Markova, M.; Maaloufova, J.; Sajdova, J.; Brezinova, J.; et al. Dynamics of telomere erosion and its association with genome instability in myelodysplastic syndromes (MDS) and acute myelogenous leukemia arising from MDS: A marker of disease prognosis? Leuk. Res. 2004, 28, 1013–1021. [Google Scholar] [CrossRef]
- Boultwood, J.; Fidler, C.; Kusec, R.; Rack, K.; Elliott, P.J.; Atoyebi, O.; Chapman, R.; Oscier, D.G.; Wainscoat, J.S. Telomere length in myelodysplastic syndromes. Am. J. Hematol. 1997, 56, 266–271. [Google Scholar] [CrossRef]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef]
- Hemann, M.T.; Rudolph, K.L.; Strong, M.A.; DePinho, R.A.; Chin, L.; Greider, C.W. Telomere dysfunction triggers developmentally regulated germ cell apoptosis. Mol. Biol. Cell 2001, 12, 2023–2030. [Google Scholar]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef]
- Bullinger, L.; Dohner, K.; Bair, E.; Frohling, S.; Schlenk, R.F.; Tibshirani, R.; Dohner, H.; Pollack, J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef]
- Alvarez, S.; Suela, J.; Valencia, A.; Fernandez, A.; Wunderlich, M.; Agirre, X.; Prosper, F.; Martin-Subero, J.I.; Maiques, A.; Acquadro, F.; et al. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PLoS One 2010, 5, e12197. [Google Scholar] [CrossRef]
- Bullinger, L.; Ehrich, M.; Dohner, K.; Schlenk, R.F.; Dohner, H.; Nelson, M.R.; van den Boom, D. Quantitative DNA methylation predicts survival in adult acute myeloid leukemia. Blood 2010, 115, 636–642. [Google Scholar] [CrossRef]
- Nolte, F.; Giehl, M.; Haass, W.; Nowak, V.; Schumann, C.; Nowak, D.; Mossner, M.; Popp, H.D.; Schulze, T.J.; Klein, S.; et al. Centrosome aberrations in bone marrow cells from patients with myelodysplastic syndromes correlate with chromosomal instability. Ann. Hematol. 2013. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Rassool, F.V.; Gaymes, T.J.; Omidvar, N.; Brady, N.; Beurlet, S.; Pla, M.; Reboul, M.; Lea, N.; Chomienne, C.; Thomas, N.S.; et al. Reactive oxygen species, DNA damage, and error-prone repair: A model for genomic instability with progression in myeloid leukemia? Cance. Res. 2007, 67, 8762–8771. [Google Scholar]
- Sallmyr, A.; Tomkinson, A.E.; Rassool, F.V. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: Consequences for the repair of DNA double-strand breaks. Blood 2008, 112, 1413–1423. [Google Scholar]
- Popp, H.D.; Bohlander, S.K. Genetic instability in inherited and sporadic leukemias. Genes Chromosomes Cancer 2010, 49, 1071–1081. [Google Scholar]
- Drummond, M.W.; Balabanov, S.; Holyoake, T.L.; Brummendorf, T.H. Concise review: Telomere biology in normal and leukemic hematopoietic stem cells. Stem Cells 2007, 25, 1853–1861. [Google Scholar]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar]
- Calado, R.T.; Regal, J.A.; Hills, M.; Yewdell, W.T.; Dalmazzo, L.F.; Zago, M.A.; Lansdorp, P.M.; Hogge, D.; Chanock, S.J.; Estey, E.H.; et al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 1187–1192. [Google Scholar]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase in normal and cancer stem cells. FEBS Lett. 2010, 584, 3819–3825. [Google Scholar]
- Martinez, P.; Blasco, M.A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11, 161–176. [Google Scholar]
- Blackburn, E.H.; Collins, K. Telomerase: An RNP enzyme synthesizes DNA. Cold Spring Harb. Perspect. Biol. 2011. [Google Scholar] [CrossRef]
- Artandi, S.E. Telomeres, telomerase, and human disease. N. Engl. J. Med. 2006, 355, 1195–1197. [Google Scholar]
- Drummond, M.W.; Hoare, S.F.; Monaghan, A.; Graham, S.M.; Alcorn, M.J.; Keith, W.N.; Holyoake, T.L. Dysregulated expression of the major telomerase components in leukaemic stem cells. Leukemia 2005, 19, 381–389. [Google Scholar]
- Murnane, J.P. Telomere loss as a mechanism for chromosome instability in human cancer. Cancer Res. 2010, 70, 4255–4259. [Google Scholar]
- Hande, M.P. DNA repair factors and telomere-chromosome integrity in mammalian cells. Cytogenet. Genome Res. 2004, 104, 116–122. [Google Scholar]
- Gisselsson, D. Mitotic instability in cancer: Is there method in the madness? Cell Cycle 2005, 4, 1007–1010. [Google Scholar]
- Briatore, F.; Barrera, G.; Pizzimenti, S.; Toaldo, C.; Casa, C.D.; Laurora, S.; Pettazzoni, P.; Dianzani, M.U.; Ferrero, D. Increase of telomerase activity and hTERT expression in myelodysplastic syndromes. Cancer Biol. Ther. 2009, 8, 883–889. [Google Scholar]
- Pettigrew, K.A.; Armstrong, R.N.; Colyer, H.A.; Zhang, S.D.; Rea, I.M.; Jones, R.E.; Baird, D.M.; Mills, K.I. Differential TERT promoter methylation and response to 5-aza-2'-deoxycytidine in acute myeloid leukemia cell lines: TERT expression, telomerase activity, telomere length, and cell death. Genes Chromosomes Cancer 2012, 51, 768–780. [Google Scholar]
- Rollison, D.E.; Epling-Burnette, P.K.; Park, J.Y.; Lee, J.H.; Park, H.; Jonathan, K.; Cole, A.L.; Painter, J.S.; Guerrier, M.; Melendez-Santiago, J.; et al. Telomere length in myelodysplastic syndromes. Leuk. Lymphoma 2011, 52, 1528–1536. [Google Scholar]
- Capraro, V.; Zane, L.; Poncet, D.; Perol, D.; Galia, P.; Preudhomme, C.; Bonnefoy-Berard, N.; Gilson, E.; Thomas, X.; El-Hamri, M.; et al. Telomere deregulations possess cytogenetic, phenotype, and prognostic specificities in acute leukemias. Exp. Hematol. 2011, 39, 195–202.e2. [Google Scholar]
- Gadji, M.; Adebayo Awe, J.; Rodrigues, P.; Kumar, R.; Houston, D.S.; Klewes, L.; Dieye, T.N.; Rego, E.M.; Passetto, R.F.; de Oliveira, F.M.; et al. Profiling three-dimensional nuclear telomeric architecture of myelodysplastic syndromes and acute myeloid leukemia defines patient subgroups. Clin. Cancer Res. 2012, 18, 3293–3304. [Google Scholar] [CrossRef]
- Calado, R.T.; Young, N.S. Telomere maintenance and human bone marrow failure. Blood 2008, 111, 4446–4455. [Google Scholar] [CrossRef]
- Young, N.S. Telomere biology and telomere diseases: Implications for practice and research. Hematology Am. Soc. Hematol. Educ. Program 2010, 2010, 30–35. [Google Scholar]
- Armanios, M. Syndromes of telomere shortening. Annu. Rev. Genomics Hum. Genet. 2009, 10, 45–61. [Google Scholar]
- Kirwan, M.; Vulliamy, T.; Marrone, A.; Walne, A.J.; Beswick, R.; Hillmen, P.; Kelly, R.; Stewart, A.; Bowen, D.; Schonland, S.O.; et al. Defining the pathogenic role of telomerase mutations in myelodysplastic syndrome and acute myeloid leukemia. Hum. Mutat. 2009, 30, 1567–1573. [Google Scholar]
- Calado, R.T.; Cooper, J.N.; Padilla-Nash, H.M.; Sloand, E.M.; Wu, C.O.; Scheinberg, P.; Ried, T.; Young, N.S. Short telomeres result in chromosomal instability in hematopoietic cells and precede malignant evolution in human aplastic anemia. Leukemia 2012, 26, 700–707. [Google Scholar]
- Yamaguchi, H.; Baerlocher, G.M.; Lansdorp, P.M.; Chanock, S.J.; Nunez, O.; Sloand, E.; Young, N.S. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood 2003, 102, 916–918. [Google Scholar] [CrossRef]
- Lange, K.; Holm, L.; Vang Nielsen, K.; Hahn, A.; Hofmann, W.; Kreipe, H.; Schlegelberger, B.; Gohring, G. Telomere shortening and chromosomal instability in myelodysplastic syndromes. Genes Chromosomes Cancer 2010, 49, 260–269. [Google Scholar]
- Gohring, G.; Lange, K.; Hofmann, W.; Nielsen, K.V.; Hellstrom-Lindberg, E.; Roy, L.; Morgan, M.; Kreipe, H.; Busche, G.; Giagounidis, A.; et al. Telomere shortening, clonal evolution and disease progression in myelodysplastic syndrome patients with 5q deletion treated with lenalidomide. Leukemia 2012, 26, 356–358. [Google Scholar] [CrossRef]
- Sobulo, O.M.; Borrow, J.; Tomek, R.; Reshmi, S.; Harden, A.; Schlegelberger, B.; Housman, D.; Doggett, N.A.; Rowley, J.D.; Zeleznik-Le, N.J. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc. Natl. Acad. Sci. USA 1997, 94, 8732–8737. [Google Scholar]
- Katsumoto, T.; Yoshida, N.; Kitabayashi, I. Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. 2008, 99, 1523–1527. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; de Ayala Alonso, A.G.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Muller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef]
- Potapova, A.; Hasemeier, B.; Romermann, D.; Metzig, K.; Gohring, G.; Schlegelberger, B.; Langer, F.; Kreipe, H.; Lehmann, U. Epigenetic inactivation of tumour suppressor gene KLF11 in myelodysplastic syndromes. Eur. J. Haematol. 2010, 84, 298–303. [Google Scholar]
- Ono, R.; Taki, T.; Taketani, T.; Taniwaki, M.; Kobayashi, H.; Hayashi, Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002, 62, 4075–4080. [Google Scholar]
- Koh, K.P.; Yabuuchi, A.; Rao, S.; Huang, Y.; Cunniff, K.; Nardone, J.; Laiho, A.; Tahiliani, M.; Sommer, C.A.; Mostoslavsky, G.; et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011, 8, 200–213. [Google Scholar]
- Matarese, F.; Carrillo-de Santa Pau, E.; Stunnenberg, H.G. 5-hydroxymethylcytosine: A new kid on the epigenetic block? Mol. Syst. Biol. 2011, 7, 562. [Google Scholar]
- Figueroa, M.E.; Skrabanek, L.; Li, Y.; Jiemjit, A.; Fandy, T.E.; Paietta, E.; Fernandez, H.; Tallman, M.S.; Greally, J.M.; Carraway, H.; et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 2009, 114, 3448–3458. [Google Scholar]
- del Rey, M.; O’Hagan, K.; Dellett, M.; Aibar, S.; Colyer, H.A.; Alonso, M.E.; Diez-Campelo, M.; Armstrong, R.N.; Sharpe, D.J.; Gutierrez, N.C.; et al. Genome-wide profiling of methylation identifies novel targets with aberrant hypermethylation and reduced expression in low-risk myelodysplastic syndromes. Leukemia 2013, 27, 610–618. [Google Scholar] [CrossRef]
- Shen, Y.; Zhu, Y.M.; Fan, X.; Shi, J.Y.; Wang, Q.R.; Yan, X.J.; Gu, Z.H.; Wang, Y.Y.; Chen, B.; Jiang, C.L.; et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood 2011, 118, 5593–5603. [Google Scholar] [CrossRef]
- Lugthart, S.; Figueroa, M.E.; Bindels, E.; Skrabanek, L.; Valk, P.J.; Li, Y.; Meyer, S.; Erpelinck-Verschueren, C.; Greally, J.; Lowenberg, B.; et al. Aberrant DNA hypermethylation signature in acute myeloid leukemia directed by EVI1. Blood 2011, 117, 234–241. [Google Scholar] [CrossRef]
- Kustikova, O.S.; Schwarzer, A.; Stahlhut, M.; Brugman, M.H.; Neumann, T.; Yang, M.; Li, Z.; Schambach, A.; Heinz, N.; Gerdes, S.; et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia 2012, 27, 1127–1138. [Google Scholar]
- Lugthart, S.; van Drunen, E.; van Norden, Y.; van Hoven, A.; Erpelinck, C.A.; Valk, P.J.; Beverloo, H.B.; Lowenberg, B.; Delwel, R. High EVI1 levels predict adverse outcome in acute myeloid leukemia: Prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood 2008, 111, 4329–4337. [Google Scholar] [CrossRef]
- Li, Z.; Dullmann, J.; Schiedlmeier, B.; Schmidt, M.; von Kalle, C.; Meyer, J.; Forster, M.; Stocking, C.; Wahlers, A.; Frank, O.; et al. Murine leukemia induced by retroviral gene marking. Science 2002, 296, 497. [Google Scholar]
- Modlich, U.; Schambach, A.; Brugman, M.H.; Wicke, D.C.; Knoess, S.; Li, Z.; Maetzig, T.; Rudolph, C.; Schlegelberger, B.; Baum, C. Leukemia induction after a single retroviral vector insertion in Evi1 or Prdm16. Leukemia 2008, 22, 1519–1528. [Google Scholar]
- Karakaya, K.; Herbst, F.; Ball, C.; Glimm, H.; Kramer, A.; Loffler, H. Overexpression of EVI1 interferes with cytokinesis and leads to accumulation of cells with supernumerary centrosomes in G0/1 phase. Cell Cycle 2012, 11, 3492–3503. [Google Scholar]
- Vajen, B.; Modlich, U.; Schienke, A.; Wolf, S.; Skawran, B.; Hofmann, W.; Busche, G.; Kreipe, H.; Baum, C.; Santos-Barriopedro, I.; et al. Histone methyltransferase Suv39h1 deficiency prevents Myc-induced chromosomal instability in murine myeloid leukemias. Genes Chromosomes Cancer 2013, 52, 423–430. [Google Scholar] [CrossRef]
- Garcia-Cao, M.; O’Sullivan, R.; Peters, A.H.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2004, 36, 94–99. [Google Scholar]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.; Moshir, S.; Mougey, V.; et al. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 9613–9618. [Google Scholar]
- Pampalona, J.; Soler, D.; Genesca, A.; Tusell, L. Whole chromosome loss is promoted by telomere dysfunction in primary cells. Genes Chromosomes Cancer 2010, 49, 368–378. [Google Scholar]
- Zhong, Z.H.; Jiang, W.Q.; Cesare, A.J.; Neumann, A.A.; Wadhwa, R.; Reddel, R.R. Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J. Biol. Chem. 2007, 282, 29314–29322. [Google Scholar]
- Fan, Q.; Zhang, F.; Barrett, B.; Ren, K.; Andreassen, P.R. A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 2009, 37, 1740–1754. [Google Scholar] [CrossRef]
- Karlsson, A.; Deb-Basu, D.; Cherry, A.; Turner, S.; Ford, J.; Felsher, D.W. Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. USA 2003, 100, 9974–9979. [Google Scholar]
- Ray, S.; Atkuri, K.R.; Deb-Basu, D.; Adler, A.S.; Chang, H.Y.; Herzenberg, L.A.; Felsher, D.W. MYC can induce DNA breaks in vivo and in vitro independent of reactive oxygen species. Cancer Res. 2006, 66, 6598–6605. [Google Scholar] [CrossRef]
- Smeenk, G.; de Groot, A.J.; Romeijn, R.J.; van Buul, P.P.; Zdzienicka, M.Z.; Mullenders, L.H.; Pastink, A.; Godthelp, B.C. Rad51C is essential for embryonic development and haploinsufficiency causes increased DNA damage sensitivity and genomic instability. Mutat. Res. 2010, 689, 50–58. [Google Scholar] [CrossRef]
- Fung, T.K.; Siu, W.Y.; Yam, C.H.; Lau, A.; Poon, R.Y. Cyclin F is degraded during G2-M by mechanisms fundamentally different from other cyclins. J. Biol. Chem. 2002, 277, 35140–35149. [Google Scholar]
- Ward, I.M.; Minn, K.; van Deursen, J.; Chen, J. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003, 23, 2556–2563. [Google Scholar]
- Goodarzi, A.A.; Noon, A.T.; Jeggo, P.A. The impact of heterochromatin on DSB repair. Biochem. Soc. Trans. 2009, 37, 569–576. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vajen, B.; Thomay, K.; Schlegelberger, B. Induction of Chromosomal Instability via Telomere Dysfunction and Epigenetic Alterations in Myeloid Neoplasia. Cancers 2013, 5, 857-874. https://doi.org/10.3390/cancers5030857
Vajen B, Thomay K, Schlegelberger B. Induction of Chromosomal Instability via Telomere Dysfunction and Epigenetic Alterations in Myeloid Neoplasia. Cancers. 2013; 5(3):857-874. https://doi.org/10.3390/cancers5030857
Chicago/Turabian StyleVajen, Beate, Kathrin Thomay, and Brigitte Schlegelberger. 2013. "Induction of Chromosomal Instability via Telomere Dysfunction and Epigenetic Alterations in Myeloid Neoplasia" Cancers 5, no. 3: 857-874. https://doi.org/10.3390/cancers5030857
APA StyleVajen, B., Thomay, K., & Schlegelberger, B. (2013). Induction of Chromosomal Instability via Telomere Dysfunction and Epigenetic Alterations in Myeloid Neoplasia. Cancers, 5(3), 857-874. https://doi.org/10.3390/cancers5030857