Epigenetic Modulating Agents as a New Therapeutic Approach in Multiple Myeloma


Abstract
:1. Introduction
2. Epigenetics
2.1. DNA Methylation
2.2. Histone Modifications
2.2.1. Histone Acetylation
2.2.2. Histone Methylation
2.2.3. Histone Phosphorylation
2.3. Cooperation between Histone Modifications and DNA Methylation
3. Epigenetic Changes in Multiple Myeloma

{kind=link}
{kind=link}
| Biological function | Gene name (symbol) | Number of MM cells lines with methylation | Methylation frequency in primary MM samples | Poor prognosis | Reference |
|---|---|---|---|---|---|
| regulation of apoptosis | XIAP-associated factor 1 (XAF-1) | 2 | - | [60] | |
| BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) | 3 | 5–13% | X | [50,51] | |
| B-cell CLL/lymphoma 7C (BCL7c) | 3 | 21% | [50] | ||
| Growth Arrest and DNA Damage inducible γ (GADD45) | 1 | 19% | [50] | ||
| Death-associated protein kinase 1 (DAPK) | 1 | 5–20% | X | [51,54,57] | |
| regulation of cell cycle | Cyclin-dependent kinase inhibitor 2A (CDKN2A, p16) | 4 | 5–40% | X | [51,54,57,58,61] |
| Cyclin-dependent kinase 4 inhibitor B (CDKN2B, p15) | - | 5–30% | [51,54,57,58] | ||
| Cyclin-A1 (CCNA1) | 3 | 8% | [50] | ||
| DNA repair | Methylated-DNA-protein-cysteine methyltransferase (MGMT) | 2 | 2–4% | [54,57] | |
| tumor suppression | Tumor protein 73 (TP73) | 2 | 12–45% | [54,57,58] | |
| Tumor protein 53 (TP53) | 4 | - | [62] | ||
| signal transduction | Suppressor of cytokine signaling 1, 3 (SOCS-1, -3) | 5 | 45–50% | [57,58,63,64] | |
| Spleen tyrosine kinase (SYK) | - | 38% | [63] | ||
| Dexamethasone-induced Ras-related protein 1 (RASD1) | 5 | 8% | [59] | ||
| Deleted in liver cancer-1 (DLC-1) | 3 | 78% | X | [53] | |
| Ras association domain-containing protein 1A (RASSF1A) | - | 2–15% | [54,58] | ||
| Stratifin (SFN) | - | 100% | [55] | ||
| Wnt pathway | Wnt inhibitory factor 1 (WIF1) | 2 | 22% | [65] | |
| Secreted frizzled-related protein 1, 2, 4, 5 (sFRP1, 2, 4, 5) | 4 | 4–50% | [65,66] | ||
| Dickkopf-related protein 1, 3 (DKK1, 3) | 2–4 | 16–32% | [65,67] | ||
| Adenomatous polyposis coli (APC) | 1 | 18% | [65] | ||
| osteogenesis | Secreted protein acidic and rich in cysteine (SPARC) | 2 | 8% | X | [50] |
| growth factor signaling | Transforming growth factor beta-receptor 2 (TGFβR2) | - | 45% | X | [55] |
| hormone signaling | Estrogen Receptor (ESR1) | - | 15–80% | [51,55] | |
| Retinoic acid receptor beta (RARβ) | 3 | 2–12% | X | [51,57] | |
| Prostaglandin-endoperoxide synthase 2 (PTGS2) | - | 100% | [55] | ||
| cell adhesion | Cadherin 1 (CDH1, E-cadherin) | - | 30–80% | X | [51,54,55] |
| Gap junction alpha-1 protein (GJA1) | 3 | 23% | [50] | ||
| CD9 antigen (CD9) | 2 | - | X | [56] | |
| A-kinase anchor protein 12 (AKAP12) | 2 | 13% | [50] | ||
| Deleted in colorectal carcinoma (DCC) | - | 45% | X | [55] | |
| coagulation | Tissue factor pathway inhibitor (TFPI2) | 2 | 10% | [50] | |
| hypoxia signaling | Egl nine homolog 3 (EGLN3) | - | 33% | X | [52] |
| transcriptional repression | Hypermethylated in cancer 1 (HIC1) | - | 70% | [55] | |
| regulation of translation | Cytoplasmic polyadenylation element-binding protein 1 (CPEB1) | 4 | 50% | [50] | |
| transcription factor | Interferon regulatory factor 8 (IRF8) | 8 | 10% | [68] |
4. Targeting Epigenetic Modifications in Multiple Myeloma
4.1. HDACi and Multiple Myeloma
| Chemical class | HDAC inhibitor | Reported targets |
|---|---|---|
| Benzamides | SNDX-275 (MS-275, Entinostat) | HDAC-1,-2,-3 |
| CI-994 (Tacedinaline) | HDAC-1, -2 | |
| MGCD-0103 | HDAC-1, -2, -3, 11 | |
| Short Chain Fatty Acids | Valproic acid (VPA) | Class I and IIa |
| Sodium butyrate | Class I and IIa, IV | |
| Phenyl butyrate (S-HDAC-42, AR-42) | Class I, II | |
| Cyclic Peptides | Depsipeptide (Romidepsin) | HDAC -1, -2 |
| Apicidin | Class I | |
| Hydroxamic Acids | JNJ-26481585 | Class I and II, IV |
| Suberoylanilide hydroxamic acid (SAHA; Vorinostat) | Class I and II, IV | |
| Trichostatin-A (TSA) | Class I and II, IV | |
| LBH589 (Panobinostat) | Class I and II, IV | |
| ITF2357 (Gavinostat) | Class I and II | |
| PXD101 (Belinostat) | Class I and II, IV | |
| NVP-LAQ824 (Dacinostat) | Class I | |
| Suberoylanilide bis-hydroxamic acid (SBHA) | HDAC-1, -3 | |
| RAS2410 (Resminostat) | HDAC-1, -3, -6 | |
| ACY-1215 (Rocilinostat) | HDAC-6 | |
| CR-2408 | Class I, II, IV | |
| Mercaptoketone | KD5170 | Class I and II |
| Others | Tubacin | HDAC-6 |
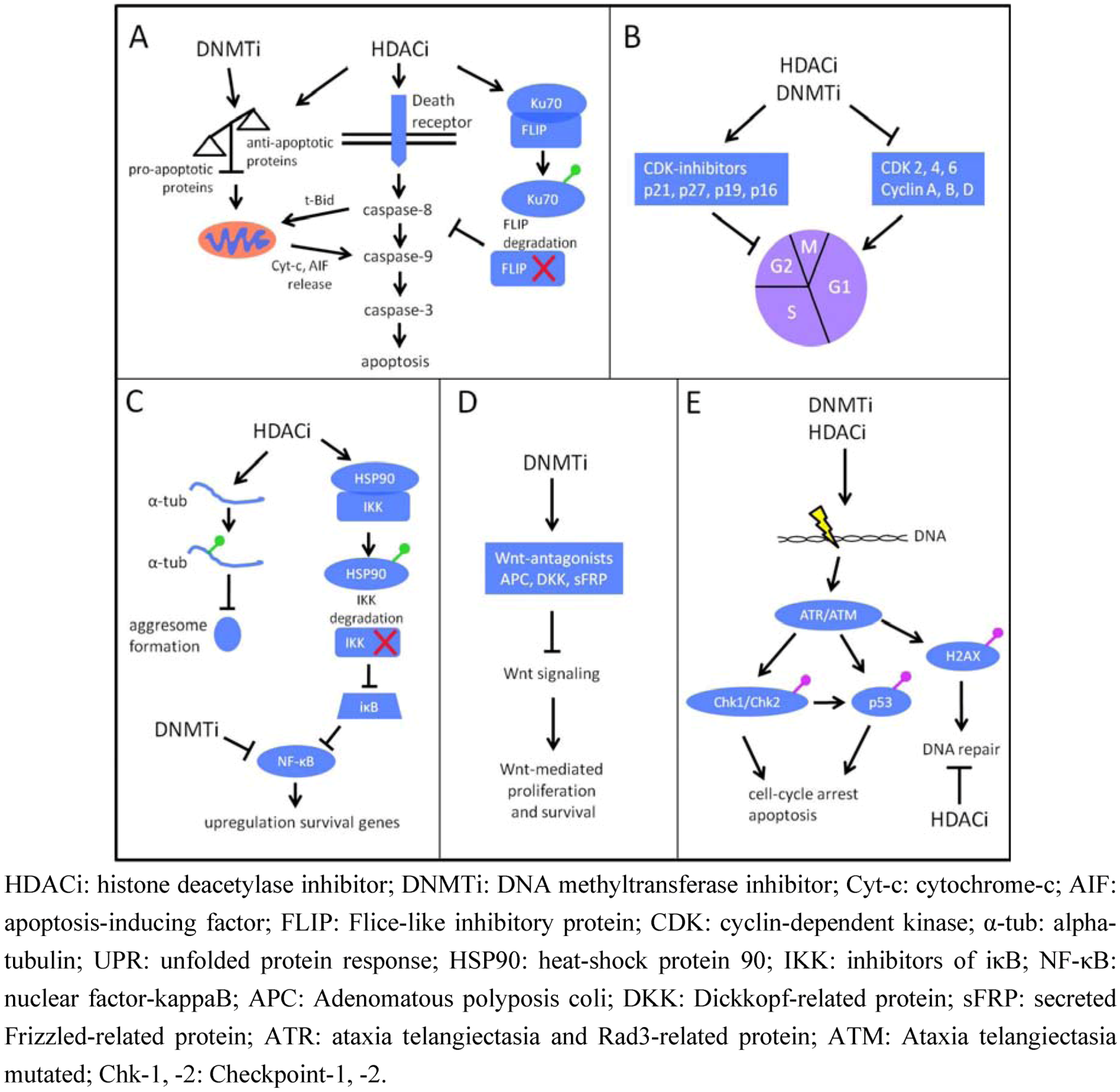
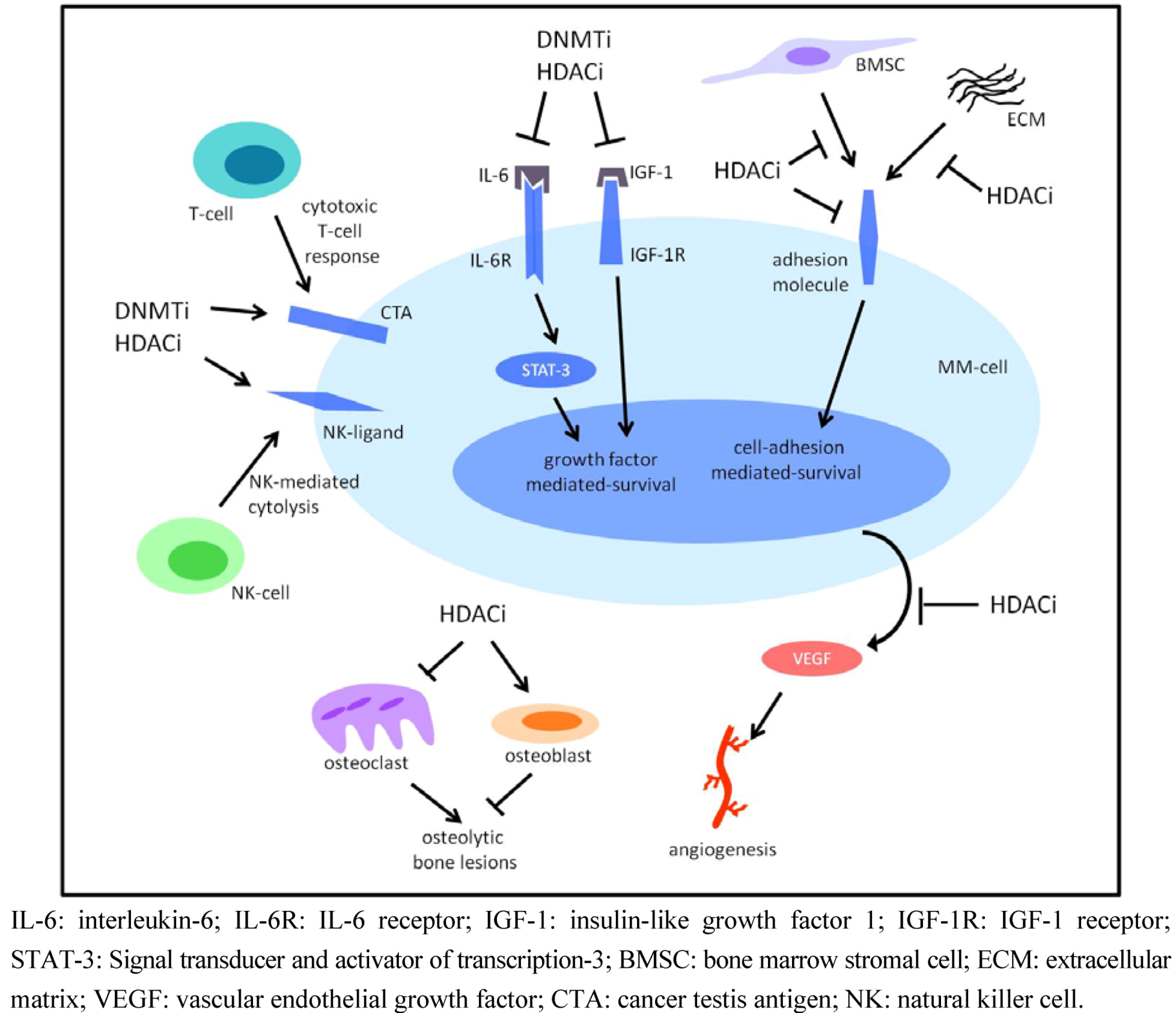
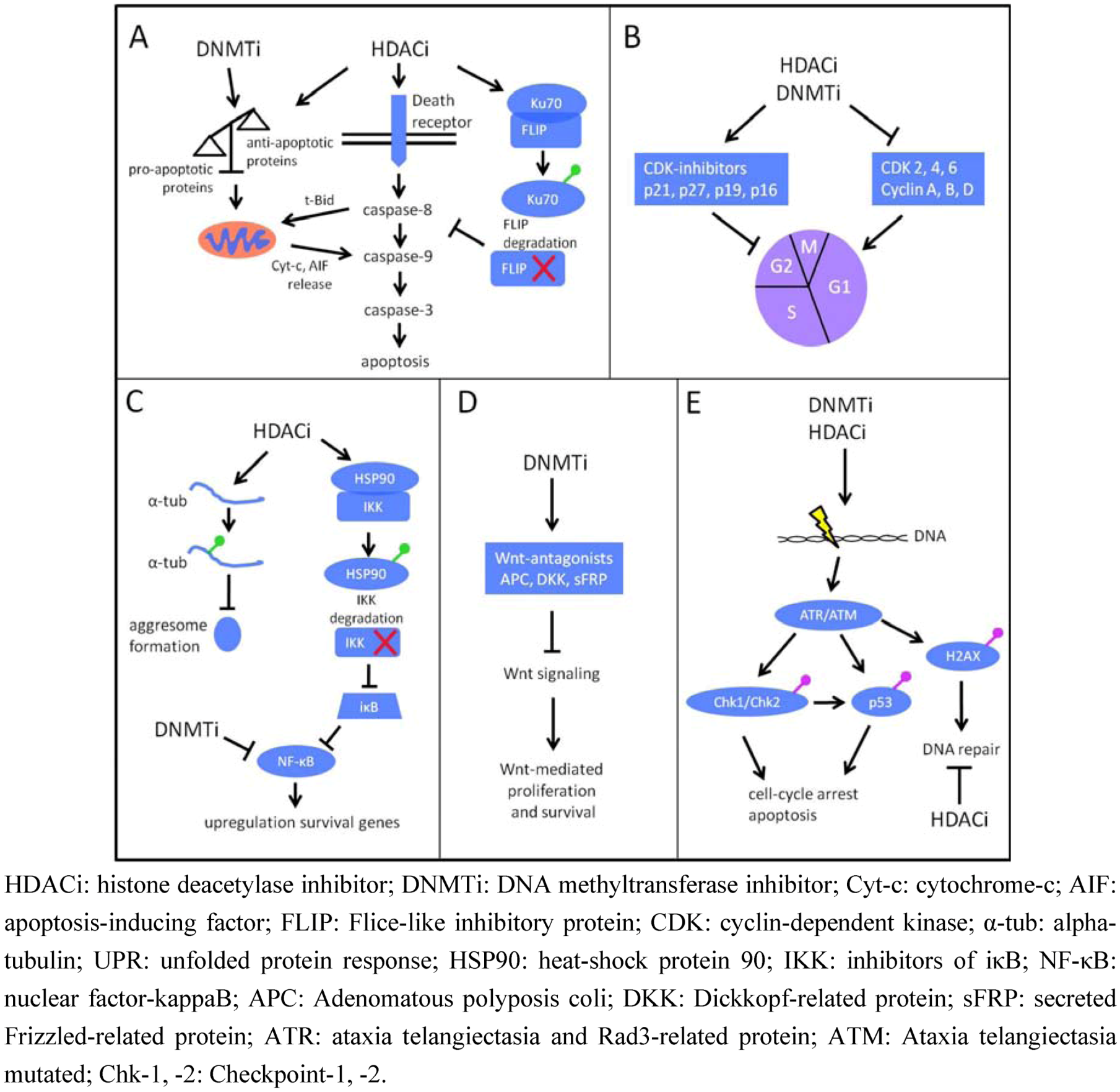
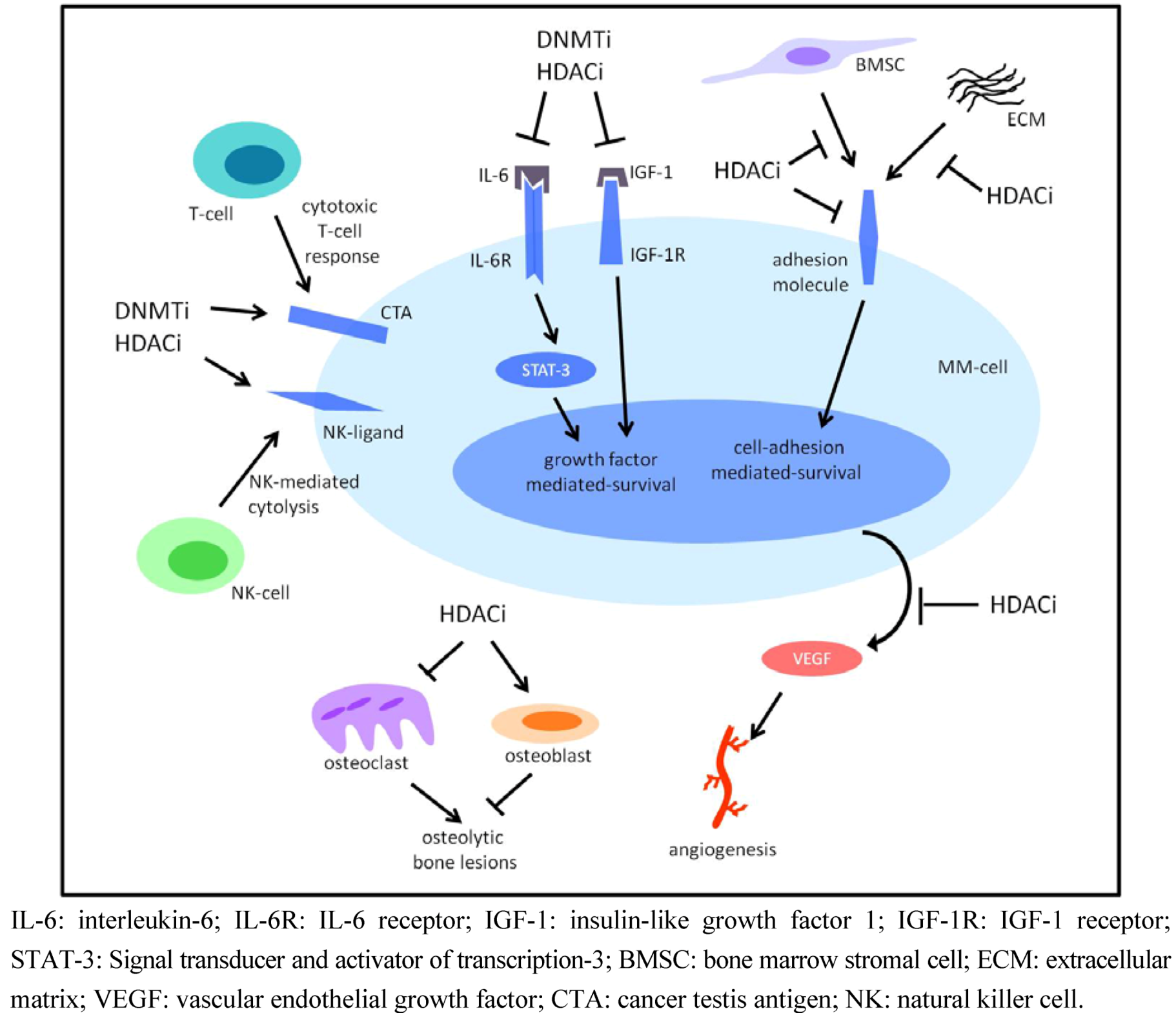
4.2. DNMTi and Multiple Myeloma
4.3. Combining HDACi and DNMTi in Multiple Myeloma
5. HDACi and DNMTi in the Clinic
| Drug | Drug | Combination with | Myeloma patients | Response | Reference |
|---|---|---|---|---|---|
| Vorinostat | I | - | relapsed/refractory (n = 13) | 1 MR | [134] |
| 9 SD | |||||
| Belinostat | I | - | relapsed/refractory (n = 4) | 1 SD | [135] |
| Panobinostat | Ia/II | - | relapsed/refractory (n = 12) | 1 PR | [131] |
| Romidepsin | II | - | relapsed/refractory (n = 13) | 4 SD | [132] |
| Gavinostat | II | - | relapsed/refractory (n = 19) | 6 SD | [133] |
| Vorinostat | I | Bortezomib | relapsed/refractory (n = 23) | 2 VGPR | [141] |
| 13 PR | |||||
| 10 SD | |||||
| Vorinostat | I | Bortezomib | relapsed/refractory (n = 6) | 1 VGPR | [140] |
| 4 MR | |||||
| 1 SD | |||||
| Vorinostat | I | Bortezomib | relapsed/refractory (n = 34) | 9 PR | [139] |
| 2 MR | |||||
| 20 SD | |||||
| Vorinostat | I | Lenalidomide | newly diagnosed (n = 30) | 10 CR | [142] |
| Bortezomib Dexamethasone | 15 VGPR | ||||
| Vorinostat | I/II | Lenalidomide Bortezomib Dexamethasone | relapsed/refractory (n = 64) | 8 CR | [143] |
| 4 VGPR | |||||
| 22 PR | |||||
| 9 MR | |||||
| 9 SD | |||||
| Panobinostat | II | Bortezomib Dexamethasone | relapsed/refractory (n = 55) | 1 CR | [144] |
| 18 PR | |||||
| 10 MR | |||||
| 20 SD | |||||
| Panobinostat | I | Carfilzomib | relapsed/refractory (n = 17) | 2 VGPR | [146] |
| 6 PR | |||||
| 1 MR | |||||
| Panobinostat | I/II | Carfilzomib | relapsed/refractory (n = 10) | ongoing | [145] |
| Romidepsin | I/II | Dexamethasone Bortezomib | previously treated (n = 25) | 2 CR | [147] |
| 13 PR | |||||
| 3 MR | |||||
| 2 SD | |||||
| Romidepsin | I/II | Bortezomib | relapsed/refractory (recruiting) | ongoing | [136] |
| Vorinostat | IIb | Bortezomib | relapsed/refractory (n = 143) | ongoing | [149] |
| Vorinostat | III | Bortezomib | relapsed/refractory (n = 637) | ongoing | [148] |
| Panobinostat | III | Bortezomib | relapsed/refractory (n = 672) | ongoing | [150] |
| Panobinostat | I/II | Lenalidomide Bortezomib Dexamethasone | newly diagnosed (recruiting) | ongoing | [136] |
| Vorinostat | III | Lenalidomide | newly diagnosed (recruiting) | ongoing | [136] |
| Thalidomide | |||||
| Bortezomib | |||||
| Vorinostat | I | Lenalidomide | post transplant (n = 16) | 4 improved responses | [152] |
| Azacytidine | II | Lenalidomide | partial remission or plateau (n = 14) | 6 CTA upregulation | [153] |
| 3 CTL responses | |||||
| Azacytidine | I | Lenalidomide | Transplantation eligible | ongoing | [136] |
| (recruiting) | |||||
| Azacytidine | I/II | Lenalidomide | relapsed/refractory | ongoing | [136] |
| Dexamethasone | (recruiting) | ||||
| Decitabine | I | - | relapsed/refractory | ongoing | [136] |
6. Conclusions and Perspectives
Conflicts of Interest
Acknowledgements
References
- Lemaire, M.; Deleu, S.; de Bruyne, E.; van Valckenborgh, E.; Menu, E.; Vanderkerken, K. The microenvironment and molecular biology of the multiple myeloma tumor. Adv. Cancer Res. 2012, 110, 19–42. [Google Scholar]
- Bruyne, E.; Maes, K.; Deleu, S.; Valckenborgh, E.; Menu, E.; Broek, I.; Fraczek, J.; Grunsven, L.; Rogiers, V.; Jernberg-Wiklund, H.; et al. Epigenetic regulation of myeloma within its bone marrow microenvironment. In Advances in Biology and Therapy of Multiple Myeloma; Munshi, N.C., Anderson, K.C., Eds.; Springer: New York, NY, USA, 2013; pp. 255–282. [Google Scholar]
- Becker, N. Epidemiology of multiple myeloma. Recent Results Cancer Res. 2011, 183, 25–35. [Google Scholar] [CrossRef]
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple myeloma. Lancet 2009, 374, 324–339. [Google Scholar]
- Mahindra, A.; Hideshima, T.; Anderson, K.C. Multiple myeloma: Biology of the disease. Blood Rev. 2011, 24, S5–S11. [Google Scholar] [CrossRef]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef]
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, W.M. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009, 113, 5418–5422. [Google Scholar]
- Kuehl, W.M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J. Clin. Invest. 2012, 122, 3456–3463. [Google Scholar] [CrossRef]
- Chng, W.J.; Glebov, O.; Bergsagel, P.L.; Kuehl, W.M. Genetic events in the pathogenesis of multiple myeloma. Best Pract. Res. Clin. Haematol. 2007, 20, 571–596. [Google Scholar] [CrossRef]
- Kumar, S.K.; Mikhael, J.R.; Buadi, F.K.; Dingli, D.; Dispenzieri, A.; Fonseca, R.; Gertz, M.A.; Greipp, P.R.; Hayman, S.R.; Kyle, R.A.; et al. Management of newly diagnosed symptomatic multiple myeloma: Updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines. Mayo Clin. Proc. 2009, 84, 1095–1110. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, K.C.; Bergsagel, P.L.; Shaughnessy, J.; Palumbo, A.; Durie, B.; Fonseca, R.; Stewart, A.K.; Harousseau, J.L.; Dimopoulos, M.; et al. Consensus recommendations for risk stratification in multiple myeloma: Report of the International Myeloma Workshop Consensus Panel 2. Blood 2012, 117, 4696–4700. [Google Scholar]
- Rajkumar, S.V. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2012, 87, 78–88. [Google Scholar]
- Avet-Loiseau, H.; Malard, F.; Campion, L.; Magrangeas, F.; Sebban, C.; Lioure, B.; Decaux, O.; Lamy, T.; Legros, L.; Fuzibet, J.G.; et al. Translocation t(14;16) and multiple myeloma: Is it really an independent prognostic factor? Blood 2012, 117, 2009–2011. [Google Scholar]
- Bird, J.M.; Owen, R.G.; D’Sa, S.; Snowden, J.A.; Pratt, G.; Ashcroft, J.; Yong, K.; Cook, G.; Feyler, S.; Davies, F.; et al. Guidelines for the diagnosis and management of multiple myeloma 2011. Br. J. Haematol. 2011, 154, 32–75. [Google Scholar]
- Palumbo, A.; Bringhen, S.; Ludwig, H.; Dimopoulos, M.A.; Blade, J.; Mateos, M.V.; Rosinol, L.; Boccadoro, M.; Cavo, M.; Lokhorst, H.; et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: A report of the European Myeloma Network (EMN). Blood 2011, 118, 4519–4529. [Google Scholar]
- Roussel, M.; Facon, T.; Moreau, P.; Harousseau, J.L.; Attal, M. Firstline treatment and maintenance in newly diagnosed multiple myeloma patients. Recent Results Cancer Res. 2011, 183, 189–206. [Google Scholar]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic mechanisms in mammals. Cell. Mol. Life Sci. 2009, 66, 596–612. [Google Scholar]
- Galm, O.; Herman, J.G.; Baylin, S.B. The fundamental role of epigenetics in hematopoietic malignancies. Blood Rev. 2006, 20, 1–13. [Google Scholar]
- Antequera, F. Structure, function and evolution of CpG island promoters. Cell. Mol. Life Sci. 2003, 60, 1647–1658. [Google Scholar]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar]
- Gardner, K.E.; Allis, C.D.; Strahl, B.D. Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 2012, 409, 36–46. [Google Scholar]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar]
- Selvi, R.B.; Kundu, T.K. Reversible acetylation of chromatin: Implication in regulation of gene expression, disease and therapeutics. Biotechnol. J. 2009, 4, 375–390. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cells 2008, 31, 449–461. [Google Scholar]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cells 2009, 33, 1–13. [Google Scholar]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar]
- Banerjee, T.; Chakravarti, D. A peek into the complex realm of histone phosphorylation. Mol. Cell. Biol. 2011, 31, 4858–4873. [Google Scholar]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar]
- Margueron, R.; Trojer, P.; Reinberg, D. The key to development: Interpreting the histone code? Curr. Opin. Genet. Dev. 2005, 15, 163–176. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Klose, R. CpG island chromatin: A platform for gene regulation. Epigenetics 2011, 6, 147–152. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar]
- Smith, E.M.; Boyd, K.; Davies, F.E. The potential role of epigenetic therapy in multiple myeloma. Br. J. Haematol. 2009, 148, 702–713. [Google Scholar] [CrossRef]
- Bollati, V.; Fabris, S.; Pegoraro, V.; Ronchetti, D.; Mosca, L.; Deliliers, G.L.; Motta, V.; Bertazzi, P.A.; Baccarelli, A.; Neri, A. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis 2009, 30, 1330–1335. [Google Scholar] [CrossRef]
- Aoki, Y.; Nojima, M.; Suzuki, H.; Yasui, H.; Maruyama, R.; Yamamoto, E.; Ashida, M.; Itagaki, M.; Asaoku, H.; Ikeda, H.; et al. Genomic vulnerability to LINE-1 hypomethylation is a potential determinant of the clinicogenetic features of multiple myeloma. Genome Med. 2012, 4, 101. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.D.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef]
- Salhia, B.; Baker, A.; Ahmann, G.; Auclair, D.; Fonseca, R.; Carpten, J. DNA methylation analysis determines the high frequency of genic hypomethylation and low frequency of hypermethylation events in plasma cell tumors. Cancer Res. 2010, 70, 6934–6944. [Google Scholar] [CrossRef]
- Sharma, A.; Heuck, C.J.; Fazzari, M.J.; Mehta, J.; Singhal, S.; Greally, J.M.; Verma, A. DNA methylation alterations in multiple myeloma as a model for epigenetic changes in cancer. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 654–669. [Google Scholar] [CrossRef]
- Heller, G.; Schmidt, W.M.; Ziegler, B.; Holzer, S.; Mullauer, L.; Bilban, M.; Zielinski, C.C.; Drach, J.; Zochbauer-Muller, S. Genome-wide transcriptional response to 5-aza-2'-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008, 68, 44–54. [Google Scholar] [CrossRef]
- Braggio, E.; Maiolino, A.; Gouveia, M.E.; Magalhaes, R.; Souto Filho, J.T.; Garnica, M.; Nucci, M.; Renault, I.Z. Methylation status of nine tumor suppressor genes in multiple myeloma. Int. J. Hematol. 2010, 91, 87–96. [Google Scholar]
- Hatzimichael, E.; Dasoula, A.; Shah, R.; Syed, N.; Papoudou-Bai, A.; Coley, H.M.; Dranitsaris, G.; Bourantas, K.L.; Stebbing, J.; Crook, T. The prolyl-hydroxylase EGLN3 and not EGLN1 is inactivated by methylation in plasma cell neoplasia. Eur. J. Haematol. 2010, 84, 47–51. [Google Scholar]
- Song, Y.F.; Xu, R.; Zhang, X.H.; Chen, B.B.; Chen, Q.; Chen, Y.M.; Xie, Y. High-frequency promoter hypermethylation of the deleted in liver cancer-1 gene in multiple myeloma. J. Clin. Pathol. 2006, 59, 947–951. [Google Scholar]
- Seidl, S.; Ackermann, J.; Kaufmann, H.; Keck, A.; Nosslinger, T.; Zielinski, C.C.; Drach, J.; Zochbauer-Muller, S. DNA-methylation analysis identifies the E-cadherin gene as a potential marker of disease progression in patients with monoclonal gammopathies. Cancer 2004, 100, 2598–2606. [Google Scholar]
- De Carvalho, F.; Colleoni, G.W.; Almeida, M.S.; Carvalho, A.L.; Vettore, A.L. TGFbetaR2 aberrant methylation is a potential prognostic marker and therapeutic target in multiple myeloma. Int. J. Cancer 2009, 125, 1985–1991. [Google Scholar]
- De Bruyne, E.; Bos, T.J.; Asosingh, K.; vande Broek, I.; Menu, E.; van Valckenborgh, E.; Atadja, P.; Coiteux, V.; Leleu, X.; Thielemans, K.; et al. Epigenetic silencing of the tetraspanin CD9 during disease progression in multiple myeloma cells and correlation with survival. Clin. Cancer Res. 2008, 14, 2918–2926. [Google Scholar]
- Galm, O.; Wilop, S.; Reichelt, J.; Jost, E.; Gehbauer, G.; Herman, J.G.; Osieka, R. DNA methylation changes in multiple myeloma. Leukemia 2004, 18, 1687–1692. [Google Scholar]
- Stanganelli, C.; Arbelbide, J.; Fantl, D.B.; Corrado, C.; Slavutsky, I. DNA methylation analysis of tumor suppressor genes in monoclonal gammopathy of undetermined significance. Ann. Hematol. 2010, 89, 191–199. [Google Scholar]
- Nojima, M.; Maruyama, R.; Yasui, H.; Suzuki, H.; Maruyama, Y.; Tarasawa, I.; Sasaki, Y.; Asaoku, H.; Sakai, H.; Hayashi, T.; et al. Genomic screening for genes silenced by DNA methylation revealed an association between RASD1 inactivation and dexamethasone resistance in multiple myeloma. Clin. Cancer Res. 2009, 15, 4356–4364. [Google Scholar]
- Chen, G.; Wang, Y.; Huang, H.; Lin, F.; Wu, D.; Sun, A.; Chang, H.; Feng, Y. Combination of DNA methylation inhibitor 5-azacytidine and arsenic trioxide has synergistic activity in myeloma. Eur. J. Haematol. 2009, 82, 176–183. [Google Scholar]
- Khong, T.; Sharkey, J.; Spencer, A. The effect of azacitidine on interleukin-6 signaling and nuclear factor-kappaB activation and its in vitro and in vivo activity against multiple myeloma. Haematologica 2008, 93, 860–869. [Google Scholar]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Reversal of p53 epigenetic silencing in multiple myeloma permits apoptosis by a p53 activator. Cancer Biol. Ther. 2006, 5, 1154–1160. [Google Scholar]
- Reddy, J.; Shivapurkar, N.; Takahashi, T.; Parikh, G.; Stastny, V.; Echebiri, C.; Crumrine, K.; Zochbauer-Muller, S.; Drach, J.; Zheng, Y.; et al. Differential methylation of genes that regulate cytokine signaling in lymphoid and hematopoietic tumors. Oncogene 2005, 24, 732–736. [Google Scholar]
- Wilop, S.; van Gemmeren, T.B.; Lentjes, M.H.; van Engeland, M.; Herman, J.G.; Brummendorf, T.H.; Jost, E.; Galm, O. Methylation-associated dysregulation of the suppressor of cytokine signaling-3 gene in multiple myeloma. Epigenetics 2012, 6, 1047–1052. [Google Scholar]
- Chim, C.S.; Pang, R.; Fung, T.K.; Choi, C.L.; Liang, R. Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia 2007, 21, 2527–2536. [Google Scholar]
- Jost, E.; Gezer, D.; Wilop, S.; Suzuki, H.; Herman, J.G.; Osieka, R.; Galm, O. Epigenetic dysregulation of secreted Frizzled-related proteins in multiple myeloma. Cancer Lett. 2009, 281, 24–31. [Google Scholar]
- Kocemba, K.A.; Groen, R.W.; van Andel, H.; Kersten, M.J.; Mahtouk, K.; Spaargaren, M.; Pals, S.T. Transcriptional silencing of the Wnt-antagonist DKK1 by promoter methylation is associated with enhanced Wnt signaling in advanced multiple myeloma. PLoS One 2012, 7, e30359. [Google Scholar]
- Tshuikina, M.; Jernberg-Wiklund, H.; Nilsson, K.; Oberg, F. Epigenetic silencing of the interferon regulatory factor ICSBP/IRF8 in human multiple myeloma. Exp. Hematol. 2008, 36, 1673–1681. [Google Scholar]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar]
- Marango, J.; Shimoyama, M.; Nishio, H.; Meyer, J.A.; Min, D.J.; Sirulnik, A.; Martinez-Martinez, Y.; Chesi, M.; Bergsagel, P.L.; Zhou, M.M.; et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 2008, 111, 3145–3154. [Google Scholar]
- Martinez-Garcia, E.; Popovic, R.; Min, D.J.; Sweet, S.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef]
- Brito, J.L.; Walker, B.; Jenner, M.; Dickens, N.J.; Brown, N.J.; Ross, F.M.; Avramidou, A.; Irving, J.A.; Gonzalez, D.; Davies, F.E.; et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 2009, 94, 78–86. [Google Scholar] [CrossRef]
- Asangani, I.A.; Ateeq, B.; Cao, Q.; Dodson, L.; Pandhi, M.; Kunju, L.P.; Mehra, R.; Lonigro, R.J.; Siddiqui, J.; Palanisamy, N.; et al. Characterization of the EZH2-MMSET Histone Methyltransferase Regulatory Axis in Cancer. Mol. Cells 2013, 49, 80–93. [Google Scholar]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef]
- Kalushkova, A.; Fryknas, M.; Lemaire, M.; Fristedt, C.; Agarwal, P.; Eriksson, M.; Deleu, S.; Atadja, P.; Osterborg, A.; Nilsson, K.; et al. Polycomb target genes are silenced in multiple myeloma. PLoS One 2010, 5, e11483. [Google Scholar] [CrossRef]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.M.; Lengauer, C.; et al. The Polycomb group protein Bmi-1 is essential for the growth of multiple myeloma cells. Cancer Res. 2010, 70, 5528–5538. [Google Scholar] [CrossRef]
- Isham, C.R.; Tibodeau, J.D.; Jin, W.; Xu, R.; Timm, M.M.; Bible, K.C. Chaetocin: A promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood 2007, 109, 2579–2588. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef]
- Federico, M.; Bagella, L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J. Biomed. Biotechnol. 2011, 2011, 475641. [Google Scholar]
- Khan, O.; La Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Lemaire, M.; Fristedt, C.; Agarwal, P.; Menu, E.; van Valckenborgh, E.; de Bruyne, E.; Osterborg, A.; Atadja, P.; Larsson, O.; Axelson, M.; et al. The HDAC Inhibitor LBH589 Enhances the Antimyeloma Effects of the IGF-1RTK Inhibitor Picropodophyllin. Clin. Cancer Res. 2012, 18, 2230–2239. [Google Scholar] [CrossRef]
- Maiso, P.; Carvajal-Vergara, X.; Ocio, E.M.; Lopez-Perez, R.; Mateo, G.; Gutierrez, N.; Atadja, P.; Pandiella, A.; San Miguel, J.F. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006, 66, 5781–5789. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Hideshima, T.; Akiyama, M.; Chauhan, D.; Munshi, N.; Gu, X.; et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: Biological and clinical implications. Proc. Natl. Acad. Sci. USA 2004, 101, 540–545. [Google Scholar] [CrossRef]
- Neri, P.; Tagliaferri, P.; di Martino, M.T.; Calimeri, T.; Amodio, N.; Bulotta, A.; Ventura, M.; Eramo, P.O.; Viscomi, C.; Arbitrio, M.; et al. In vivo anti-myeloma activity and modulation of gene expression profile induced by valproic acid, a histone deacetylase inhibitor. Br. J. Haematol. 2008, 143, 520–531. [Google Scholar]
- Todoerti, K.; Barbui, V.; Pedrini, O.; Lionetti, M.; Fossati, G.; Mascagni, P.; Rambaldi, A.; Neri, A.; Introna, M.; Lombardi, L.; et al. Pleiotropic anti-myeloma activity of ITF2357: Inhibition of interleukin-6 receptor signaling and repression of miR-19a and miR-19b. Haematologica 2010, 95, 260–269. [Google Scholar] [CrossRef]
- Deleu, S.; Menu, E.; Valckenborgh, E.V.; Camp, B.V.; Fraczek, J.; Broek, I.V.; Rogiers, V.; Vanderkerken, K. Histone deacetylase inhibitors in multiple myeloma. Hematol. Rev. 2009, 1, 46–56. [Google Scholar]
- Baumann, P.; Junghanns, C.; Mandl-Weber, S.; Strobl, S.; Oduncu, F.; Schmidmaier, R. The pan-histone deacetylase inhibitor CR2408 disrupts cell cycle progression, diminishes proliferation and causes apoptosis in multiple myeloma cells. Br. J. Haematol. 2012, 156, 633–642. [Google Scholar] [CrossRef]
- Feng, R.; Ma, H.; Hassig, C.A.; Payne, J.E.; Smith, N.D.; Mapara, M.Y.; Hager, J.H.; Lentzsch, S. KD5170, a novel mercaptoketone-based histone deacetylase inhibitor, exerts antimyeloma effects by DNA damage and mitochondrial signaling. Mol. Cancer Ther. 2008, 7, 1494–1505. [Google Scholar] [CrossRef]
- Mandl-Weber, S.; Meinel, F.G.; Jankowsky, R.; Oduncu, F.; Schmidmaier, R.; Baumann, P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br. J. Haematol. 2010, 149, 518–528. [Google Scholar] [CrossRef]
- Stuhmer, T.; Arts, J.; Chatterjee, M.; Borawski, J.; Wolff, A.; King, P.; Einsele, H.; Leo, E.; Bargou, R.C. Preclinical anti-myeloma activity of the novel HDAC-inhibitor JNJ-26481585. Br. J. Haematol. 2010, 149, 529–536. [Google Scholar] [CrossRef]
- Zhang, S.; Suvannasankha, A.; Crean, C.D.; White, V.L.; Chen, C.S.; Farag, S.S. The novel histone deacetylase inhibitor, AR-42, inhibits gp130/STAT3 pathway and induces apoptosis and cell cycle arrest in multiple myeloma cells. Int. J. Cancer 2010, 129, 204–213. [Google Scholar]
- De Bruyne, E.; Bos, T.J.; Schuit, F.; van Valckenborgh, E.; Menu, E.; Thorrez, L.; Atadja, P.; Jernberg-Wiklund, H.; Vanderkerken, K. IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood 2010, 115, 2430–2440. [Google Scholar] [CrossRef]
- Chen, S.; Dai, Y.; Pei, X.Y.; Grant, S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: Evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol. Cell. Biol. 2009, 29, 6149–6169. [Google Scholar] [CrossRef]
- Fandy, T.E.; Shankar, S.; Ross, D.D.; Sausville, E.; Srivastava, R.K. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia 2005, 7, 646–657. [Google Scholar] [CrossRef]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Tai, Y.T.; Atadja, P.; Remiszewski, S.; Hideshima, T.; Mitsiades, N.; Shringarpure, R.; LeBlanc, R.; Chauhan, D.; et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood 2003, 102, 2615–2622. [Google Scholar] [CrossRef]
- Cheriyath, V.; Kuhns, M.A.; Kalaycio, M.E.; Borden, E.C. Potentiation of apoptosis by histone deacetylase inhibitors and doxorubicin combination: Cytoplasmic cathepsin B as a mediator of apoptosis in multiple myeloma. Br. J. Cancer 2011, 104, 957–967. [Google Scholar] [CrossRef]
- Lee, C.K.; Wang, S.; Huang, X.; Ryder, J.; Liu, B. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010, 296, 233–240. [Google Scholar] [CrossRef]
- Sanchez, E.; Shen, J.; Steinberg, J.; Li, M.; Wang, C.; Bonavida, B.; Chen, H.; Li, Z.W.; Berenson, J.R. The histone deacetylase inhibitor LBH589 enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Leuk. Res. 2010, 35, 373–379. [Google Scholar]
- Lavelle, D.; Chen, Y.H.; Hankewych, M.; DeSimone, J. Histone deacetylase inhibitors increase p21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am. J. Hematol. 2001, 68, 170–178. [Google Scholar] [CrossRef]
- Deleu, S.; Lemaire, M.; Arts, J.; Menu, E.; van Valckenborgh, E.; King, P.; vande Broek, I.; de Raeve, H.; van Camp, B.; Croucher, P.; et al. The effects of JNJ-26481585, a novel hydroxamate-based histone deacetylase inhibitor, on the development of multiple myeloma in the 5T2MM and 5T33MM murine models. Leukemia 2009, 23, 1894–1903. [Google Scholar] [CrossRef]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef]
- Bai, L.Y.; Omar, H.A.; Chiu, C.F.; Chi, Z.P.; Hu, J.L.; Weng, J.R. Antitumor effects of (S)-HDAC42, a phenylbutyrate-derived histone deacetylase inhibitor, in multiple myeloma cells. Cancer Chemother. Pharmacol. 2010, 35, 373–379. [Google Scholar]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef]
- Deleu, S.; Lemaire, M.; Arts, J.; Menu, E.; van Valckenborgh, E.; vande Broek, I.; de Raeve, H.; Coulton, L.; van Camp, B.; Croucher, P.; et al. Bortezomib alone or in combination with the histone deacetylase inhibitor JNJ-26481585: Effect on myeloma bone disease in the 5T2MM murine model of myeloma. Cancer Res. 2009, 69, 5307–5311. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2012, 10, 2034–2042. [Google Scholar]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Qu, X.; Du, J.; Zhang, C.; Fu, W.; Xi, H.; Zou, J.; Hou, J. Arsenic trioxide exerts antimyeloma effects by inhibiting activity in the cytoplasmic substrates of histone deacetylase 6. PLoS One 2012, 7, e32215. [Google Scholar]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar]
- Feng, R.; Oton, A.; Mapara, M.Y.; Anderson, G.; Belani, C.; Lentzsch, S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br. J. Haematol. 2007, 139, 385–397. [Google Scholar] [CrossRef]
- Chen, X.; Wong, P.; Radany, E.H.; Stark, J.M.; Laulier, C.; Wong, J.Y. Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of RAD51 protein and inhibition of homology-directed repair in multiple myeloma. Mol. Cancer Res. 2012, 10, 1052–1064. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Kremer, M.; Specht, K.; Calzada-Wack, J.; Nathrath, M.; Schaich, R.; Hofler, H.; Fend, F. Analysis of signal transducer and activator of transcription 3 (Stat 3) pathway in multiple myeloma: Stat 3 activation and cyclin D1 dysregulation are mutually exclusive events. Am. J. Pathol. 2003, 162, 1449–1461. [Google Scholar] [CrossRef]
- Kitazoe, K.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Harada, T.; Nakano, A.; Takeuchi, K.; Hashimoto, T.; Ozaki, S.; et al. Valproic acid exerts anti-tumor as well as anti-angiogenic effects on myeloma. Int. J. Hematol. 2009, 89, 45–57. [Google Scholar] [CrossRef]
- Kaiser, M.; Zavrski, I.; Sterz, J.; Jakob, C.; Fleissner, C.; Kloetzel, P.M.; Sezer, O.; Heider, U. The effects of the histone deacetylase inhibitor valproic acid on cell cycle, growth suppression and apoptosis in multiple myeloma. Haematologica 2006, 91, 248–251. [Google Scholar]
- Wu, X.; Tao, Y.; Hou, J.; Meng, X.; Shi, J. Valproic Acid Upregulates NKG2D Ligand Expression through an ERK-dependent Mechanism and Potentially Enhances NK Cell-mediated Lysis of Myeloma. Neoplasia 2012, 14, 1178–1189. [Google Scholar]
- Ewald, B.; Sampath, D.; Plunkett, W. Nucleoside analogs: Molecular mechanisms signaling cell death. Oncogene 2008, 27, 6522–6537. [Google Scholar] [CrossRef]
- Lavelle, D.; DeSimone, J.; Hankewych, M.; Kousnetzova, T.; Chen, Y.H. Decitabine induces cell cycle arrest at the G1 phase via p21(WAF1) and the G2/M phase via the p38 MAP kinase pathway. Leuk. Res. 2003, 27, 999–1007. [Google Scholar] [CrossRef]
- Khong, T.; Spencer, A. Targeting HSP 90 induces apoptosis and inhibits critical survival and proliferation pathways in multiple myeloma. Mol. Cancer Ther. 2012, 10, 1909–1917. [Google Scholar]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2009, 23, 10–24. [Google Scholar] [CrossRef]
- Moreaux, J.; Reme, T.; Leonard, W.; Veyrune, J.L.; Requirand, G.; Goldschmidt, H.; Hose, D.; Klein, B. Development of gene expression-based score to predict sensitivity of multiple myeloma cells to DNA methylation inhibitors. Mol. Cancer Ther. 2012, 11, 2685–2692. [Google Scholar] [CrossRef]
- Klco, J.M.; Spencer, D.H.; Lamprecht, T.L.; Sarkaria, S.M.; Wylie, T.; Magrini, V.; Hundal, J.; Walker, J.; Varghese, N.; Erdmann-Gilmore, P.; et al. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood 2013, 121, 1633–1643. [Google Scholar] [CrossRef]
- Suarez, L.; Gore, S.D. Demethylation demystification. Blood 2013, 121, 1488–1489. [Google Scholar] [CrossRef]
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol. Cancer Ther. 2007, 6, 1718–1727. [Google Scholar] [CrossRef]
- Du, H.L.; Ren, L.M.; Chen, H.; Zhu, Y.; Qi, Y. Re-expression of p16 gene in the myeloma cell line U266 induced by synergy of sodium butyrate and 5-Aza-2'-deoxycytidine. Di Yi Jun Yi Da Xue Xue Bao 2002, 22, 981–984. [Google Scholar]
- Lu, Q.; Lin, X.; Feng, J.; Zhao, X.; Gallagher, R.; Lee, M.Y.; Chiao, J.W.; Liu, D. Phenylhexyl isothiocyanate has dual function as histone deacetylase inhibitor and hypomethylating agent and can inhibit myeloma cell growth by targeting critical pathways. J. Hematol. Oncol. 2008, 1, 6. [Google Scholar] [CrossRef]
- Goodyear, O.; Agathanggelou, A.; Novitzky-Basso, I.; Siddique, S.; McSkeane, T.; Ryan, G.; Vyas, P.; Cavenagh, J.; Stankovic, T.; Moss, P.; et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood 2010, 116, 1908–1918. [Google Scholar] [CrossRef]
- Moreno-Bost, A.; Szmania, S.; Stone, K.; Garg, T.; Hoerring, A.; Szymonifka, J.; Shaughnessy, J., Jr.; Barlogie, B.; Prentice, H.G.; van Rhee, F. Epigenetic modulation of MAGE-A3 antigen expression in multiple myeloma following treatment with the demethylation agent 5-azacitidine and the histone deacetlyase inhibitor MGCD0103. Cytotherapy 2011, 13, 618–628. [Google Scholar] [CrossRef]
- Deangelo, D.J.; Spencer, A.; Bhalla, K.N.; Prince, H.M.; Fischer, T.; Kindler, T.; Giles, F.J.; Scott, J.W.; Parker, K.; Liu, A.; et al. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia 2013. [Google Scholar] [CrossRef]
- Niesvizky, R.; Ely, S.; Mark, T.; Aggarwal, S.; Gabrilove, J.L.; Wright, J.J.; Chen-Kiang, S.; Sparano, J.A. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer 2011, 117, 336–342. [Google Scholar] [CrossRef]
- Galli, M.; Salmoiraghi, S.; Golay, J.; Gozzini, A.; Crippa, C.; Pescosta, N.; Rambaldi, A. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann. Hematol. 2010, 89, 185–190. [Google Scholar] [CrossRef]
- Richardson, P.; Mitsiades, C.; Colson, K.; Reilly, E.; McBride, L.; Chiao, J.; Sun, L.; Ricker, J.; Rizvi, S.; Oerth, C.; et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk. Lymphoma 2008, 49, 502–507. [Google Scholar] [CrossRef]
- Gimsing, P.; Hansen, M.; Knudsen, L.M.; Knoblauch, P.; Christensen, I.J.; Ooi, C.E.; Buhl-Jensen, P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur. J. Haematol. 2008, 81, 170–176. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: http://www.clinicaltrials.gov/ (accessed on 14 February 2013).
- Moreau, P. The future of therapy for relapsed/refractory multiple myeloma: Emerging agents and novel treatment strategies. Semin. Hematol. 2012, 1, S33–S46. [Google Scholar] [CrossRef]
- Prince, H.M. Pioneering studies of histone deacetylase inhibitors in myeloma: Signals of activity set the stage for combination therapy trials. Leuk. Lymphoma 2012, 53, 1658–1659. [Google Scholar] [CrossRef]
- Weber, D.M.; Graef, T.; Hussein, M.; Sobecks, R.M.; Schiller, G.J.; Lupinacci, L.; Hardwick, J.S.; Jagannath, S. Phase I trial of vorinostat combined with bortezomib for the treatment of relapsing and/or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2012, 12, 319–324. [Google Scholar] [CrossRef]
- Mazumder, A.; Vesole, D.H.; Jagannath, S. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: A case series illustrating utility in clinical practice. Clin. Lymphoma Myeloma Leuk. 2010, 10, 149–151. [Google Scholar] [CrossRef]
- Badros, A.; Burger, A.M.; Philip, S.; Niesvizky, R.; Kolla, S.S.; Goloubeva, O.; Harris, C.; Zwiebel, J.; Wright, J.J.; Espinoza-Delgado, I.; et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer Res. 2009, 15, 5250–5257. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Shah, J.J.; Laubach, J.P.; Mitchell, A.R.; Sharp, C.; Lewis, C.; Harvey, R.D.; Gleason, C.; Casbourne, D.; Nooka, A.K.; et al. Lenalidomide, Bortezomib, and Dexamethasone (RVD) in Combination with Vorinostat As Front-Line Therapy for Patients with Multiple Myeloma (MM): Results of a Phase 1 Study. ASH Annu. Meet. Abstr. 2012, 120, 336. [Google Scholar]
- Shah, J.J.; Orlowski, R.Z.; Thomas, S.K.; Alexanian, R.; Wang, M.; Qazilbash, M.H.; Popat, U.R.; Parmar, S.; Shah, N.; Bashir, Q.; et al. Final Results of a Phase I/II Trial of the Combination of Concurrent Lenalidomide, Thalidomide and Dexamethasone in Patients with Relapsed and/or Refractory Myeloma. ASH Annu. Meet. Abstr. 2012, 120, 75. [Google Scholar]
- Richardson, P.G.; Alsina, M.; Weber, D.; Coutre, S.E.; Lonial, S.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.; Khan, M.; Paley, C.; et al. PANORAMA 2: Panobinostat Combined with Bortezomib and Dexamethasone in Patients with Relapsed and Bortezomib-Refractory Multiple Myeloma. ASH Annu. Meet. Abstr. 2012, 120, 1852. [Google Scholar]
- Berdeja, J.G.; Hart, L.; Lamar, R.; Murphy, P.; Morgan, S.; Flinn, I.W. Phase I/II Study of Panobinostat and Carfilzomib in Patients (pts) with Relapsed or Refractory Multiple Myeloma (MM), Interim Phase I Safety Analysis. ASH Annu. Meet. Abstr. 2012, 120, 4048. [Google Scholar]
- Shah, J.J.; Thomas, S.K.; Weber, D.M.; Wang, M.; Alexanian, R.; Qazilbash, M.H.; Bashir, Q.; Parmar, S.; Shah, N.; Popat, U.R.; et al. Phase 1/1b Study of the Efficacy and Safety of the Combination of Panobinostat + Carfilzomib in Patients with Relapsed and/or Refractory Multiple Myeloma. ASH Annu. Meet. Abstr. 2012, 120, 4081. [Google Scholar]
- Harrison, S.J.; Quach, H.; Link, E.; Seymour, J.F.; Ritchie, D.S.; Ruell, S.; Dean, J.; Januszewicz, H.; Johnstone, R.; Neeson, P.; et al. A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood 2011, 118, 6274–6283. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Jagannath, S.; Yoon, S.-S.; Siegel, D.S.; Lonial, S.; Hajek, R.; Facon, T.; Rosinol, L.; Blacklock, H.A.; Goldschmidt, H.; et al. Vantage 088: Vorinostat in Combination with Bortezomib in Patients with Relapsed/Refractory Multiple Myeloma: Results of a Global, Randomized Phase 3 Trial. ASH Annu. Meet. Abstr. 2011, 118, 811. [Google Scholar]
- Siegel, D.S.; Dimopoulos, M.A.; Yoon, S.-S.; Laubach, J.P.; Kaufman, J.L.; Goldschmidt, H.; Reece, D.E.; Leleu, X.; Durrant, S.; Offner, F.C.; et al. Vantage 095: Vorinostat in Combination with Bortezomib in Salvage Multiple Myeloma Patients: Final Study Results of a Global Phase 2b Trial. ASH Annu. Meet. Abstr. 2011, 118, 480. [Google Scholar]
- San-Miguel, J.F.; de Moraes Hungria, V.T.; Yoon, S.-S.; Wiktor-Jedrzejczak, W.; Elghandour, A.; Siritanaratkul, N.; Dimopoulos, M.A.; Corradini, P.; Nakorn, T.N.; Shelekhova, T.; et al. Update on a Phase III Study of Panobinostat with Bortezomib and Dexamethasone in Patients with Relapsed Multiple Myeloma: PANORAMA 1. ASH Annu. Meet. Abstr. 2011, 118, 3976. [Google Scholar]
- Morgan, G. Future drug developments in multiple myeloma: An overview of novel lenalidomide-based combination therapies. Blood Rev. 2010, 24, S27–S32. [Google Scholar]
- Hofmeister, C.C.; Bowers, M.A.; Efebera, Y.A.; Humphries, K.; Benson, D.M., Jr.; Greenfield, C.N.; Sell, M.; Devine, S.M. Phase I Trial of Lenalidomide + Vorinostat After Autologous Transplant in Multiple Myeloma. ASH Annu. Meet. Abstr. 2012, 120, 3114. [Google Scholar]
- Toor, A.A.; Payne, K.K.; Chung, H.M.; Sabo, R.T.; Hazlett, A.F.; Kmieciak, M.; Sanford, K.; Williams, D.C.; Clark, W.B.; Roberts, C.H.; et al. Epigenetic induction of adaptive immune response in multiple myeloma: Sequential azacitidine and lenalidomide generate cancer testis antigen-specific cellular immunity. Br. J. Haematol. 2012, 158, 700–711. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maes, K.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Vanderkerken, K.; De Bruyne, E. Epigenetic Modulating Agents as a New Therapeutic Approach in Multiple Myeloma. Cancers 2013, 5, 430-461. https://doi.org/10.3390/cancers5020430
Maes K, Menu E, Van Valckenborgh E, Van Riet I, Vanderkerken K, De Bruyne E. Epigenetic Modulating Agents as a New Therapeutic Approach in Multiple Myeloma. Cancers. 2013; 5(2):430-461. https://doi.org/10.3390/cancers5020430
Chicago/Turabian StyleMaes, Ken, Eline Menu, Els Van Valckenborgh, Ivan Van Riet, Karin Vanderkerken, and Elke De Bruyne. 2013. "Epigenetic Modulating Agents as a New Therapeutic Approach in Multiple Myeloma" Cancers 5, no. 2: 430-461. https://doi.org/10.3390/cancers5020430
APA StyleMaes, K., Menu, E., Van Valckenborgh, E., Van Riet, I., Vanderkerken, K., & De Bruyne, E. (2013). Epigenetic Modulating Agents as a New Therapeutic Approach in Multiple Myeloma. Cancers, 5(2), 430-461. https://doi.org/10.3390/cancers5020430