Epidermal to Mesenchymal Transition and Failure of EGFR-Targeted Therapy in Glioblastoma

 ,
, {kind=link}
Abstract
:1. Introduction
2. E-Cadherin and EMT
3. “Go or Grow”
4. Matrix Metalloproteases (MMPs) and TGF-β as EMT Inducers
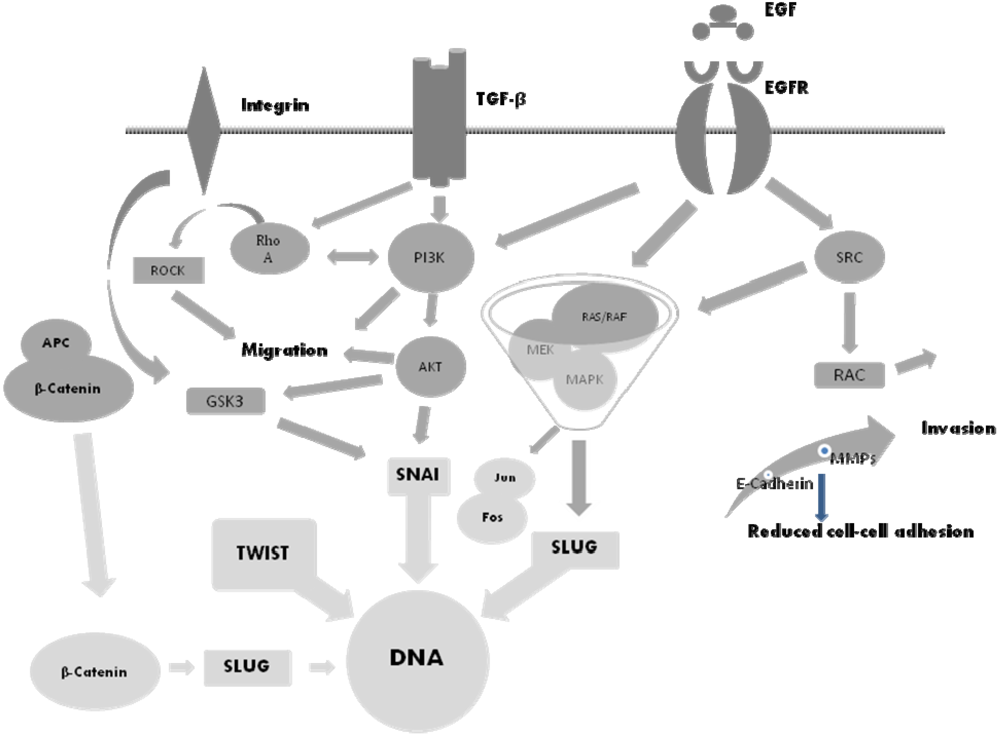
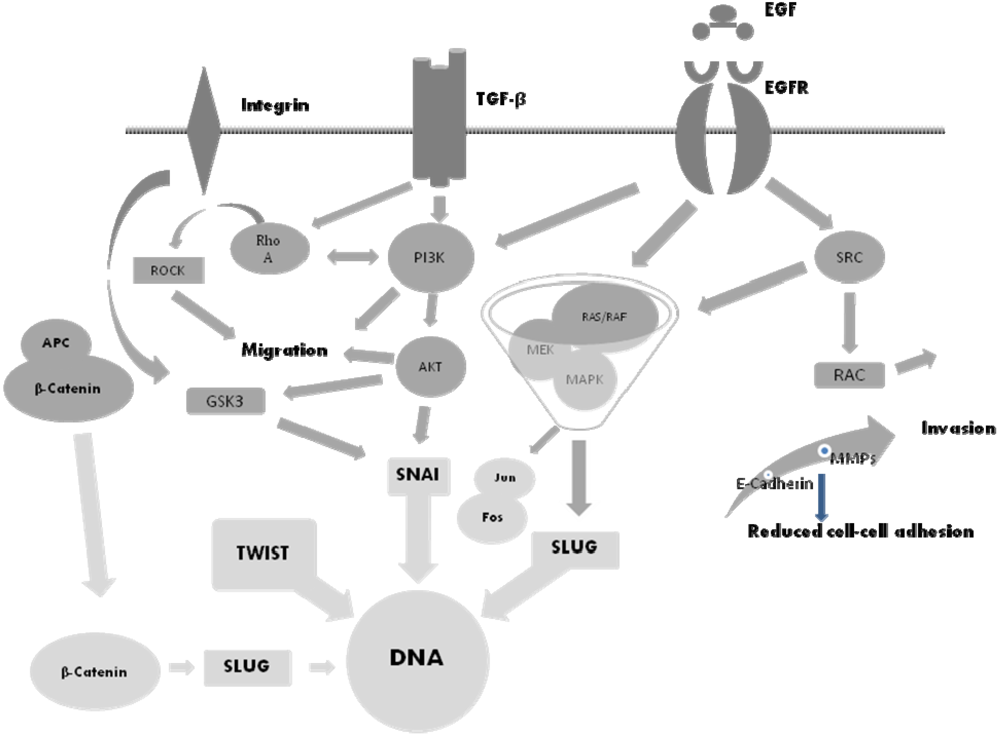
5. The Molecular Basis of EMT and Potential Therapeutic Targets
6. The SNAI Family
7. Conclusions
Conflict of Interest
References and Notes
- Suda, K.; Tomizawa, K.; Fujii, M; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudami, T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. 2011, 6, 1152–1161. [Google Scholar] [CrossRef]
- Chung, J.H.; Rho, J.K.; Xu, X.; Lee, J.S.; Yoon, H.I.; Lee, C.T.; Choi, Y.J.; Kim, H.R.; Kim, C.H.; Lee, J.C. Clinical and molecular evidence of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011, 2, 176–182. [Google Scholar]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metastasis 2008, 8, 843–854. [Google Scholar]
- Rho, J.K.; Chou, I.J.; Lee, J.K.; Ryoo, B.Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer 2008, 2, 219–226. [Google Scholar]
- Sathornsumetee, S.; Reardon, D.A.; Desjardins, A.; Jennifer, Q.A.; James, V.J.; Jeremy, R.N. Molecular targeted therapy for malignant glioma. Cancer 2007, 110, 13–24. [Google Scholar]
- Löw, S.; Schmidt, U.; Unterberg, A.; Halatsch, M.-E. The epidermal growth factor receptor as a therapeutic target in glioblastoma multiforme and other malignant neoplasms. Anticancer Agents Med. Chem. 2009, 9, 703–715. [Google Scholar]
- Haddad, Y.; Woonyoung, C.; McConkey, J.D. Delta-crystallin enhancer binding factor 1 controls the epithelial to mesenchymal transition phenotype and resistance to the epidermal growth factor receptor inhibitor erlotinib in human head and neck squamous cell carcinoma lines. Clin. Cancer Res. 2009, 2, 532–542. [Google Scholar]
- Krakstad, C.; Chekenya, M. Survival signaling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135–149. [Google Scholar]
- Deng, Q.-F.; Zhou, C.-C.; Su, C.-X. Clinicopathological features and epidermal growth factor receptor mutations associated with epithelial-mesenchymal transition in non-small cell lung cancer. Respirology 2009, 14, 371–376. [Google Scholar]
- Fuchs, B.C.; Fujii, T.; Dorfman, J.D.; Goodwin, J.M.; Zhu, A.X.; Lanuti, M.; Tanabe, K.K. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008, 7, 2391–2399. [Google Scholar]
- Wever, O.D.; Pauwels, P.; Craene, B.D.; Michèle, S.; Emami, S.; Redeuilh, G.; Gespach, C.; Berx, G. Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell Biol. 2008, 130, 481–494. [Google Scholar]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3 kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. Twist 1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194–212. [Google Scholar]
- Motta, F.J.; Valera, E.T.; Lucio-Eterovic, A.K.; Queiroz, R.G.; Neder, L.; Scrideli, C.A.; Machado, H.R.; Carlotti-Junior, C.G.; Marie, S.K. Differential expression of E-cadherin gene in human neuroepithelial tumors. Genet. Mol. Res. 2008, 7, 295–304. [Google Scholar]
- Tektonidis, M.; Haralambos, H.; Chauviere, A.; Simon, M.; Schaller, K.; Deutsch, A. Identification of intrinsic in vitro cellular mechanisms for glioma invasion. J. Theor. Biol. 2011, 287, 131–147. [Google Scholar] [CrossRef]
- Qiao, B.; Johnson, N.W.; Gao, J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-β1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Oncology 2010, 37, 663–668. [Google Scholar]
- Han, S.P.; Kim, J.H.; Han, M.E.; Sim, H.E.; Kim, K.S.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Oh, S.O. Snai 1 is involved in the proliferation and migration of glioblastoma cells. Cell. Mol. Neurobiol. 2011, 31, 489–496. [Google Scholar]
- Xun, J.; Hee-Young, J.; Kveung, M.J.; Kim, J.K.; Jin, J.; Kim, S.H.; Kang, B.G.; Beck, S.; Lee, S.J.; Kim, J.K.; et al. Frizzled 4 regulates stemness and invasiveness of migrating glioma cells established by serial intracranial transplantation. Cancer Res. 2011, 71, 3066–3075. [Google Scholar]
- Katoh, M.; Katoh, M. Comparative genomics on SNAI 1, SNAI 2, and SNAI 3 orthologs. Oncol. Rep. 2005, 14, 1083–1086. [Google Scholar]
- Xia, M.; Hu, M.; Wang, J.; Xu, Y.; Chen, X.; Ma, Y.; Su, L. Identification of the role of Smad interacting protein 1 (SIP 1) in glioma. J. Neurooncol. 2010, 97, 225–232. [Google Scholar]
- Yang, H.W.; Menon, L.G.; Black, P.M.; Carroll, R.S.; Johnson, M.D. SNAI2/SLUG promotes growth and invasion in human gliomas. BMC Cancer 2010, 10, 301. [Google Scholar]
- Lee, M.Y.; Chou, C.Y.; Tang, M.J.; Shen, M.R. Epithelial-mesenchymal transition in cervical cancer: Correlation with tumor progression, epidermal growth factor receptor overexpression, and Snail up-regulation. Clin. Cancer Res. 2008, 15, 4743–4750. [Google Scholar]
- Koppikar, P.; Lui, V.W.Y.; Man, D.; Xi, S.; Chai, R.L.; Nelson, E.; Tobey, A.B.J.; Grandis, J.R. Constitutive activation of signal tranducer and activator of transcription 5 contributes to tumor growth, epithelial-mesenchymal transition and resistance to epidermal growth factor receptor targeting. Clin. Cancer Res. 2008, 14, 7682–7690. [Google Scholar]
- Chi, A.S. Rapid radiographic and clinical improvement after treatment of MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 2012, 30, 30–33. [Google Scholar]
- Moon, Y.-W.; Weil, R.J.; Pack, S.D.; Park, W.-S.; Park, E.; Pham, T.; Karkera, J.D.; Kim, H.-K.; Vortmeyer, A.O.; Fuller, B.G.; et al. Missense mutation of the MET gene detected in human glioma. Mod. Pathol. 2000, 13, 973–977. [Google Scholar] [CrossRef] [Green Version]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pala, A.; Karpel-Massler, G.; Kast, R.E.; Wirtz, C.R.; Halatsch, M.-E. Epidermal to Mesenchymal Transition and Failure of EGFR-Targeted Therapy in Glioblastoma. Cancers 2012, 4, 523-530. https://doi.org/10.3390/cancers4020523
Pala A, Karpel-Massler G, Kast RE, Wirtz CR, Halatsch M-E. Epidermal to Mesenchymal Transition and Failure of EGFR-Targeted Therapy in Glioblastoma. Cancers. 2012; 4(2):523-530. https://doi.org/10.3390/cancers4020523
Chicago/Turabian StylePala, Andrej, Georg Karpel-Massler, Richard Eric Kast, Christian Rainer Wirtz, and Marc-Eric Halatsch. 2012. "Epidermal to Mesenchymal Transition and Failure of EGFR-Targeted Therapy in Glioblastoma" Cancers 4, no. 2: 523-530. https://doi.org/10.3390/cancers4020523
APA StylePala, A., Karpel-Massler, G., Kast, R. E., Wirtz, C. R., & Halatsch, M.-E. (2012). Epidermal to Mesenchymal Transition and Failure of EGFR-Targeted Therapy in Glioblastoma. Cancers, 4(2), 523-530. https://doi.org/10.3390/cancers4020523