Remodeling of Tumor Stroma and Response to Therapy

{kind=link}
Abstract
:1. Tumor Stroma: The Players
2. Tumor Blood Vessels: Kill or Not To Kill
3. The Concept of Vascular Normalization
4. What Is Tumor Vessel Normalization?
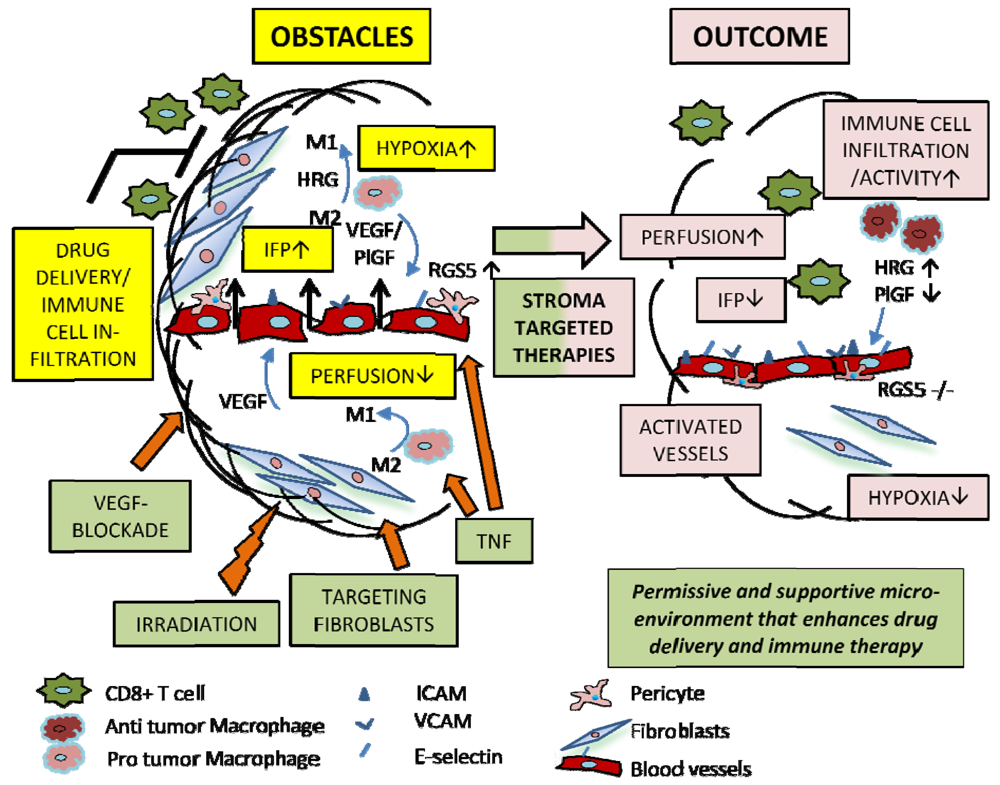
5. The Role of Stromal Cells in Regulating Vascular Normalization and Tumor Progression
5.1. Pericytes
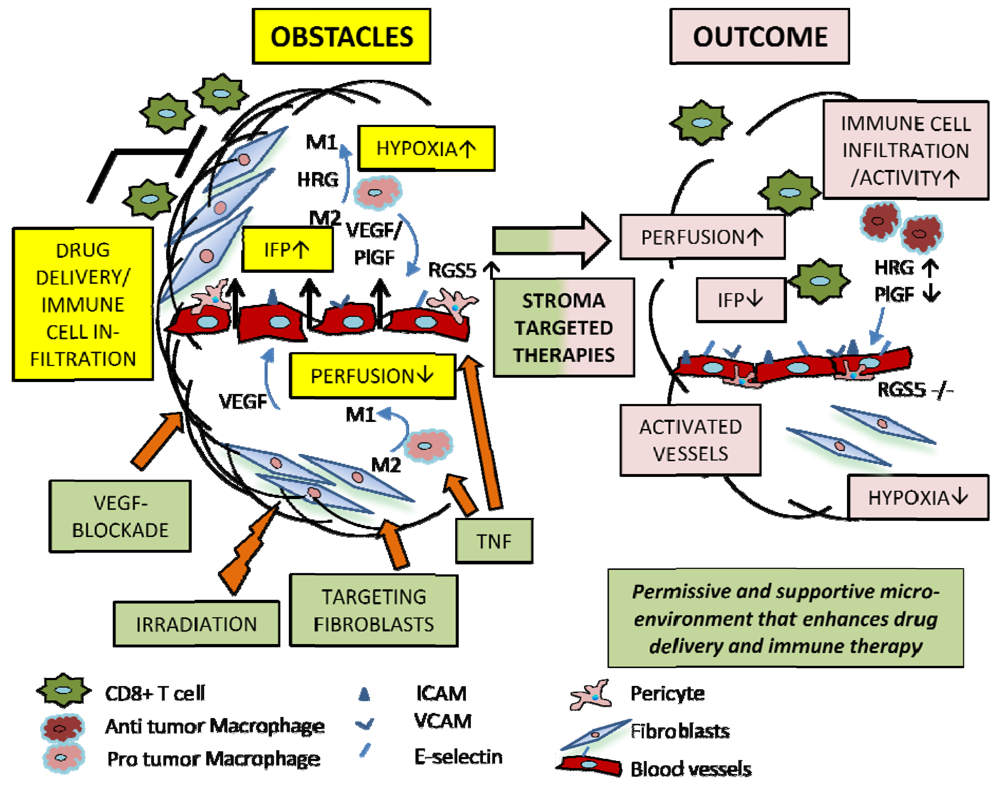
5.2. Macrophages
5.3. Fibroblasts
6. Conclusions
Acknowledgements
References
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar]
- Ganss, R. Tumor stroma fosters neovascularization by recruitment of progenitor cells into the tumor bed. J. Cell. Mol. Med. 2006, 10, 857–865. [Google Scholar]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar]
- Mantovani, A.; Bottazzi, B.; Colotta, F.; Sozzani, S.; Ruco, L. The origin and function of tumor-associated macrophages. Immunol. Today 1992, 13, 265–270. [Google Scholar]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar]
- Pages, F.; Galon, J.; Dieu-Nosjean, M.C.; Tartour, E.; Sautes-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikstrom, P.; et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am. J. Pathol. 2010, 177, 1031–1041. [Google Scholar] [CrossRef]
- Shiao, S.L.; Ganesan, A.P.; Rugo, H.S.; Coussens, L.M. Immune microenvironments in solid tumors: New targets for therapy. Genes Dev. 2011, 25, 2559–2572. [Google Scholar]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Zhang, L.; Yang, N.; Park, J.W.; Katsaros, D.; Fracchioli, S.; Cao, G.; O'Brien-Jenkins, A.; Randall, T.C.; Rubin, S.C.; Coukos, G. Tumor-derived vascular endothelial growth factor up-regulates angiopoietin-2 in host endothelium and destabilizes host vasculature, supporting angiogenesis in ovarian cancer. Cancer Res. 2003, 63, 3403–3412. [Google Scholar]
- Tsuzuki, Y.; Fukumura, D.; Oosthuyse, B.; Koike, C.; Carmeliet, P.; Jain, R.K. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha → hypoxia response element → VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 2000, 60, 6248–6252. [Google Scholar]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure-An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar]
- Manzur, M.; Hamzah, J.; Ganss, R. Modulation of G protein signaling normalizes tumor vessels. Cancer Res. 2009, 69, 396–399. [Google Scholar]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar]
- Le Serve, A.W.; Hellmann, K. Metastases and the normalization of tumour blood vessels by ICRF 159: A new type of drug action. Br. Med. J. 1972, 1, 597–601. [Google Scholar]
- Yuan, F.; Chen, Y.; Dellian, M.; Safabakhsh, N.; Ferrara, N.; Jain, R.K. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc. Natl. Acad. Sci. USA 1996, 93, 14765–14770. [Google Scholar]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 2004, 6, 553–563. [Google Scholar]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar]
- van der Veldt, A.A.; Lubberink, M.; Bahce, I.; Walraven, M.; de Boer, M.P.; Greuter, H.N.; Hendrikse, N.H.; Eriksson, J.; Windhorst, A.D.; Postmus, P.E.; et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: Implications for scheduling of anti-angiogenic drugs. Cancer Cell 2012, 21, 82–91. [Google Scholar] [CrossRef]
- Ganss, R.; Ryschich, E.; Klar, E.; Arnold, B.; Hammerling, G.J. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002, 62, 1462–1470. [Google Scholar]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar]
- Ganss, R.; Arnold, B.; Hammerling, G.J. Mini-review: Overcoming tumor-intrinsic resistance to immune effector function. Eur. J. Immunol. 2004, 34, 2635–2641. [Google Scholar]
- Garbi, N.; Arnold, B.; Gordon, S.; Hammerling, G.J.; Ganss, R. CpG motifs as proinflammatory factors render autochthonous tumors permissive for infiltration and destruction. J. Immunol. 2004, 172, 5861–5869. [Google Scholar]
- Griffioen, A.W. Anti-angiogenesis: Making the tumor vulnerable to the immune system. Cancer Immunol. Immunother. 2008, 57, 1553–1558. [Google Scholar]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O'Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar]
- Mazzone, M.; Dettori, D.; Leite de Oliveira, R.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; Ruiz de Almodovar, C.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef]
- de Bock, K.; Mazzone, M.; Carmeliet, P. Antiangiogenic therapy, hypoxia, and metastasis: Risky liaisons, or not? Nat. Rev. Clin. Oncol. 2011, 8, 393–404. [Google Scholar] [CrossRef]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Grone, H.J.; Hammerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar]
- Song, S.; Ewald, A.J.; Stallcup, W.; Werb, Z.; Bergers, G. PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat. Cell Biol. 2005, 7, 870–879. [Google Scholar]
- Berger, M.; Bergers, G.; Arnold, B.; Hammerling, G.J.; Ganss, R. Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood 2005, 105, 1094–1101. [Google Scholar]
- Manzur, M.; Hamzah, J.; Ganss, R. Modulation of the “blood-tumor” barrier improves immunotherapy. Cell Cycle 2008, 7, 2452–2455. [Google Scholar]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar]
- Hamzah, J.; Nelson, D.; Moldenhauer, G.; Arnold, B.; Hammerling, G.J.; Ganss, R. Vascular targeting of anti-CD40 antibodies and IL-2 into autochthonous tumors enhances immunotherapy in mice. J. Clin. Invest. 2008, 118, 1691–1699. [Google Scholar]
- Stockmann, C.; Doedens, A.; Weidemann, A.; Zhang, N.; Takeda, N.; Greenberg, J.I.; Cheresh, D.A.; Johnson, R.S. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 2008, 456, 814–818. [Google Scholar]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef]
- Yonenaga, Y.; Mori, A.; Onodera, H.; Yasuda, S.; Oe, H.; Fujimoto, A.; Tachibana, T.; Imamura, M. Absence of smooth muscle actin-positive pericyte coverage of tumor vessels correlates with hematogenous metastasis and prognosis of colorectal cancer patients. Oncology 2005, 69, 159–166. [Google Scholar]
- O'Keeffe, M.B.; Devlin, A.H.; Burns, A.J.; Gardiner, T.A.; Logan, I.D.; Hirst, D.G.; McKeown, S.R. Investigation of pericytes, hypoxia, and vascularity in bladder tumors: Association with clinical outcomes. Oncol. Res. 2008, 17, 93–101. [Google Scholar]
- Stefansson, I.M.; Salvesen, H.B.; Akslen, L.A. Vascular proliferation is important for clinical progress of endometrial cancer. Cancer Res. 2006, 66, 3303–3309. [Google Scholar]
- Cooke, V.G.; Lebleu, V.S.; Keskin, D.; Khan, Z.; O'Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte Depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by Met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S.; Meyer-Morse, N.; Bergsland, E.; Hanahan, D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 2003, 111, 1287–1295. [Google Scholar]
- Helfrich, I.; Scheffrahn, I.; Bartling, S.; Weis, J.; von Felbert, V.; Middleton, M.; Kato, M.; Ergun, S.; Schadendorf, D. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J. Exp. Med. 2010, 207, 491–503. [Google Scholar] [CrossRef]
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood 2011, 118, 2906–2917. [Google Scholar]
- Nisancioglu, M.H.; Betsholtz, C.; Genove, G. The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor-A blockade. Cancer Res. 2010, 70, 5109–5115. [Google Scholar]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar]
- Hagemann, T.; Biswas, S.K.; Lawrence, T.; Sica, A.; Lewis, C.E. Regulation of macrophage function in tumors: The multifaceted role of NF-kappaB. Blood 2009, 113, 3139–3146. [Google Scholar]
- DeNardo, D.G.; Andreu, P.; Coussens, L.M. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010, 29, 309–316. [Google Scholar]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar]
- de Palma, M.; Venneri, M.A.; Galli, R.; Sergi Sergi, L.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar]
- Mazzieri, R.; Pucci, F.; Moi, D.; Zonari, E.; Ranghetti, A.; Berti, A.; Politi, L.S.; Gentner, B.; Brown, J.L.; Naldini, L.; et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011, 19, 512–526. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Chen, Y.Y.; Muthana, M.; Welford, A.F.; Tal, A.O.; Scholz, A.; Plate, K.H.; Reiss, Y.; Murdoch, C.; de Palma, M.; et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J. Immunol. 2011, 186, 4183–4190. [Google Scholar]
- Johansson, A.C.; Hamzah, J.; Payne, C.J.; Ganss, R. Tumor targeted TNFα stabilizes tumor vessels and enhances active immunotherapy. 2012; Submitted for publication. [Google Scholar]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar]
- Welford, A.F.; Biziato, D.; Coffelt, S.B.; Nucera, S.; Fisher, M.; Pucci, F.; di Serio, C.; Naldini, L.; de Palma, M.; Tozer, G.M.; et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J. Clin. Invest. 2011, 121, 1969–1973. [Google Scholar]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar]
- Pietras, K.; Sjoblom, T.; Rubin, K.; Heldin, C.H.; Ostman, A. PDGF receptors as cancer drug targets. Cancer Cell 2003, 3, 439–443. [Google Scholar]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar]
- Nakagawa, H.; Liyanarachchi, S.; Davuluri, R.V.; Auer, H.; Martin, E.W., Jr.; de la Chapelle, A.; Frankel, W.L. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 2004, 23, 7366–7377. [Google Scholar]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar]
- Bauer, M.; Su, G.; Casper, C.; He, R.; Rehrauer, W.; Friedl, A. Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene 2010, 29, 1732–1740. [Google Scholar]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar]
- Qin, Z.; Schwartzkopff, J.; Pradera, F.; Kammertoens, T.; Seliger, B.; Pircher, H.; Blankenstein, T. A critical requirement of interferon gamma-mediated angiostasis for tumor rejection by CD8+ T cells. Cancer Res. 2003, 63, 4095–4100. [Google Scholar]
- Qin, Z.; Blankenstein, T. CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity 2000, 12, 677–686. [Google Scholar]
- Lu, Y.; Yang, W.; Qin, C.; Zhang, L.; Deng, J.; Liu, S.; Qin, Z. Responsiveness of stromal fibroblasts to IFN-gamma blocks tumor growth via angiostasis. J. Immunol. 2009, 183, 6413–6421. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Johansson, A.; Ganss, R. Remodeling of Tumor Stroma and Response to Therapy. Cancers 2012, 4, 340-353. https://doi.org/10.3390/cancers4020340
Johansson A, Ganss R. Remodeling of Tumor Stroma and Response to Therapy. Cancers. 2012; 4(2):340-353. https://doi.org/10.3390/cancers4020340
Chicago/Turabian StyleJohansson, Anna, and Ruth Ganss. 2012. "Remodeling of Tumor Stroma and Response to Therapy" Cancers 4, no. 2: 340-353. https://doi.org/10.3390/cancers4020340
APA StyleJohansson, A., & Ganss, R. (2012). Remodeling of Tumor Stroma and Response to Therapy. Cancers, 4(2), 340-353. https://doi.org/10.3390/cancers4020340