Aberrant Signaling Pathways in Glioma

Abstract
: Glioblastoma multiforme (GBM), a WHO grade IV malignant glioma, is the most common and lethal primary brain tumor in adults; few treatments are available. Median survival rates range from 12–15 months. The biological characteristics of this tumor are exemplified by prominent proliferation, active invasiveness, and rich angiogenesis. This is mainly due to highly deregulated signaling pathways in the tumor. Studies of these signaling pathways have greatly increased our understanding of the biology and clinical behavior of GBM. An integrated view of signal transduction will provide a more useful approach in designing novel therapies for this devastating disease. In this review, we summarize the current understanding of GBM signaling pathways with a focus on potential molecular targets for anti-signaling molecular therapies.1. Introduction
Despite advances in our understanding of the molecular biology of glioblastoma multiforme (GBM), one of the most aggressive human cancers, these tumors are still incurable. GBMs almost universally recur after conventional therapies, including maximal surgical resection, radiation, and chemotherapy, because GBMs are a heterogeneous group of tumors with variable natural history and treatment responses. Recent advances in research techniques have given rise to a wealth of new information regarding the distinct molecular subgroups of GBM with aberrant signaling pathways that drive the tumor phenotype. According to the genomic analysis by The Cancer Genome Atlas (TCGA) network, GBM has four distinct molecular subtypes: Proneural, neural, classical, and mesenchymal [1,2].
The pathogenesis of GBM is complex due to a highly deregulated tumor genome with opportunistic deletion of tumor suppressor genes, amplification, and/or mutational hyper-activation of oncogenes, which are involved in a network of interconnected signaling pathways [2]. The signaling pathway has been described in systems biology terms as a complex biological network of three steps: (1) an input step in which membrane receptors and their ligands trigger the signal coming from outside the cell; (2) a core system processing step in which protein kinases transmit the signal to the nucleus; (3) and an output step in which transcription factors regulate genes that affect various cellular functions. GBM characteristics are derived from the activation of these pathways, including uncontrolled proliferation, invasion, and angiogenesis. Current efforts therefore focus on the development of new molecular approaches targeting these genetic lesions [3,4]. In this review, we provide an overview of the most recent developments and current understanding of GBM signaling and the therapeutic potential of molecular targeted therapy in GBM.
2. The Character of Glioma
2.1. General Knowledge of GBM
Diffuse astrocytic tumors are the most frequent intracranial neoplasms and account for more than 60% of all primary brain tumors. The major characteristic by which astrocytic tumors are grouped is the histological resemblance of the tumor cells to astrocytes. Therefore, the presence of cells with fine fibrillary processes, expressing glial fibrillary acidic protein (GFAP), is a major diagnostic feature in these tumors. A proportion of the cells demonstrating astrocytic features and GFAP staining is important for identifying the astrocytic tumor cells, even in the more undifferentiated and anaplastic type of tumor. Usually, the tumor cells diffusely infiltrate the adjacent brain parenchyma and are thus termed as “diffuse” astrocytic tumors [5].
According to WHO classification and grading, diffuse astrocytic tumors are subdivided into three categories: (1) diffuse astrocytoma (grade II); (2) anaplastic astrocytoma (grade III); and (3) the most undifferentiated GBM (grade IV). Previously termed “glioblastoma multiforme” because of the heterogeneity of its macroscopic appearance, the name ‘glioblastoma’ is now considered less complicated. GBM is the most malignant astrocytic tumor, composed of poorly differentiated neoplastic astrocytes. The presence of microvascular proliferation and/or necrosis is essential for histo-pathological diagnosis of GBM. Two subgroups of GBMs have been established based on clinical experience and have been affiliated with distinct genetic mechanisms of tumorigenesis, although these two groups are histologically indistinguishable. Primary GBMs seem to develop rapidly and manifest high-grade lesion from the outset. In contrast, secondary GBMs develop slowly through progression from grade II or grade III.
2.2. Clinical Features of GBM
2.2.1. Incidence and site
GBMs are the most common primary brain tumors in adult neurosurgical practice, and account for 40%–60% of all diffuse astrocytic tumors and 10%–15% of all intracranial neoplastic lesions. Although they may occur at any age, GBMs have a peak incidence between 50 and 70 years. The cerebral hemispheres are the most common site for GBM, with 95% arising in the supratentorial region, and less commonly appearing in the brain stem, cerebellum, and spinal cord.
2.2.2. Radiology
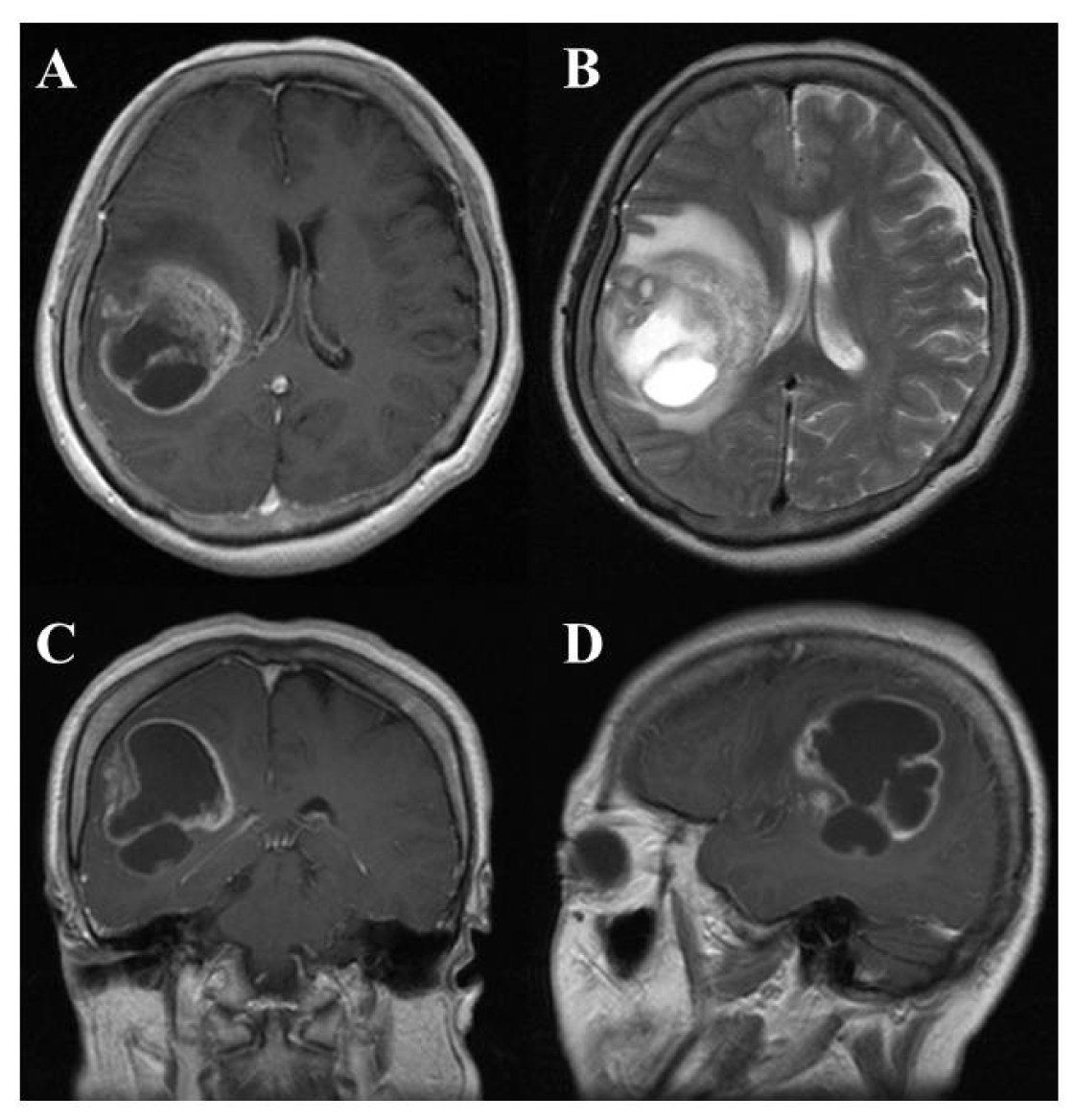
On computed tomography (CT) and magnetic resonance imaging (MRI), many GBMs present a typical appearance of ring enhancement indicating a central necrosis surrounded by enhancing viable tumor (Figure 1). Peritumoral edema, usually with distortion of the surrounding brain and ventricles (mass effect), is well visualized by MRI. Despite the typical appearance, other enhancing lesions within the cerebral hemisphere, especially abscess, metastasis, lymphoma, or even acute multiple sclerosis plaques and subacute infarctions, should be considered as notable differential diagnoses.
2.2.3. Therapy and prognosis of GBM
The prognosis for GBM is worse than that for diffuse astrocytoma and anaplastic astrocytoma. Most clinical studies suggest that 50% of patients with GBM will survive no longer than one year. The outcome of GBM depends largely upon the patient's preoperatively scored neurological state, scored by the Karnofsky Performance Status (KPS), age, and tumor site [6,7]. Generally, individuals under the age of 50 have a slightly longer survival time than those over 50, and patients over the age of 65 have a particularly poor prognosis. The location of the tumor is closely related with radical surgical removal and it is an important factor in tumor management. GBMs deeply seated in the thalamus and basal ganglia, and thus eloquent cortices (motor cortex, sensory cortex, language cortex, etc.) inaccessible to surgical excision, have a poor prognosis. GBMs in the non-eloquent frontal and temporal lobes, which are accessible to excision, may have a slightly longer survival time. The recent advent of intraoperative surgical modalities including neuronavigation, intraoperative monitoring, and 5-aminolevulinic acid (5-ALA) fluorescence-guided tumor resection, have improved the rate of gross total excision and prolonged survival time [2].
Corticosteroid treatment is often very effective initially in patients with GBM. They reduce intracranial pressure and reduce the severity of symptoms due to early-stage tumors. After surgical resection, radiotherapy also has some beneficial effect on the overall prognosis and may increase the quality of life for the patient; many hospitals employ chemotherapy after/with radiotherapy. In particular, chemotherapy with the recently introduced chemo-agent temozolomide plus radiotherapy has been proven effective on newly diagnosed GBM patients and has provided a new standard of care after tumor excision [8]. However, despite such combined therapy with surgical resection with adjuvant chemo/radiotherapy, overall survival time in GBM patients is still never more than two years.
2.3. GBM Cell Biology
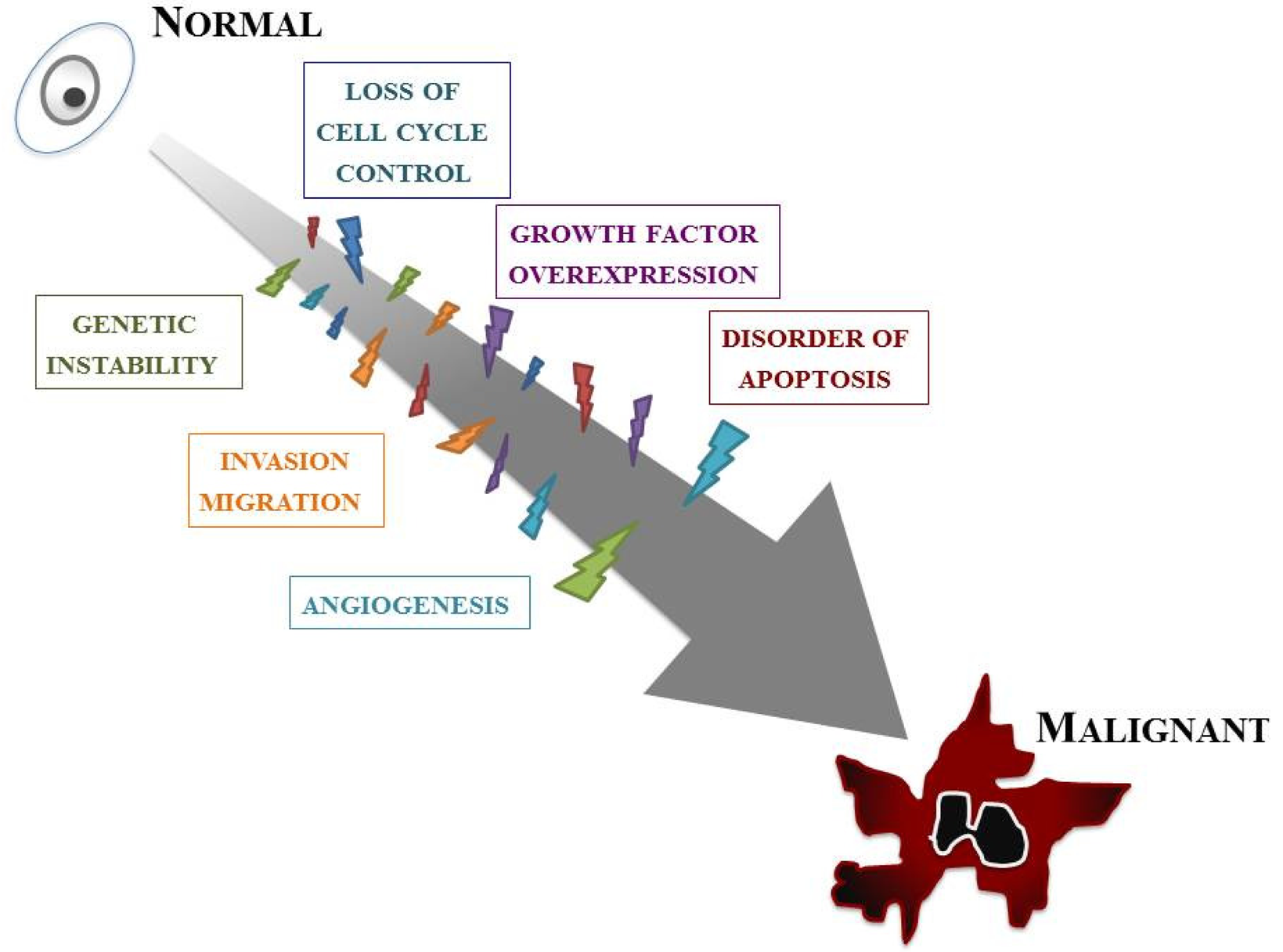
Our knowledge of the molecular biology of GBM has increased markedly over the last two decades. GBM cells are subjected to many kinds of cellular dysfunction, which make themselves resistant to various anti-GBM therapies. Six typical intracellular events that characterize glioma are overviewed below, but it should be stressed that such events do not arise in isolation; a combination of multiple tumorigenic events cause and sustain GBM (Figure 2).
The first event is loss of cell cycle control. The progression of the normal cell cycle is under strict control. However, glioma cells develop means for evading such control, giving them a growth benefit. The cell cycle checkpoint that has received the most attention is the G1–S phase transition. p16INK4a/cyclin-dependent kinase (CDK)-4/RB (retinoblastoma) 1 pathway, one of the major pathways controlling this checkpoint, involves p16, CDK-4, cyclin D1, and RB1 [9]. The CDK/cyclin D1 complex phosphorylates RB1, thereby inducing release of the E2F transcription factor that activates genes involved in the G1/S transition [10]. Many of the genetic defects in growth regulatory molecules occur preferentially in malignant, rather than low-grade, gliomas. Alterations of at least one component of this pathway occur in many anaplastic astrocytomas and in the vast majority of GBMs [11,12]. Nearly all high-grade tumors have impairments of this single critical cell cycle control pathway. It is likely as well that less profound defects in cell cycle regulation occur in low-grade gliomas; for instance, p53 gene mutations may affect both the G1–S and G2–M checkpoints.
The second event is overexpression of growth factors and their receptors. A variety of growth factors such as epidermal growth factor receptor (EGFR), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF, FGF-2), transforming growth factor (TGF)-α, and insulin-like growth factor (IGF)-1 are overexpressed in GBM and thus provide a growth advantage to neoplastic cells. Generally, glioma cells express both the growth factor ligands and their receptors, setting up an autocrine growth-promoting loop. Of these, EGFR and PDGF have been best characterized in GBMs [13,14]. EGFR is a transmembrane receptor responsible for sensing extracellular ligands, such as EGF and TGF-α, and for transducing this proliferation signal.
Angiogenesis is also an important feature. A dramatic sequence of angiogenic alterations occurs in the progression to GBMs, which in MRI appears as ring-like contrast enhancements that surround rapidly growing tumors [15]. Malignant gliomas are vascular tumors, and the histological presence of microvascular proliferation indicates a high grade. Angiogenic molecules have been found in malignant gliomas, primarily in GBMs [16]. Vascular endothelial growth factor (VEGF) is the most clearly implicated and an endothelial cell mitogenic factor that is expressed most often adjacent to areas of necrosis but not in low-grade astrocytomas. This suggests that the malignant progression from low-grade astrocytomas to GBMs includes an “angiogenic switch”.
One of the key features of GBMs, invasion and migration, is their diffuse infiltration of the surrounding neural net. The expression of several extracellular matrix (ECM) molecules and cell surface receptors may modulate signal transduction and influence invasion and migration in GBMs [5,17,18]. These include cytoskeletal proteins; signaling molecules that mediate interactions between the microenvironment and the cytoskeleton; cell surface receptors involved in cell migration such as transmembrane adhesion molecules; and components of ECM, including proteases.
Abnormality of apoptosis should be mentioned. Apoptosis refers to cell death that occurs in a programmed manner, characterized by non-inflammatory cellular condensation. Glioma cells may develop means not only for increasing proliferation but for abrogating apoptosis as well. A number of genes involved in gliomagenesis have roles in apoptosis, most notably p53 [19,20]. p53 mutations disturb the normal glial apoptotic response that would follow growth factor overexpression in low-grade gliomas, allowing further progression [21].
The last intracellular event is genetic instability. An essential feature of low-grade gliomas is their nearly universal progression to higher-grade lesions over time. Such malignant progression is related to the emergence of more malignant clones. Genomic instability, a feature of many tumors, encourages further genomic damage, thus allowing the eventual selection of more malignant clones [22]. Mutations in p53, also known as “guardian of the genome”, may therefore lead to tumor progression through genomic instability. Patients with syndromes of genomic instability, such as the Turcot syndrome, have an increased susceptibility to malignant gliomas [23].
3. Proliferation Signaling
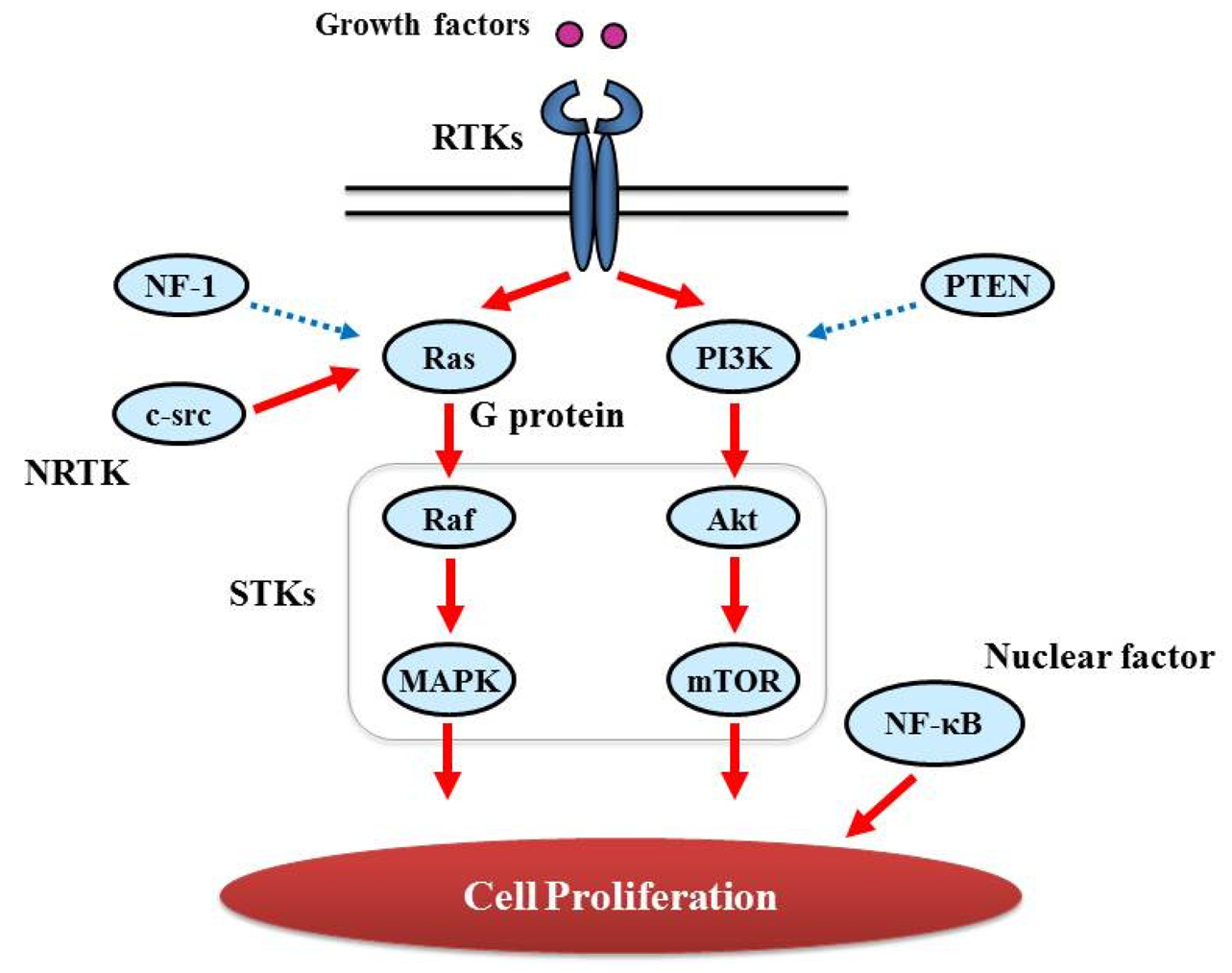
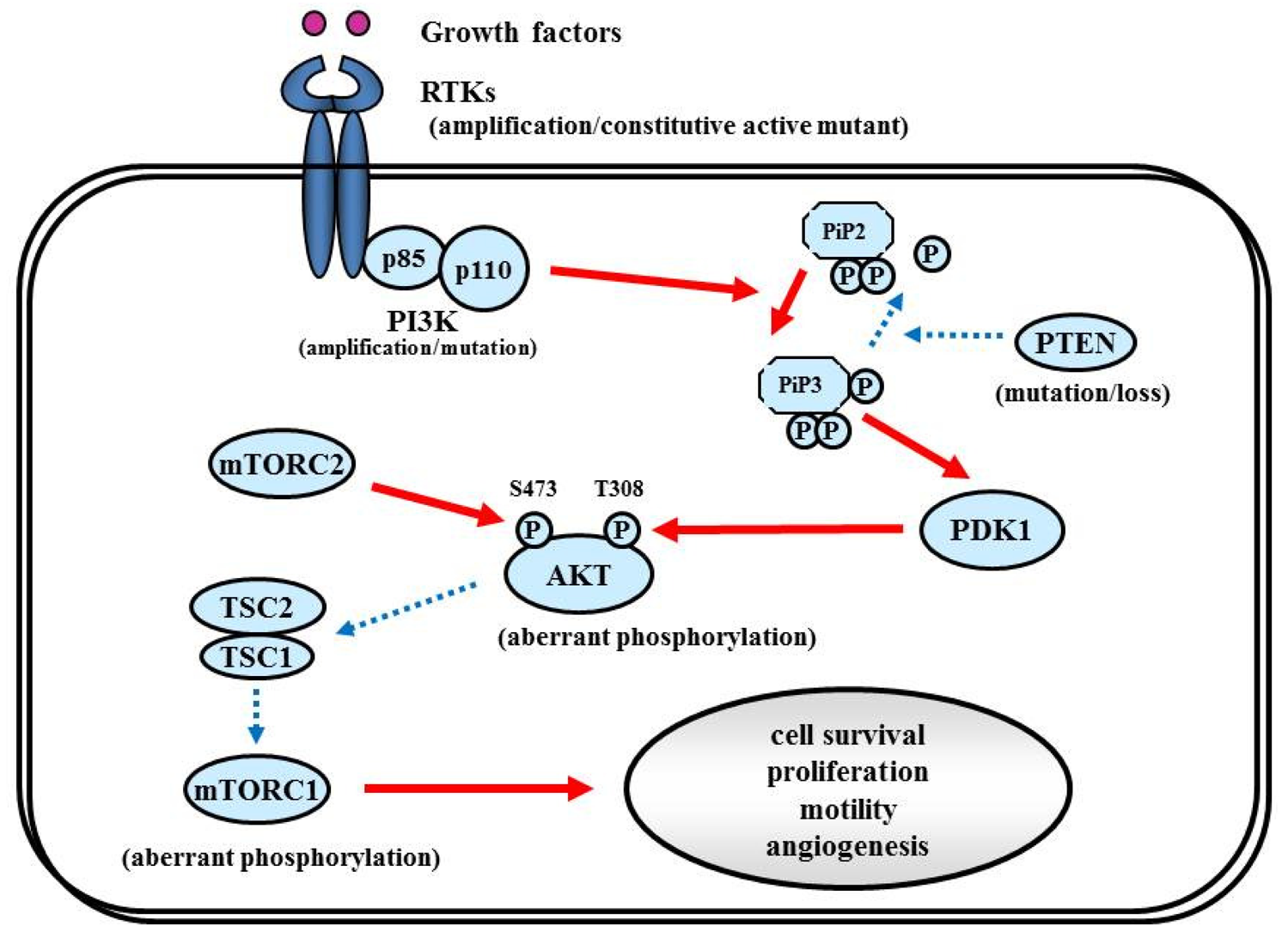
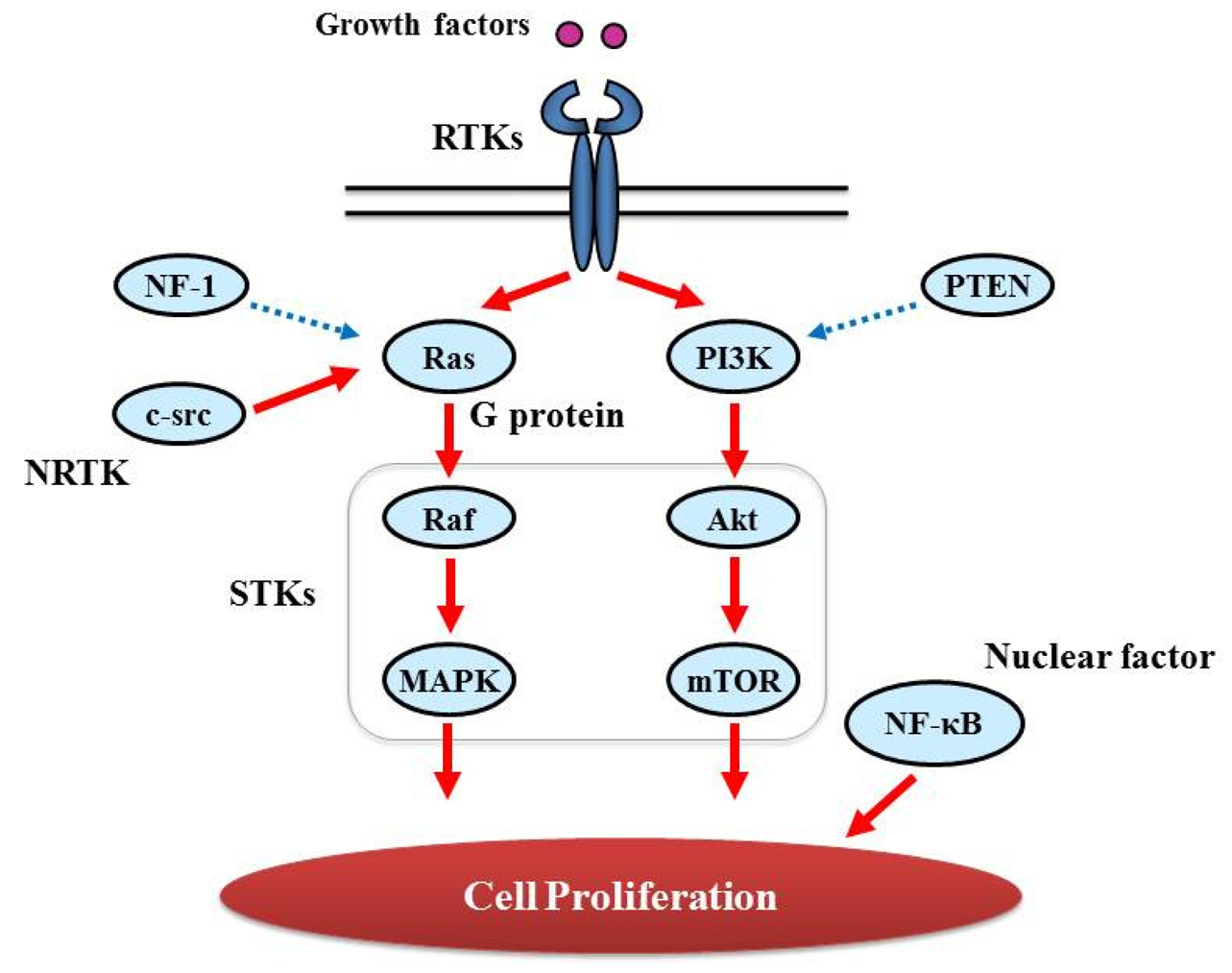
Proliferative activity with histopathologically detectable mitoses is prominent in almost all GBM cases. It is essential to define the contribution of proliferation signaling (or growth factor-mediated signaling) to the GBM cellular phenotype in order to understand the biology of transformation in the CNS. Two of the most important signaling cascades frequently deregulated in glioma are the PI3K/Akt/mTOR and Ras/MEK/MAPK pathways (Figure 3).
3.1. Tyrosine Kinase (TK) and Its Ligand
In glioma growth and progression, proliferation signaling depends largely upon the activity of cell surface membrane receptors that control the intracellular signal transduction pathways regulating proliferation; in other words, the leading actor is tyrosine kinase.
3.1.1. Receptor Tyrosine Kinase (RTK)
RTKs are glycoproteins that possess an N-terminal extracellular ligand-binding domain, a single anchoring transmembrane α helix, and a cytosolic C-terminal domain that contains the catalytic domain [24]. For instance, EGFR is a transmembrane glycoprotein member of the ErbB receptor family. In response to binding of ligands EGF and TGF-α, the receptor homodimerizes and/or forms heterodimers with other similar but not identical kinases of the same subfamily, especially ErbB-2 (HER2), allowing a highly varied response to the extracellular signal [25,26]. RTK activation in glioma can be achieved through protein overexpression or genetic amplification of several RTKs, and through gene mutations that lead to constitutive activity. In GBM, EGFR is dysregulated through overexpression, which arises because of EGFR gene amplification or activating mutations such as EGFRvIII that lead to ligand-independent signaling; this is the case in half of all GBM patients [27,28]. Increased transcription of PDGFRα has been observed in astrocytic tumors, but gene amplification is detected only in a limited subset of GBMs (13%) [29,30]. This also creates autocrine or paracrine loops that promote tumor cell proliferation. These receptors are described in detail in “Major signaling and related molecules”. Other RTKs such as FGFR, IGFR, and mesenchymal-epithelial transition factor (c-Met) are abnormally expressed in GBMs [31-34]. Although the mechanisms for such growth factor abnormality, mainly overexpression, vary between growth factors, they are thought to affect glioma proliferation or transformation.
3.1.2. Non-RTK (NRTK)
NRTKs lack transmembrane domains and are found at various intracellular locations, including the cytosol, nucleus, and the inner surface of the plasma membrane. c-src [v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (avian)] TK includes SH2, SH3, and TK domains. c-src mediates intracellular signaling pathways that control key biologic/oncogenic processes. Increased c-src activity in GBM has been reported [35].
3.1.3. Growth factor
It is well known that various kinds of growth factors, such as PDGF (PDGFA, PDGFB), bFGF (bFGF, FGF-2), TGF (TGF-α, TGF-β), and IGF-1 are overexpressed in GBM [36-39]. However, their role in GBM progression is not always fixed. For instance, TGF-β acts as a tumor suppressor in normal epithelial cells and early-stage tumors and becomes an oncogenic factor in advanced tumors [40].
3.2. Others
Molecules that play important roles in GBM proliferation, other than RTK and its ligands, are described below.
3.2.1. GTPase activating protein (G protein)
Ras genes, an abbreviation of Rat Sarcoma, encode a protein family of small GTPases that cycle between inactive GDP-bound and active GTP-bound conformations by coupling cell membrane growth factor receptors such as EGFR; they regulate cellular signal transduction by acting as a one-way switch for the transmission of extracellular growth signals to the nucleus [41]. Because these signals result in cell growth and division, dysregulated Ras signaling can lead to oncogenesis and cancer [42]. Ras activates a number of signaling pathways that include stem cell factor (SCF)/c-kit signaling, mammalian target of rapamycin (mTOR), and mitogen-activated protein (MAP) kinases pathways. The role of Ras in GBM is described in detail in “Major signaling and related molecules”.
3.2.2. Serine/threonine specific protein kinase (STK)
The protein kinase family of enzymes plays a pivotal role in signal transduction across the cell membrane as well as inside cells [43]. Both types of protein kinases, TK and STK, share a high sequence similarity in their catalytic domains, which suggests that they descended from a common ancestral gene. STK have received comparatively little attention, vis-a-vis TKs, in terms of their involvement in cancers [44]. In contrast to the TKs, most of the known STKs are cytoplasmic proteins.
STK belongs to the family of transferases, specifically those that transfer phosphorus-containing groups as protein-serine/threonine kinases. STK expression is altered in various cancers. Akt is one of the STKs that play a key role in cellular proliferation. GBM frequently contains alterations in PTEN (Phosphatase and Tensin Homolog Deleted from Chromosome 10) [45] that is a lipid phosphatase and lead to activation of the Akt/mTOR pathway [46]. BRAF, v-RAF murine sarcoma viral oncogene homolog B1, is also an STK and a member of the RAS/RAF/MEK (MAPK kinase)/MAPK pathway that is commonly activated by somatic point mutation V600E in pilocytic astrocytoma [47]. In contrast, this mutation is rarely reported in GBM [48].
TGF-β receptor is predicted to receptor-type STK [49], albeit most growth factor receptors are transmembrane TKs or are associated with cytoplasmic TKs, and plays an important role in growth and progression of GBM, via Smad phosphorylation [50].
3.2.3. Nuclear Factor
NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells, is a protein complex that controls DNA transcription and regulates genes that control cell proliferation and cell survival [51]. Aberrant constitutive activation of NF-κB in GBM in response to PDGF overexpression promotes glioma cell proliferation [51]. The PI3K pathway has been implicated in mediating the activation of NF-κB by PDGF, and inactivation of PTEN and PDGF expression may contribute to the high levels of NF-κB [52].
4. Angiogenesis Signaling
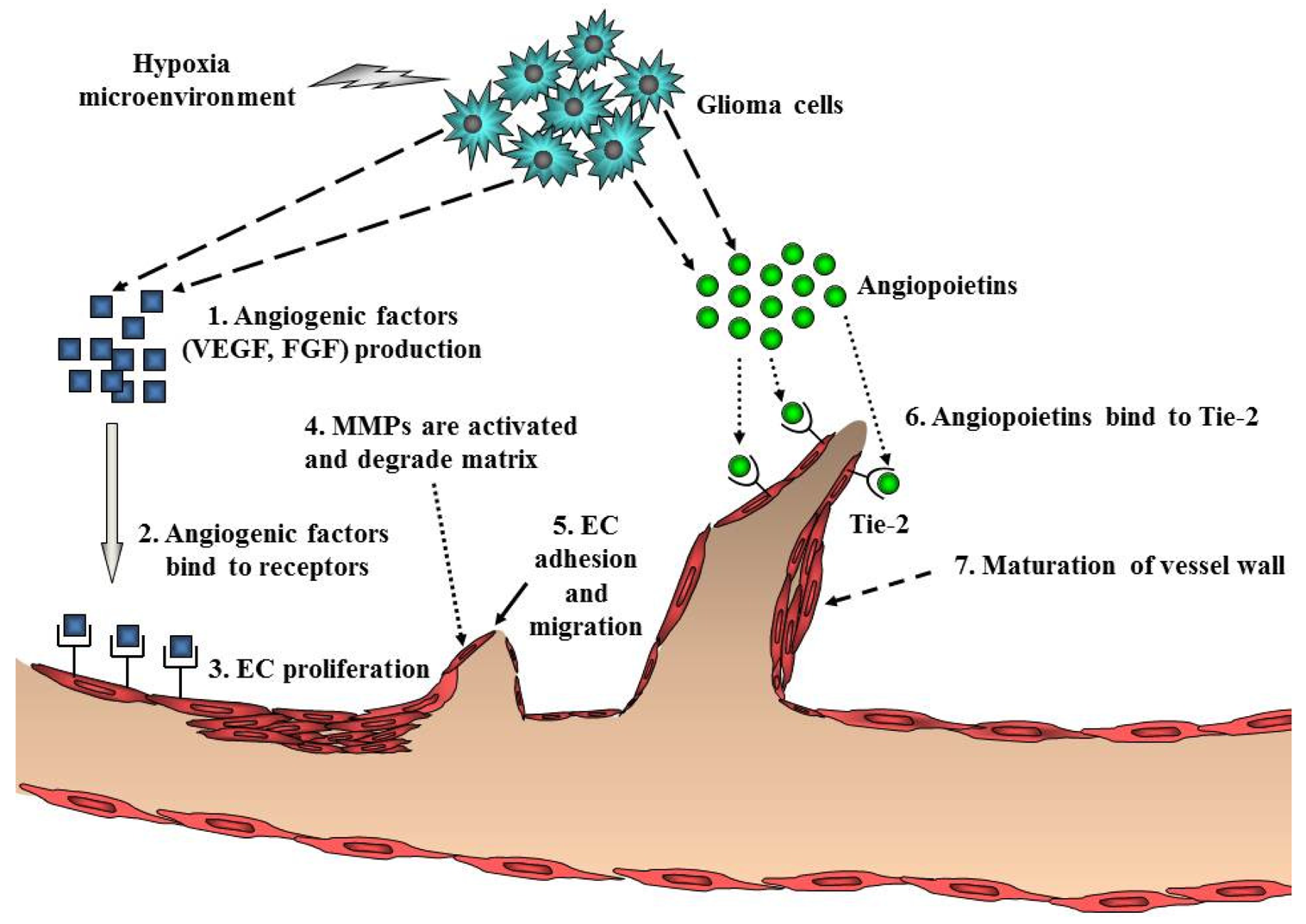
Ubiquitous angiogenesis is a hallmark of GBMs [53]. The degree of vascularization is significantly correlated with glioma malignancy, tumor aggressiveness, and clinical prognosis [54]. A number of scientists have aggressively investigated angiogenesis signaling and the function of angiogenic factors in malignant glioma. Many studies have implicated pro-angiogenic pathways including a sequence of coordinated events that is initiated by the expression of angiogenic factors such as VEGF with subsequent binding to its cognate receptors on endothelial cells (Figure 4). VEGF/VEGFR (VEGF receptor) participates in the formation of primitive blood vessels from progenitors, hemangioblasts/angioblasts, and the further development of blood vessels in glioma tissue [55,56]. Many other factors increase VEGF expression, including acidosis, nitric oxide, altered oncogenes, and tumor suppressor genes, cytokines such as bFGF, PDGF, and EGF, and activated intracellular signaling pathways such as PI3K/Akt and Ras/MAPK [57] (Table 1). Indeed, increased levels of VEGF and VEGFR in GBM have been reported [58,59].
Endothelial cell migration and proliferation are triggered by VEGF binding to its cognate receptors on endothelial cells [60]. Endothelial cells near the tumor strongly express VEGFR2, which establishes a paracrine signaling loop that stimulates the growth and proliferation of endothelial cells. Local degradation and breakdown of ECM occurs simultaneously, paving the way for newly sprouting vessels. Then, angiopoietins (Ang) are involved in the stability and maintenance of the tumor vasculature. Binding of Ang-2 to its cognate receptor, Tie-2, serves to destabilize vessels, which is a requirement for angiogenesis of glioma [2] (Figure 4). Another key angiogenic process involves cathepsin B and matrix metalloproteinases (MMPs), as well as the expression of matrix proteins such as fibronectin, laminin, tenascin-C, and vitronectin [61]. Formation of the new blood vessel is accomplished by endothelial cells that form a lumen. Individual sprouts are then connected to form vascular loops, through which blood begins to flow. Maturation of the vessel wall then begins by recruitment of pericytes to assemble along the endothelial cells outside the new vessel. The angiogenic process of glioma is completed by the formation of a new basement membrane [62].
5. Invasion Signaling
GBM is highly invasive by nature but does not metastasize to other organs like carcinomas in other organs, whereas metastatic brain tumors from other organs usually do not exhibit invasive behavior in the brain. Migrating glioma cells tend to move along the vessels, dendrites, and fibers in white matter. These characteristics suggest that GBM possesses specific biological mechanisms that mediate its invasive nature. The highly infiltrative nature of human gliomas recapitulates the migratory behavior of glial progenitors during development of the CNS, suggesting that the activators, receptors, and signaling proteins that contribute to neural crest cell migration are key players in glioma invasion. Glioma invasion is a complex process involving (1) detachment from the original site; (2) adhesion to the ECM; (3) remodeling of the ECM; and (4) cell migration [5]. Many molecules are involved in each step.
5.1 Membrane Type Protein
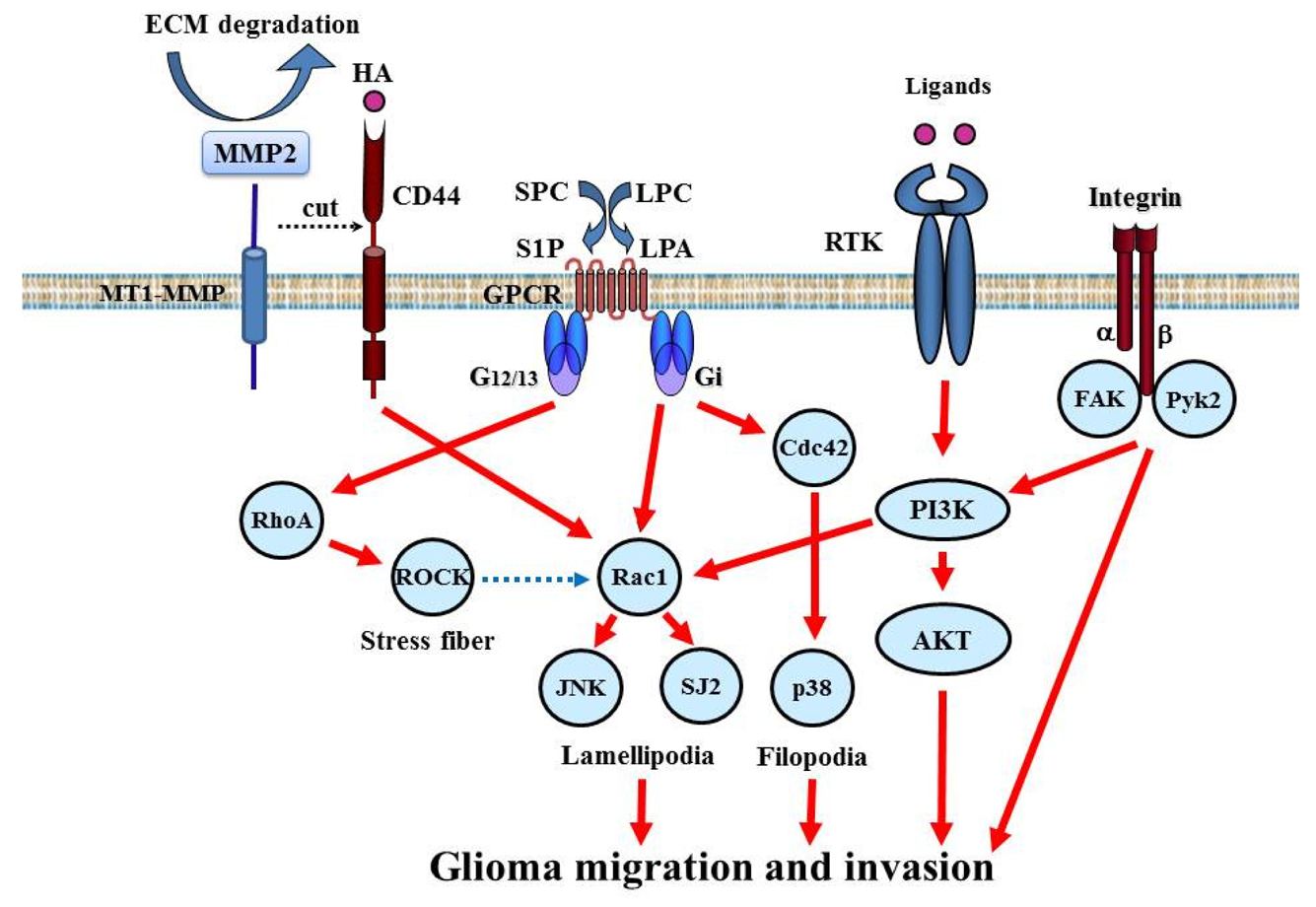
Accumulating studies have shown that invasion signaling is induced by several kinds of membrane-type protein such as RTK, integrin, CD44, and G protein-coupled receptor (GPCR, Figure 5).
5.1.1. RTK
RTKs such as EGFR, PDGFR, c-Met, Tie, DDR1 (discoidin domain receptor), Eph, and Axl are expressed at very high levels in GBM and play a major role in glioma invasion [5,63]. Multiple RTKs are simultaneously activated in GBM cells and accelerate invasion signaling. RTK signaling links to intracellular invasion signaling. Additionally, EGFR, Tie, and DDR1 signaling induce the expression and activation of MMP-2, which degrades and remodels the ECM surrounding tumor cells [64-66].
5.1.2. Integrin
Integrins are transmembrane heterodimer receptors consisting of α and β subunits. The combination of α and β subunits determines ligand specificity. Integrins serve to create a link between the ECM and the cytoskeleton actin filaments. This is integral to the maintenance of cell shape and architecture and the regulation of cell migration. Among the 8 β-subunit members, β1 is important to glioma biology and its expression has been correlated with the invasive behavior of glioma [67]. Integrin β5 expression is correlated with in vitro invasiveness and migration of glioma cells [68]. Although the β1 subunit can collaborate with several α subunits, α3β1 is consistently over-expressed and is a key regulator of glioma cell migration [69].
5.1.3. CD44
CD44 is a transmembrane glycoprotein expressed in glioma and serves as a surface receptor for components of the ECM such as hyaluronic acid (HA). The standard form of CD44 consists of a cytoplasmic tail, a transmembrane region, and a large (90 amino acid-long) extracellular domain. Under physiological conditions, CD44 in tumor cells is cleaved by a membrane-associated MMP at the membrane-proximal region of the ectodomain. This shedding of CD44 plays a critical role in efficient cell detachment from the HA substrate and promotes cell migration. Overexpression of CD44 in glioma is related to invasion [70]. CD44 can be cleaved by ADAM (a disintegrin and metalloproteinase) 10 and 17, and both the extracellular and intracellular cleaved components of CD44 promote cell migration [71,72]. The CD44 cleavage product is detected in 60% of gliomas. Rac1 activation is linked to CD44 shedding [73].
5.2. Intracellular Signaling Molecules
Many invasion signaling pathways converge on the common intracellular signaling molecules PI3K/Akt and small GTPases, such as Rac1, cdc42, and RhoA (Figure 5).
5.2.1. Small GTPases
Small GTP-binding proteins of the Rho family are central regulators of dynamic reorganization of the actin-based cytoskeleton and are key mediators of several cellular processes, including cell migration and polarity [74]. Rac belongs to the Rho GTPases, a family of proteins best known for their regulation of the cytoskeleton and cell migration. Rac1 play key roles in regulating actin polymerization and promoting lamellipodial formation at the front edges of migrating cells. Rac1 is ubiquitously expressed in various tissues. In glioma cells, depletion of Rac1 expression by small interfering RNA reduces cell migration and invasion and strongly inhibits lamellipodia formation [75]. In contrast, Rho activation has been associated with stress fiber formation. RhoA plays a key role in the regulation of actomyosin contractility. Activated Rho relays extracellular signals to a number of downstream effectors, which include Rho kinase (ROCK). Inhibition of ROCK induces glioma cell migration through Rac1 activation, suggesting that the relative degree of activation between RhoA and Rac1 regulates glioma cell movement [76]. Cdc42 plays a critical role in filopodia formation and cell polarity in most eukaryotic organisms. The function of cdc42 in glioma invasion is not completely understood.
6. Crosstalk
These signaling elements can contribute to proliferation, survival, migration, invasion, and angiogenesis in GBM cells. A hallmark of signaling networks is the presence of crosstalk, and multiple cross talks can cooperate during glioma initiation and progression to aggressive, invasive, and hyper-vascularized disease stages.
TKs, which are major inducers in GBM, exhibit crosstalk. The c-Met receptor is strongly phosphorylated as a function of EGFRvIII receptor levels, suggesting the presence of crosstalk between c-Met and EGFRvIII signaling, although the intermediary molecule has yet to be elucidated [77]. Axl RTK also follows a similar phosphorylation response as a function of EGFRvIII levels [77]. EGFR and EphA2 RTK are expressed in GBM and co-localize to the cell surface. EphA2 phosphorylation is dependent on EGFR activity and EphA2 downregulation inhibits EGFR phosphorylation, downstream signaling, and EGF-induced cell viability [78].
Inactivation of either Ras/Raf/MAPK or PI3K/Akt/mTOR triggered activation of the other, suggesting that there may be mutually inhibitory crosstalk between them [79] (Figure 3). Generally, Ras/Raf/MAPK is the dominant signaling pathway associated with proliferation, whereas PI3K/Akt/mTOR is the invasion control pathway. The experimental evidence indicates that there may be an inherent and inverse correlation between cell motility and proliferation of a cell population. Consistent with this observation, cDNA microarray data showed that stimulated migration is accompanied by a concomitant down-regulation of genes responsible for proliferation in vitro [80].
Although the signaling mechanisms underlying these complex crosstalk phenomena remain to be elucidated, recent studies suggest that signaling crosstalk in GBM may aid in the design of treatment protocols targeting dominant signaling pathways that are preferentially activated.
7. Major Signaling Pathways and Related Molecules
7.1. RTK/PI3K/Akt Pathway
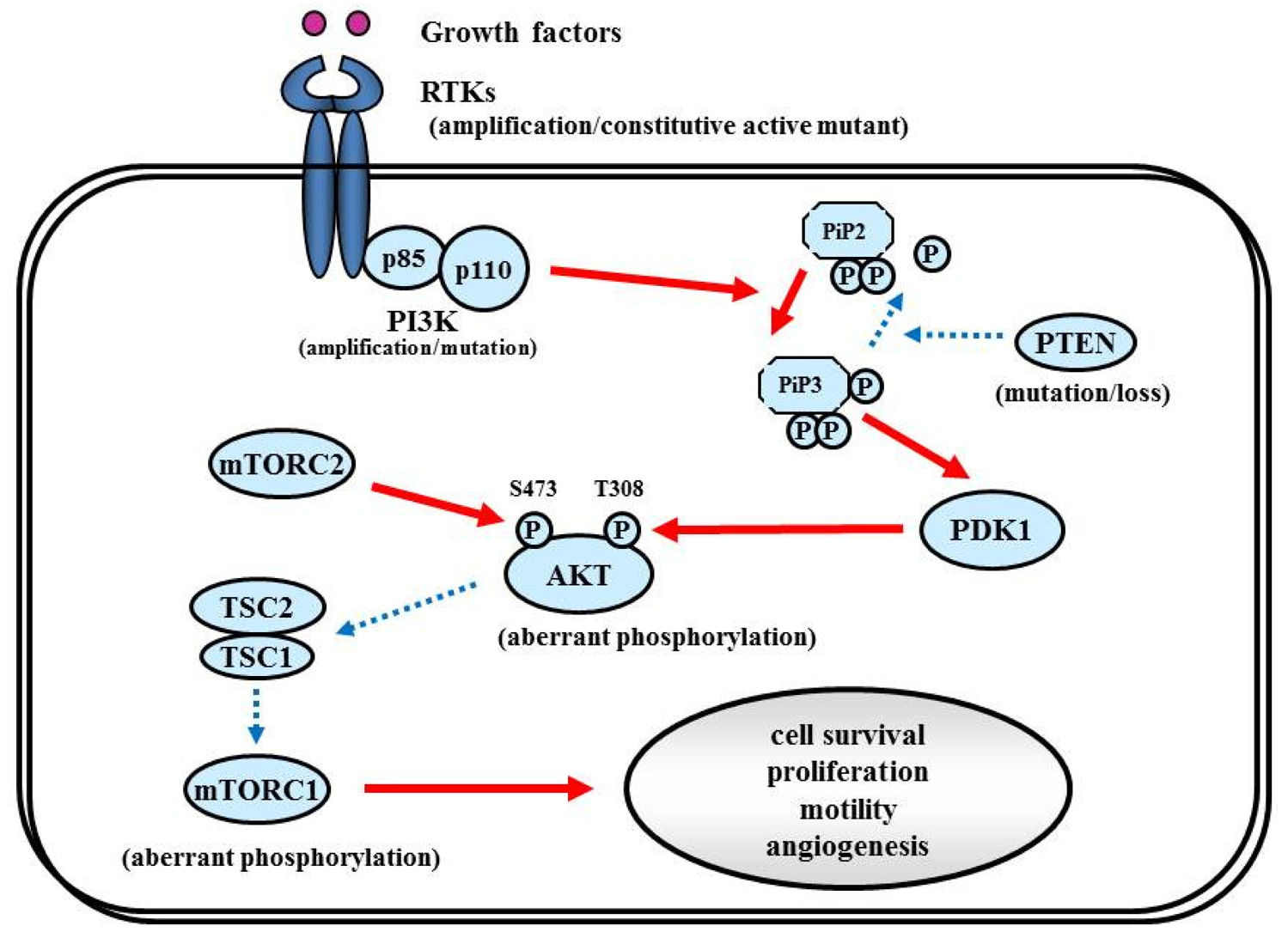
The RTK/PI3K/Akt pathway regulates various cellular processes such as proliferation, growth, apoptosis, and cytoskeletal rearrangement. The pathway involves RTKs (EGFR, PDGFR, and VEGFR, etc.) as well as tumor suppressor protein phosphatase PTEN, and protein kinases PI3K, Akt, and mTOR (Figure 6). Aberrant activation of RTK/PI3K/Akt is frequently observed in malignant gliomas because of the alterations of these proteins [81].
7.1.1. RTK
Among all the RTK alterations, EGFR gene amplification is the most frequent alteration (approximately 40%) in GBM [82-84]. Amplification of the EGFR gene is also associated with structural alteration, and the most common is called EGFRvIII, a mutant that can send ligand-independent constitutive growth signals [13,85,86]. Some reports have shown that GBM patients with EGFR overexpression or mutants have shorter survival, suggesting that alterations of EGFR may be correlated with increased aggressiveness of GBM [87-89]. According to TCGA, EGFR aberrations are correlated with a classical subtype of GBM [1,90]. Despite the high frequency of amplification and rearrangement of the EGFR gene, EGFR inhibitors (e.g., Gefinitib, Erlotinib) have not elicited clinical responses in patients with GBMs in clinical trials [91-93].
Overexpression of PDGFR (especially PDGFR-α) and PDGF has been observed in astrocytic tumors of all grades, possibly associated with malignant progression [29,36,94,95]. PDGFRA amplification (14%), as well as IDH1 mutation, is a hallmark of the proneural subtype of GBM according to the TCGA consortium, suggesting the association of this subtype and secondary GBM [1,90]. Despite deep association of this molecule with GBM, anti-PDGFR therapy using Imatinib yields only limited clinical responses [96,97].
7.1.2. PI3K
The PI3Ks are widely expressed lipid kinases that promote diverse biological functions. The binding of PI3Ks and RTKs results in activation of Akt through PiP3 and PDK1, which affects multiple cellular processes including cell survival, proliferation, and motility [98] (Figure 6). The PI3K complex is comprised of a catalytically active protein, p110α, encoded by PIK3CA, and a regulatory protein, p85α, encoded by PIK3R1. Oncogenic mutations or gene amplification of PIK3CA has been reported in various neoplasms including human brain tumors [99]. In primary GBM, PIK3CA mutations and amplification are observed in 5% and 13% of cases [81]. Unlike PIK3CA, PIK3R1 has rarely been reported as mutated in cancers. The TCGA analysis revealed that PIK3R1 mutations were found in 10% (9/91) of GBM cases. According to the integrated genomic classification of GBM, PI3K mutations (15%) are associated with the proneural subtype [1,90]. First generation of the PI3K inhibitors, LY294002 and wortmannin were not applied for clinical trials because of problems in solubility, selectivity, and toxicity [100]. To date, lots of PI3K inhibitors (many of them are PI3K/mTOR dual inhibitors) such as XL147, XL765 (Exelixis and Sanofi-Aventis), BKM120, BEZ235, BGT226 (Novartis), GDC0980 (Genentech), PKI587, PF04691502 (Pfizer), GSK2126458 (Glaxo-Smith-Kline) are currently undergoing Phase I/II trials for GBM patients.
7.1.3. PTEN
Decreased PTEN activity can activate the RTKs/PI3K/Akt pathway since PTEN negatively regulates the pathway by antagonizing PI3K function [101]. Decreased expression of PTEN and activation of Akt have been shown in a variety of cancers, including GBM [102]. Homozygous deletion or mutation of PTEN is a common genetic feature in GBM (∼40%) [1,103,104], resulting in constitutive activation of the RTKs/PI3K/Akt pathway. Some reports implied that GBMs with EGFRvIII and intact PTEN are more likely to respond to EGFR inhibitors [105,106]. PTEN loss is associated with both classical and mesenchymal subtypes of GBM, according to the TCGA study [1].
7.1.4. Akt
Akt is an STK that regulates cell growth, proliferation, and apoptosis. Akt activation has been reported in approximately 80% in human GBMs [46,105], well correlated with the fact that RTKs/PI3K/Akt signaling is altered in 88% of GBM [1]. Oncogenic Akt mutations have not been detected in GBM [107]. Akt inhibitor perifosine is undergoing clinical evaluation in malignant gliomas (NCT00590954) [108].
7.1.5. mTOR
mTOR is an STK downstream from Akt. mTOR can be activated by the Akt and RAS pathways. Rapamycin (sirolimus; Wyeth Pharmaceuticals) and its synthetic analogs Torisel (temsirolimus, Wyeth) and Afinitor (everolimus, Novartis), have been intensively evaluated in clinical trials of recurrent malignant gliomas, demonstrating modest efficacy [109,110].
7.2. p53
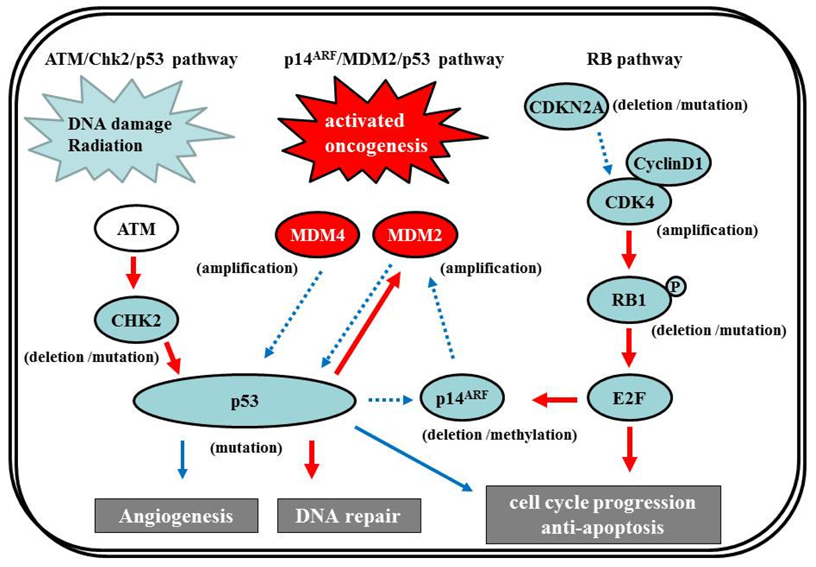
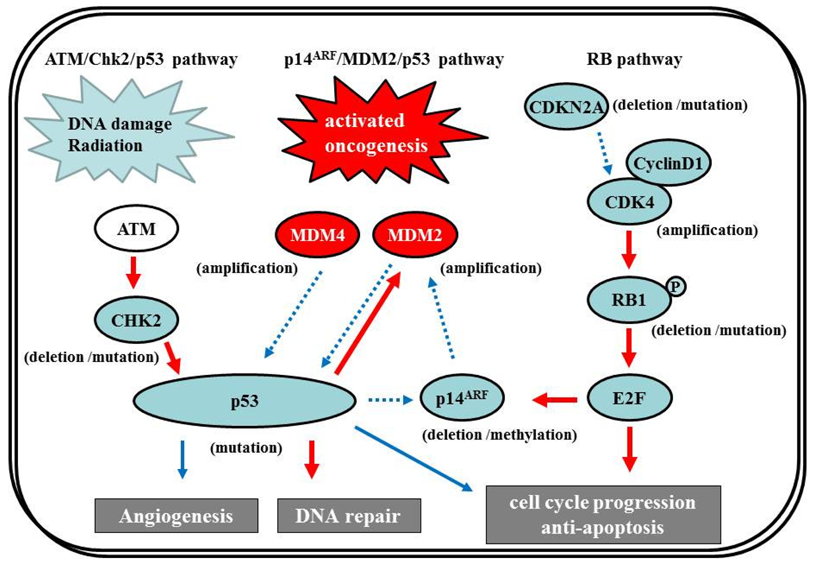
7.2.1. p14ARF/MDM2/p53 pathway
The p53 gene, at chromosome 17q13.1, encodes a protein that responds to diverse cellular stresses to regulate target genes that induce cell cycle arrest, cell death, cell differentiation, senescence, DNA repair, and neovascularization [111]. Following DNA damage, p53 is activated and induces transcription of genes such as p21Waf1/Cip1 that function as regulators of cell cycle progression at G1 phase [10,112,113]. The MDM2 gene, at chromosome 12q14.3-q15, encodes a putative transcription factor and enhances the tumorigenic potential of cells when it is overexpressed. Forming a tight complex with the p53 gene, the MDM2 oncogene inhibits p53 transcriptional activity by binding to the N-terminal transactivation domain, and participates in the ubiquitination and proteasomal degradation of p53 [114,115]. Conversely, the transcription of the MDM2 gene is induced by wild-type p53 [116,117]. This autoregulatory feedback loop regulates the expression of MDM2 and the activity of p53. The p14ARF (a part of the complex CDKN2A locus on chromosome 9p21) gene encodes a protein that directly binds to MDM2 and inhibits MDM2-mediated p53 degradation and transactivational silencing [113,118-120]. In turn, p14ARF expression is negatively regulated by p53 [113]. Thus, inactivation of p14ARF/MDM2/p53 is caused by altered expression of any of the p53, MDM2, or p14ARF genes (Figure 7).
The p53 pathway plays a crucial role in the development of secondary GBMs. IDH1 gene mutations at chromosome 2q33, which were identified in an analysis of 20,661 protein-coding genes in GBMs [1], are early events in the development of astrocytomas and constitute a remarkably reliable molecular signature of secondary GBMs as well as their precursor lesions [122-124]. The additional acquisition of a p53 mutation, which is a genetic event subsequent to the IDH1/2 mutations except in the case of Li-Fraumeni syndrome [125], may lead to astrocytic differentiation, while subsequent loss of 1p/19q favors the acquisition of an oligodendroglial phenotype [123,126]. GBMs with IDH1/2 mutations frequently have p53 mutations, which are significantly associated [122]. p53 mutations are also common in two-thirds of precursor low-grade diffuse astrocytomas; this frequency is similar to that in anaplastic astrocytomas and secondary GBMs [28,84,127]. p53 mutations also occur in primary GBMs, but at a significantly lower frequency (approx. 25%) [128]. In secondary GBMs, 57% of p53 mutations are located in the hotspot codons 248 and 273; however, in primary GBMs, mutations are more equally distributed through all exons, with only 17% occurring in codons 248 and 273. The less-specific pattern of p53 mutations may constitute, at least in part, secondary events owing to increasing genomic instability during tumor development.
The p53 gene is the most commonly mutated p53 pathway gene in glioma; however, molecular abnormalities involving other genes in the pathway—Such as p14ARF, MDM2, or MDM4—have also been described. MDM2 amplification is observed in about 10% of GBMs, exclusively in primary GBMs that lack a p53 mutation [12,129]. Loss of p14ARF expression is frequently present in GBMs. p14ARF appears to be associated mostly with aberrant promoter methylation or hemizygous deletion, whereas mutational inactivation is rare [130,131]. Promoter methylation of p14ARF is more frequent in secondary than primary GBMs, but there is no significant difference in the overall frequency of p14ARF alterations between GBM subtypes [131].
7.2.2 ATM/Chk2/p53 pathway
Recently, in addition to the p14ARF/MDM2/p53 pathway, the ATM/Chk2/p53 pathway has come under the spotlight. Squatrito et al. reported that loss of the ATM/Chk2/p53 pathway components accelerates glioma development and contributes to radiation resistance [132]. In response to ionizing radiation, cells activate the sensor kinases ATM, ATR, and DNA-PK, a DNA-dependent protein kinase [133,134] that phosphorylates multiple downstream mediators, including the checkpoint kinases Chk1 and Chk2 [135,136], resulting in cell-cycle checkpoint initiation and/or apoptosis. Chk2, at chromosome 22q12.1, can regulate p53-dependent apoptosis in an ATM-independent manner as a tumor suppressor [137,138] (Figure 7). Although previous studies reported no or low frequency of Chk2 mutations (approx. 6%) [139,140], 22% of glioma patients in the TCGA study presented single-copy loss of the chromosomal region containing Chk2, with a significant reduction of Chk2 mRNA, suggesting that it might represent an important tumor suppressor in a subset of glioma patients [1,132].
7.3. RB Pathway
The RB pathway suppresses cell cycle entry and progression, as well as the p53 pathway. The 107-kDa RB1 protein encoded by RB1 (at 13q14) controls progression through G1 into the S-phase of the cell cycle [9]. The CDKN2A (p16INK4a) protein binds to CDK4 and inhibits the CDK4/cyclin D1 complex, thus inhibiting cell cycle transition from G1 to S phase (Figure 7) [10,12]. Thus, alteration of RB1, CDK4, or CDKN2A can cause dysregulation of the G1-S phase transition. Inactivation of this pathway is commonly observed in both primary and secondary GBMs [12]. Genetic loss of RB1 (40%), homozygous deletion of CDKN2A (40%), and/or CDK4 amplification (15%) are detected in the majority of GBM (80%) [11,141], and appear to be roughly mutually exclusive [11,12,142-144]. The TCGA pilot project revealed that the frequency of genetic alterations in the RB signaling pathway was 77%, containing CDKN2A homozygous deletion or mutations (52%), CDKN2B (p15INK4b) homozygous deletion (47%), CDKN2C (p18INK4c) homozygous deletion (2%), CDK4 amplification (18%), CCND2 (cyclin D2) amplification (2%), CDK6 amplification (1%), and RB1 mutation or homozygous deletion (11%) [1]. Alteration of only the Rb pathway is insufficient to induce tumor formation. EGFR amplification enhances the PI3K pro-growth pathway and is typically associated with CDKN2A deletions [145,146]. CDKN2A loss is associated with the classical subtype of GBM, according to the TCGA study [1].
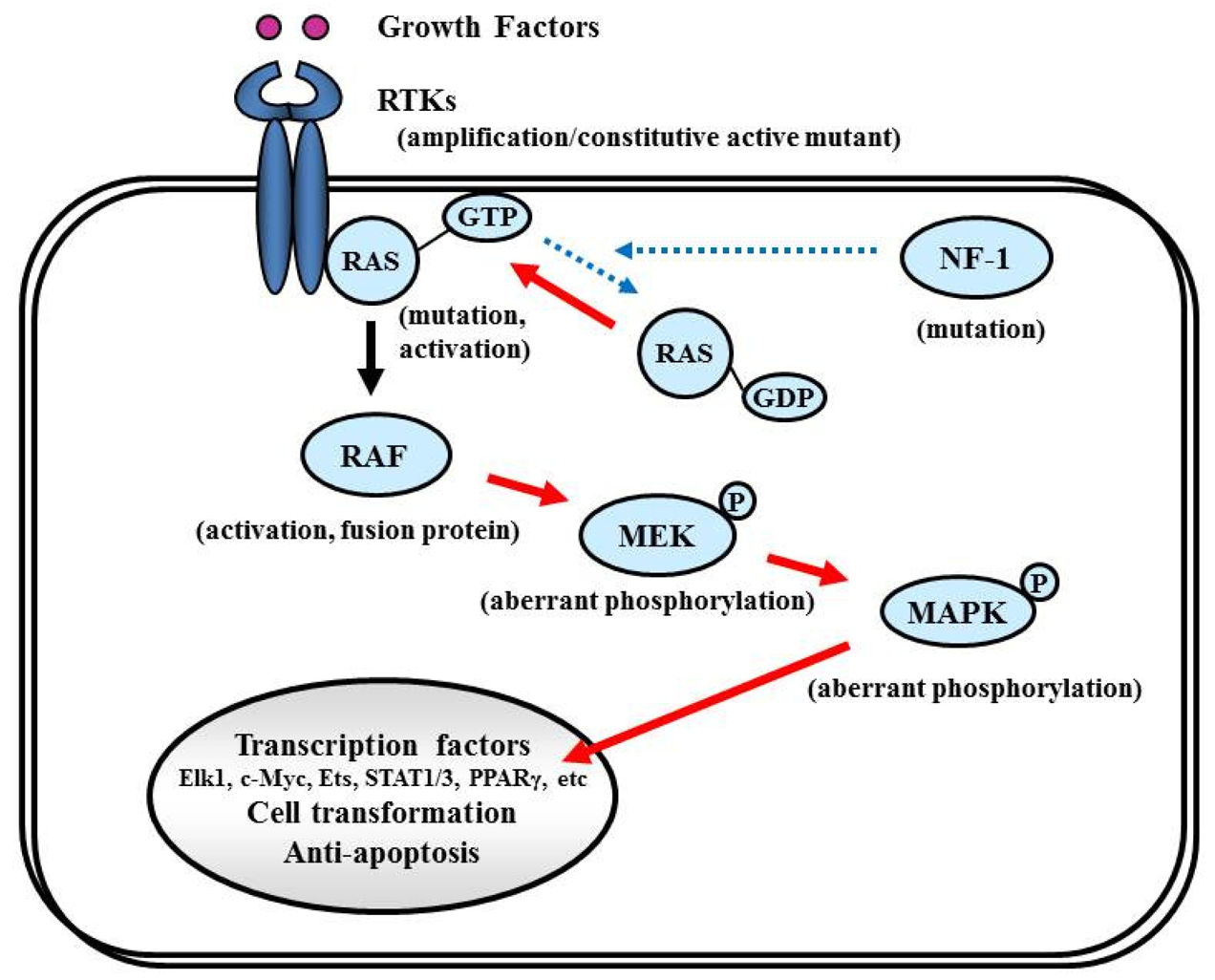
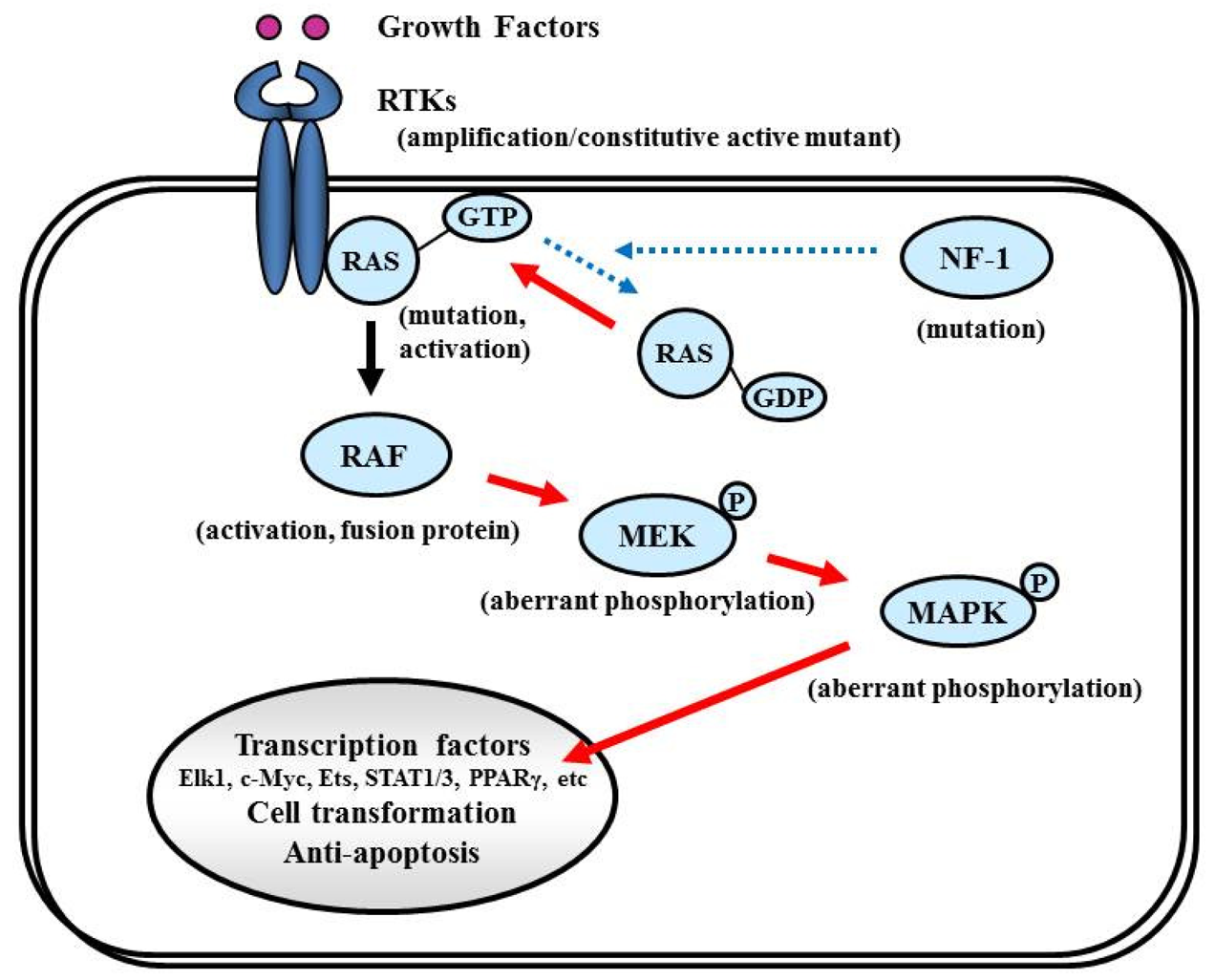
7.4. Ras/MAPK Pathway
7.4.1. NF-1
The NF-1 tumor suppressor gene encodes neurofibromin, which functions primarily as a RAS negative regulator and plays a role in adenylate cyclase- and Akt-mTOR-mediated pathways [147,148] (Figure 8). There is increasing evidence that the NF-1 gene is involved in the tumorigenesis of not only NF-1-related but also sporadically occurring gliomas. In the TCGA pilot study, NF-1 mutation/homozygous deletions were identified in 18% of GBM [1]. Mesenchymal GBMs, having frequent inactivation of the NF-1 (37%), p53 (32%), and PTEN genes, respond to aggressive chemo-radiation adjuvant therapies [90].
7.4.2. RAS
RAS activates the STKs Raf, MAPK (ERK1 and ERK2), PI3K, and a number of proteins that translocate to the nucleus to promote cell proliferation, differentiation, and survival (Figure 8). The RAS gene family comprises four different genes that encode HRAS, NRAS, KRAS4A, and KRAS4B [149,150]. Combined activation of RAS and Akt in neural progenitors induces GBM formation in a mouse model [46]. Although activating RAS mutations are observed in approximately 30% of human cancers [149], the RAS mutations are rare in human GBM (2%, according to the TCGA study [1]). Therefore, the observed deregulation of the Ras/RAF/MAPK signaling pathway in GBM is attributed to its upstream positive regulators, including EGFR and PDGFR, which are highly active in the majority of malignant gliomas [87,151].
7.4.3. RAF
Activated RAS recruits RAF kinase to the membrane, where it is activated by multiple phosphorylation events (Figure 8). The RAF family of STKs, which consist of ARAF, BRAF, and CRAF1, play an important role in proliferative signaling. In pediatric low-grade glioma, consisting of pilocytic astrocytoma and fibrillary astrocytoma, chromosome 7q34 rearrangements result in an in-frame KIAA1549:BRAF fusion gene that possesses constitutive BRAF kinase activity resembling oncogenic BRAF (V600E) [152]. In contrast, RAF mutations or rearrangements are rare in malignant gliomas in adults. A phase I/II clinical trial is currently underway for recurrent GBM with combined Raf and mTOR inhibiters, Sorafenib (NexavarR, Bayer, and Onyx, Emeryville, CA, USA) and temsirolimus (CCI-779) (NCT00335764).
7.4.4. MEK/MAPK
Activated RAF phosphorylates and activates MAPK kinase (MAPKK), also called MEK, which in turn phosphorylates and activates MAPK [153]. Activated MAPK translocates into the nucleus to activate several transcription factors such as Elk1, c-myc, Ets, STAT (signal transducers and activators of transcription) 1/3, and PPARγ (peroxisome proliferator-activated receptor γ), which induce cell cycle progression and anti-apoptosis genes [154,155] (Figure 8). Aberrant signaling through this pathway leads to cell transformation and resistance to apoptosis; thus, this pathway is an attractive target for malignant glioma therapy [156].
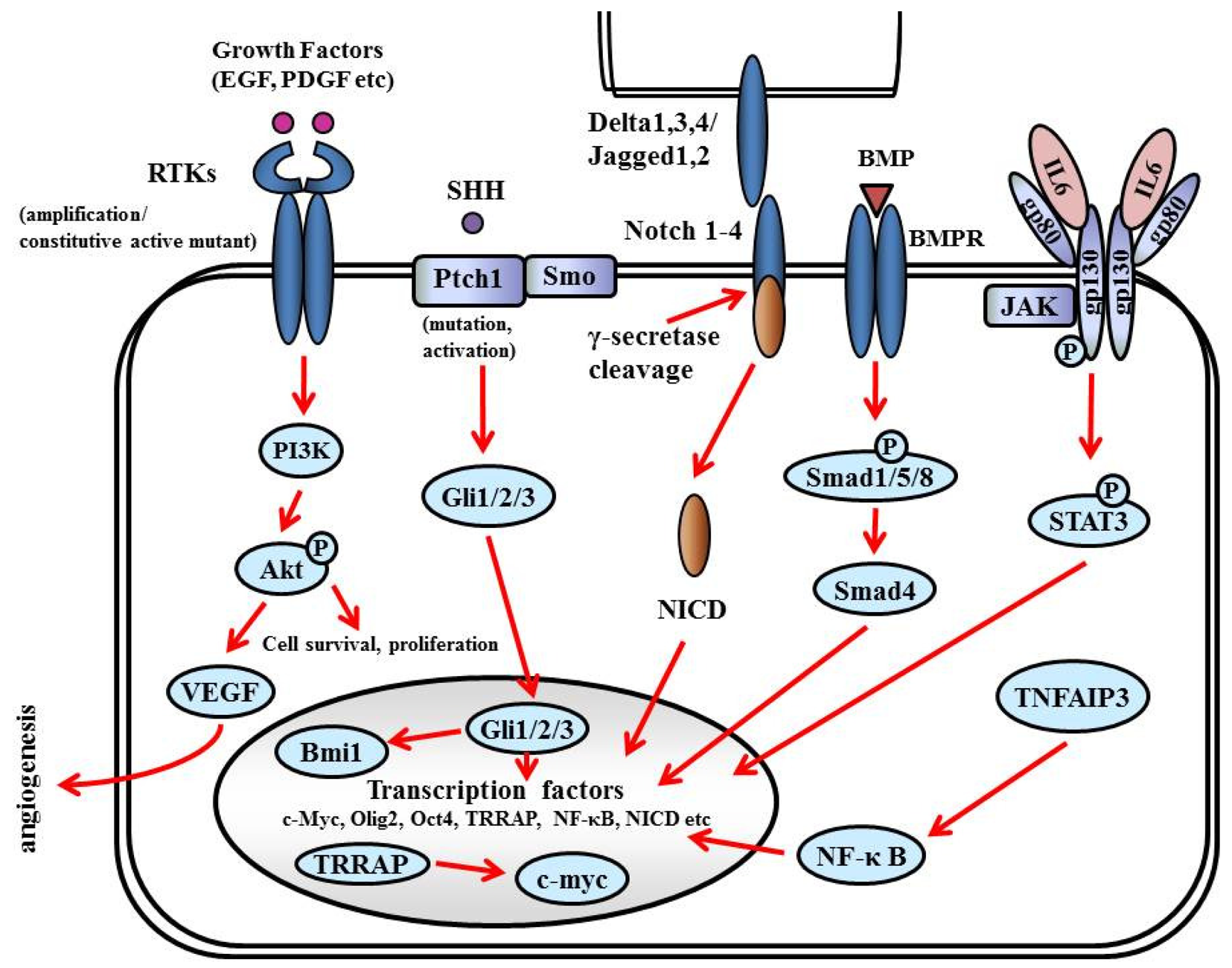
7.5. Glioma Stem Cell (GSC) Pathway
GSCs are distinguished by (i) the ability to self-renew, (ii) the ability to initiate brain tumors upon orthotopic implantation in immunodeficient mice, (iii) the expression of neural stem cell markers, and (iv) multipotency—The capacity to differentiate into cells with a neuronal, astrocytic, or oligodendroglial phenotype. GSCs express antigens specific for neural stem and progenitor cells: Nestin, CD133 (prominin-1), Musashi-1, and Bmi-1 [157-159]. CD133+ cells comprise 5%–30% of the tumor cell population, and as few as 100 cells can reproduce tumors in animal models [160]. Sonic hedgehog homolog (SHH) and Notch are key regulators of neural progenitors and have been found to be altered or overexpressed in GSCs [161] (Figure 9).
7.5.1. RTKs
RTKs mediate the effects of multiple oncogenic growth factor pathways. Ligand binding to RTK activates PI3K. Activated PI3K activates PDK1 which phosphorylates Akt, which transduces signals for cell survival, proliferation, motility, and angiogenesis as shown in Figure 6. PDGF signaling in neural stem cells is required for oligodendrogenesis, and amplification of this signal induces the aberrant proliferation of neural stem cells and the formation of large glioma-like lesions [162].
7.5.2. SHH
SHH is a critical mitogen for medulloblastoma precursor cells. Hereditary loss of function mutations in the SHH receptor, Patched (PTCH) lead to constitutive activation of the SHH pathway and predisposition to medulloblastoma in Gorlin syndrome. The SHH pathway is also activated in GSCs; the Hedgehog inhibitor cyclopamin depleted clonogenic GBM stem cells in vitro and inhibited tumor initiation in vivo [163]. The binding of SHH ligands to their receptors activates transducers termed Gli (named for their discovery in gliomas), which translocate into the nucleus and activate (Gli1/2) or repress (Gli3) downstream targets. The SHH pathway is also connected to cell cycling since it inactivates RB1, facilitating the over-expression of cell cycle regulators such as N-myc. Smoothened (SMO) is a downstream protein in the SHH pathway. In the absence of ligand, SMO is inhibited by PTCH1. A current theory suggests that PTCH regulates SMO by removing oxysterols. The binding of SHH relieves SMO inhibition, leading to activation of the GLI transcription factors. Bmi1, a promoter of neural stem cell self-renewal and neural development, is expressed in most gliomas and promotes self-renewal of GSC [164].
7.5.3. Notch
Notch (Notch1-4 in mammals) is a family of transmembrane receptors that regulate intercellular signaling [165]. Notch ligands (Delta-like1, Delta-like3, and Delta-like4, and Jagged1–2 in mammals) are also transmembrane proteins and, when bound to Notch, expose the receptor to proteolytic activation. Presenilins cleave Notch to generate a Notch1 intracellular domain (NICD), which translocates to the nucleus to act as a transcriptional activator. Notch activation induces expression of downstream target genes, such as p53, and promotes neural stem cell growth [166]. The Notch pathway has been targeted using γ-secretase inhibitors (GSIs), depleting CD133-positive GSCs, and inhibiting tumor initiation and growth in vivo [167].
7.5.4. Bone Morphogenetic Proteins (BMPs)
BMPs are a family of growth factors named for their central role in bone and cartilage formation. Most BMPs elicit their actions through binding to cell-surface receptor kinases (BMPRs). The effectors of BMPRs are the Smad proteins. Activating phosphorylation of Smad1/5/8 enables these proteins to bind to the co-activator Smad4, translocate into the nucleus, and regulate transcription. BMP ligands deplete the GSC population by inducing the differentiation of GSCs into astroglial and neuron-like cells. Treating GSCs with BMPs in vivo delays tumor growth and reduces tumor invasion [168].
7.5.5. IL6
The IL6 signal transduction pathway is initiated by IL6 ligand binding to heteromeric plasma membrane receptor complexes formed from a specific IL6 binding receptor, IL6 receptor α (IL6Rα, gp80), and the common signal transducing receptor glycoprotein 130 (gp130). Upon receptor activation, intracellular signaling is propagated by Janus kinase (JAK) tyrosine kinase family members, leading to activation of transcription factors of the signal transducers and activators of transcription (STAT) family, particularly STAT3 [169]. STAT3 activation, as indicated by phosphorylation at tyrosine705, is present in glioma patient samples and increases with tumor grade [170]. IL6 signals promote STAT3 activation in GBM cells in vitro, and targeting either STAT3 or IL6 decreases GBM cell survival [171]. STAT3 is also linked to stem cell biology, as STAT3 is required to maintain pluripotency of normal embryonic stem cells and neural stem cells [172].
7.5.6. Tumor Necrosis Factor alpha-Induced Protein (TNFAIP) 3
TNFAIP3 is a regulator of the NF-κB pathway that is overexpressed in GBM stem cells in comparison to non-stem tumor cells. Knockdown of TNFAIP3 by lentiviral shRNA reduces glioma stem cell self-renewal, growth, and apoptotic resistance [173].
7.5.7. Transcription factors
Transcription factor Olig2 is expressed in oligodendroglia and in mitogen-treated “transit-amplifying cells” of the subventricular zone, the presumed site of most adult neural stem/progenitor cells, and a hypothesized site for the origin of gliomas [162]. Olig2 is almost universally found in NG2-positive glia and is required for development of these cells [174]. NG2 is a chondroitin sulfate proteoglycan that is thought to be another marker of oligodendrocyte progenitor cells. Olig2 promotes the proliferation of both neural progenitors and GBM stem cells by repressing the p21 tumor suppressor [175].
Transformation/Transcription Domain-Associated Protein (TRRAP) is an ATM/R-related pseudokinase that scaffolds several histone acetyltransferase (HAT) complexes. TRRAP plays a central role in c-myc transcription activation; it is required for p53-mediated transcription activation. Knockdown of TRRAP sensitizes GSCs to apoptotic stimuli, decreases self-renewal and proliferation, and suppresses tumor formation in vivo, suggesting that TRRAP maintains GSCs in a stem cell-like state [176].
7.5.8. MicroRNA (miRNA)
miRNA is a small non-coding RNA that post-transcriptionally downregulates gene expression during apoptosis, differentiation, and development. Several studies identified aberrant miRNA (microRNA-21, miR-326, microRNA-34a) expression in gliomas, and linked some of them to GSC maintenance and growth [177-179].
8. Molecular Targets
Based on the recent progress of molecular biology research in GBM, efforts are currently underway to examine therapeutic targeting of molecules in signaling pathways with the hope of developing effective novel treatments for malignant glioma. Because of their frequent activation or mutation in human GBM and their paramount role in the maintenance of proliferation signaling, both EGFR and PDGFR are plausible targets for molecular therapies. However, clinical trials with drugs targeting EGFR or PDGFR, so far, have yielded disappointing results. However, according to the individual genetic analysis in this clinical trial, the carriers of variant EGFR (EGFRvIII) and wild-type PTEN showed good response to the inhibitors [106]. This example demonstrates why molecular targeting therapies represent a therapeutic strategy of great clinical interest for the treatment of GBM, especially when the inhibitors are rationally selected. So far, VEGF inhibitor (bevacizumab) and integrin inhibitor (cilengitide) have yielded good results in Phase II clinical trials and have moved ahead to Phase III trials.
8.1. Bevacizumab
Because of its prominent role in glioma angiogenesis, the VEGF pathway is widely identified as an attractive therapeutic target. Treatment strategies targeting angiogenesis signaling cascades have demonstrated promising results in preclinical studies. In these strategies, targeting the VEGF pathway has yielded the most clinically advanced anti-angiogenic drugs [180]. Several VEGF-targeting approaches are under clinical investigation in malignant gliomas.
One of the most well established anti-angiogenic medicines is bevacizumab, which is an IgG1 monoclonal antibody against free VEGF-A in the circulation, preventing attachment to the VEGF receptor and activation of a pro-angiogenic stimulus. Bevacizumab was originally developed for use in colorectal cancer, and has been approved by the European Medicines Agency for treatment in metastatic breast and kidney cancers [181,182]. Bevacizumab, alone or in combination with chemotherapy, has been associated with reduction in vasogenic brain edema and prolonged progression-free survival. Vredenburgh et al. reported the Phase II trial data of recurrent GBM patients who received bevacizumab plus irinotecan every two weeks. The 6-month progression-free survival among all 35 patients was 46%; the 6-month overall survival was 77% [183]. According to the good radiological response and decreasing corticosteroid use, the Food and Drug Administration (FDA) approved bevacizumab in 2009 as a therapy for recurrent GBM [184]. A recent clinical trial reported a 16% 2-year survival rate in recurrent GBM with bevacizumab monotherapy [3]. An international clinical trial is ongoing comparing radiochemotherapy with temozolomide and radiochemotherapy with temozolomide and bevacizumab. Based on the result, bevacizumab may be added as the standard therapy. However, recent data suggest that not all vessel formation in glioma depends on VEGF; non-endothelial cell-lined blood vessels are formed by tumor cells, indicating that some GBMs may grow in the absence of endothelial cell recruitment [185,186].
Although the response rate to anti-angiogenic agents targeting VEGF for GBM is 40%–60%, effective responses are commonly transient. Some glioma patients may exhibit primary refractoriness to the agents targeting VEGF. As yet, anti-angiogenic therapy is one of the most effective treatments for glioma patients; further understanding of the mechanisms of resistance to anti-angiogenic therapies and better patient selection will be crucial to improve outcomes for patients with GBM.
8.2. Cilengitide
The integrin family of cell adhesion receptors is emerging as a promising target of anticancer therapy. α3β1, αvβ1, αvβ3, and αvβ5 integrins are overexpressed on both glioma cells and tumor vasculature. Cilengitide, the most advanced specific αvβ3 and αvβ5 integrin inhibitor in oncology, has shown antitumor activity against glioma in early clinical trials.
Obvious benefits have been observed in phase I and phase II trials with cilengitide. A phase II clinical trial using cilengitide for recurrent GBM reported 23% 2-year survival and 10% 4-year survival [3]. In the clinical trial using cilengitide in conjunction with standard chemoradiotherapy for newly diagnosed GBM, progression-free survival was eight months and overall survival was 16.1 months [187]. Cilengitide is now being explored in prospective randomized phase III (CENTRIC) trials in the first-line setting in combination with standard radiochemotherapy.
8.3. Multi-Kinase Inhibitors
Multiple RTKs are concomitantly activated in GBMs in vitro and in vivo. Redundant inputs drive and maintain downstream signaling [188]. This finding can explain the weak clinical responses to RTK-inhibitor monotherapy. There are commonly multiple target molecules in individual GBM. Multi-kinase inhibitors are being developed with the expectation of more favorable outcomes against GBM. Dasatinib is an inhibitor targeting c-Src, c-kit, PDGFR, and Bcr-Abl. Enzastaurin is an STK inhibitor that targets protein kinase C and PI3K/Akt. Both agents are now in clinical trials.
9. Conclusions and Perspectives
Recent studies have highlighted the critical pathological roles played by various signaling networks and complex signaling crosstalk in the progression of glioma. It is important to clarify the dominant signaling pathways and crosstalk in order to establish effective molecular targeted therapies against GBM.
Future studies are necessary to delineate the signaling elements implicated in the positive and negative regulation of the malignant phenotype of GBM. In particular, it will be important to identify the factors involved in the enhancement of oncogenic signaling and potential interactive crosstalk with other oncogenic pathways during glioma progression. These studies should lead to the identification of new potential targets, optimized choice of therapeutic regimens, and personalized medicine, as well as the development of novel effective molecular targeted strategies that could be used to improve current GBM therapies.
The number of clinical trials for GBM patients will increase as new targets are discovered. Most importantly, well-designed clinical trials should be planned because the incidence of GBM is relatively low compared with other carcinomas. Furthermore, surgical tissue should be accessed at relevant time points to analyze pathway activation biomarkers. Ideally, GBM will be categorized by its molecular profile, to identify patients who may be expected to respond to certain molecular targeted drugs will be appropriately selected for clinical trials. In the future, the concept of “tailor-made molecular targeted therapy” against GBM will become reality.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Angiogenic factors | Function |
|---|---|
| VEGF-A, -B, -C and -D | Angiogenesis, promotes endothelial proliferation and migration, mitosis of endothelial cells and creation of blood vessel lumen |
| VEGFR1 | Hematopoiesis, promotes tumor angiogenesis, participates in the activation of MMP |
| VEGFR2 | Mediates the mitogenic, angiogenic and permeability-enhancing effects of VEGF-A |
| Angiopoietin | Induces stabilization, remodeling and maturation of blood vessels |
| Acidic FGF and basic FGF | Resists apoptosis of endothelial cells, induces endothelial cell proliferation and migration |
| PDGF | Recruits of pericytes and facilitates migration of pericytes into newly formed blood vessels |
| EGF | Stimulates VEGF production in glioma cells |
| TGF-β | Participates in regulator of proliferation, migration, differentiation and ECM synthesis in endothelial cells |
| SF/HGF | Promotes tumor growth and angiogenesis in vivo |
| IL-6 | Induces transcriptional activation of VEGF and regulates VEGF promoter activity |
| IL-8 | Exerts potent angiogenic properties on endothelial cells via the interaction with the CXC chemokine receptor 1 and 2 |
| TNF-α | Involved in systemic inflammation and acute phase reaction, induces tubular morphogenesis in vitro |
| IGF-1 | Involved in immunoreactivity in the tumor cells, promotes microvascular proliferation |
| Integrins | Responsible for the interaction of endothelial and tumor cells with ECM |
| MMP-2 and MMP-9 | Involved in the proteolytic degradation of ECM components and facilitate cell motility during invasion and angiogenesis |
HGF: hepatocyte growth factor; IGF, insulin-like growth factor; IL: interleukin; MMP: matrix metalloproteinase; PDGF: platelet-derived growth factor; SF: scatter factor; TGF: transforming growth factor; TNF: tumor necrosis factor; VEGF: vascular endothelial growth factor.
References
- Network, C.G.A.R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar]
- Stupp, R.; Weller, M. 2010: Neuro-oncology is moving! Curr. Opin. Neurol. 2010, 23, 553–555. [Google Scholar]
- Nagane, M. Neuro-oncology: Continuing multidisciplinary progress. Lancet Neurol. 2011, 10, 18–20. [Google Scholar]
- Nakada, M.; Nakada, S.; Demuth, T.; Tran, N.L.; Hoelzinger, D.B.; Berens, M.E. Molecular targets of glioma invasion. Cell. Mol. Life Sci. 2007, 64, 458–478. [Google Scholar]
- Kita, D.; Ciernik, I.F.; Vaccarella, S.; Franceschi, S.; Kleihues, P.; Lutolf, U.M.; Ohgaki, H. Age as a predictive factor in glioblastomas: Population-based study. Neuroepidemiology 2009, 33, 17–22. [Google Scholar]
- Ohgaki, H. Epidemiology of brain tumors. Methods Mol. Biol. 2009, 472, 323–342. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar]
- Ueki, K.; Ono, Y.; Henson, J.W.; Efird, J.T.; von Deimling, A.; Louis, D.N. CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res. 1996, 56, 150–153. [Google Scholar]
- Biernat, W.; Tohma, Y.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 1997, 94, 303–309. [Google Scholar]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar]
- Westermark, B.; Heldin, C.H.; Nister, M. Platelet-derived growth factor in human glioma. Glia 1995, 15, 257–263. [Google Scholar]
- Brat, D.J.; Mapstone, T.B. Malignant glioma physiology: Cellular response to hypoxia and its role in tumor progression. Ann. Int. Med. 2003, 138, 659–668. [Google Scholar]
- Louis, D.N. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. 2006, 1, 97–117. [Google Scholar]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar]
- Onishi, M.; Ichikawa, T.; Kurozumi, K.; Date, I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011, 28, 13–24. [Google Scholar]
- Chao, C.; Saito, S.; Kang, J.; Anderson, C.W.; Appella, E.; Xu, Y. p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. EMBO J. 2000, 19, 4967–4975. [Google Scholar]
- Sarkar, C.; Karak, A.K.; Nath, N.; Sharma, M.C.; Mahapatra, A.K.; Chattopadhyay, P.; Sinha, S. Apoptosis and proliferation: Correlation with p53 in astrocytic tumours. J. Neurooncol. 2005, 73, 93–100. [Google Scholar]
- Gomez-Manzano, C.; Fueyo, J.; Kyritsis, A.P.; McDonnell, T.J.; Steck, P.A.; Levin, V.A.; Yung, W.K. Characterization of p53 and p21 functional interactions in glioma cells en route to apoptosis. J. Natl. Cancer Inst. 1997, 89, 1036–1044. [Google Scholar]
- Leung, S.Y.; Yuen, S.T.; Chan, T.L.; Chan, A.S.; Ho, J.W.; Kwan, K.; Fan, Y.W.; Hung, K.N.; Chung, L.P.; Wyllie, A.H. Chromosomal instability and p53 inactivation are required for genesis of glioblastoma but not for colorectal cancer in patients with germline mismatch repair gene mutation. Oncogene 2000, 19, 4079–4083. [Google Scholar]
- Hamilton, S.R.; Liu, B.; Parsons, R.E.; Papadopoulos, N.; Jen, J.; Powell, S.M.; Krush, A.J.; Berk, T.; Cohen, Z.; Tetu, B.; et al. The molecular basis of Turcot's syndrome. N. Engl. J. Med. 1995, 332, 839–847. [Google Scholar]
- Hubbard, S.R. Structural analysis of receptor tyrosine kinases. Prog. Biophys. Mol. Biol. 1999, 71, 343–358. [Google Scholar]
- Hirata, A.; Hosoi, F.; Miyagawa, M.; Ueda, S.; Naito, S.; Fujii, T.; Kuwano, M.; Ono, M. HER2 overexpression increases sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, through inhibition of HER2/HER3 heterodimer formation in lung cancer cells. Cancer Res. 2005, 65, 4253–4260. [Google Scholar]
- Zwick, E.; Bange, J.; Ullrich, A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr.-Relat. Cancer 2001, 8, 161–173. [Google Scholar]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar]
- Watanabe, K.; Tachibana, O.; Sata, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996, 6, 217–223, discussion 223-214. [Google Scholar]
- Fleming, T.P.; Saxena, A.; Clark, W.C.; Robertson, J.T.; Oldfield, E.H.; Aaronson, S.A.; Ali, I.U. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992, 52, 4550–4553. [Google Scholar]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar]
- Baserga, R. The insulin-like growth factor I receptor: A key to tumor growth? Cancer Res. 1995, 55, 249–252. [Google Scholar]
- Morrison, R.S.; Yamaguchi, F.; Bruner, J.M.; Tang, M.; McKeehan, W.; Berger, M.S. Fibroblast growth factor receptor gene expression and immunoreactivity are elevated in human glioblastoma multiforme. Cancer Res. 1994, 54, 2794–2799. [Google Scholar]
- Hirose, Y.; Kojima, M.; Sagoh, M.; Hayashi, T.; Murakami, H.; Shimazaki, K.; Yoshida, K.; Kawase, T. Clinical importance of c-Met protein expression in high grade astrocytic tumors. Neurol. Med. Chir. (Tokyo) 1998, 38, 851–858, discussion 858-859. [Google Scholar]
- Macaulay, V.M. Insulin-like growth factors and cancer. Br. J. Cancer 1992, 65, 311–320. [Google Scholar]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.M.; Gladson, C.L. Targeting SRC in glioblastoma tumors and brain metastases: Rationale and preclinical studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nister, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Murphy, G.M., Jr.; Lee, Y.L.; Jia, X.C.; Yu, A.C.; Majewska, A.; Song, Y.; Schmidt, K.; Eng, L.F. Tumor necrosis factor-alpha and basic fibroblast growth factor decrease glial fibrillary acidic protein and its encoding mRNA in astrocyte cultures and glioblastoma cells. J. Neurochem. 1995, 65, 2716–2724. [Google Scholar]
- Takahashi, J.A.; Fukumoto, M.; Kozai, Y.; Ito, N.; Oda, Y.; Kikuchi, H.; Hatanaka, M. Inhibition of cell growth and tumorigenesis of human glioblastoma cells by a neutralizing antibody against human basic fibroblast growth factor. FEBS Lett. 1991, 288, 65–71. [Google Scholar]
- Sandberg-Nordqvist, A.C.; Stahlbom, P.A.; Reinecke, M.; Collins, V.P.; von Holst, H.; Sara, V. Characterization of insulin-like growth factor 1 in human primary brain tumors. Cancer Res. 1993, 53, 2475–2478. [Google Scholar]
- Platten, M.; Wick, W.; Weller, M. Malignant glioma biology: Role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech. 2001, 52, 401–410. [Google Scholar]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar]
- Bredel, M.; Pollack, I.F. The p21-Ras signal transduction pathway and growth regulation in human high-grade gliomas. Brain Res. Brain Res. Rev. 1999, 29, 232–249. [Google Scholar]
- ten Dijke, P.; Ichijo, H.; Franzen, P.; Schulz, P.; Saras, J.; Toyoshima, H.; Heldin, C.H.; Miyazono, K. Activin receptor-like kinases: A novel subclass of cell-surface receptors with predicted serine/threonine kinase activity. Oncogene 1993, 8, 2879–2887. [Google Scholar]
- Capra, M.; Nuciforo, P.G.; Confalonieri, S.; Quarto, M.; Bianchi, M.; Nebuloni, M.; Boldorini, R.; Pallotti, F.; Viale, G.; Gishizky, M.L.; et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res. 2006, 66, 8147–8154. [Google Scholar]
- Smith, J.S.; Tachibana, I.; Passe, S.M.; Huntley, B.K.; Borell, T.J.; Iturria, N.; O'Fallon, J.R.; Schaefer, P.L.; Scheithauer, B.W.; James, C.D.; et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J. Natl. Cancer Inst. 2001, 93, 1246–1256. [Google Scholar]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar]
- Basto, D.; Trovisco, V.; Lopes, J.M.; Martins, A.; Pardal, F.; Soares, P.; Reis, R.M. Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol. 2005, 109, 207–210. [Google Scholar]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocana, A.; Penuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11, 147–160. [Google Scholar]
- Smith, D.; Shimamura, T.; Barbera, S.; Bejcek, B.E. NF-kappaB controls growth of glioblastomas/astrocytomas. Mol. Cell. Biochem. 2008, 307, 141–147. [Google Scholar]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar]
- Plate, K.H.; Risau, W. Angiogenesis in malignant gliomas. Glia 1995, 15, 339–347. [Google Scholar]
- Roberts, W.G.; Palade, G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57, 765–772. [Google Scholar]
- Shibuya, M. Brain angiogenesis in developmental and pathological processes: Therapeutic aspects of vascular endothelial growth factor. FEBS J. 2009, 276, 4636–4643. [Google Scholar]
- Chi, A.S.; Sorensen, A.G.; Jain, R.K.; Batchelor, T.T. Angiogenesis as a therapeutic target in malignant gliomas. Oncologist 2009, 14, 621–636. [Google Scholar]
- Lamszus, K.; Ulbricht, U.; Matschke, J.; Brockmann, M.A.; Fillbrandt, R.; Westphal, M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clin. Cancer Res. 2003, 9, 1399–1405. [Google Scholar]
- Salmaggi, A.; Eoli, M.; Frigerio, S.; Silvani, A.; Gelati, M.; Corsini, E.; Broggi, G.; Boiardi, A. Intracavitary VEGF, bFGF, IL-8, IL-12 levels in primary and recurrent malignant glioma. J. Neurooncol. 2003, 62, 297–303. [Google Scholar]
- Ausprunk, D.H.; Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977, 14, 53–65. [Google Scholar]
- Zagzag, D.; Friedlander, D.R.; Dosik, J.; Chikramane, S.; Chan, W.; Greco, M.A.; Allen, J.C.; Dorovini-Zis, K.; Grumet, M. Tenascin-C expression by angiogenic vessels in human astrocytomas and by human brain endothelial cells in vitro. Cancer Res. 1996, 56, 182–189. [Google Scholar]
- Wong, M.L.; Prawira, A.; Kaye, A.H.; Hovens, C.M. Tumour angiogenesis: Its mechanism and therapeutic implications in malignant gliomas. J. Clin. Neurosci. 2009, 16, 1119–1130. [Google Scholar]
- Vajkoczy, P.; Knyazev, P.; Kunkel, A.; Capelle, H.H.; Behrndt, S.; von Tengg-Kobligk, H.; Kiessling, F.; Eichelsbacher, U.; Essig, M.; Read, T.A.; et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. USA 2006, 103, 5799–5804. [Google Scholar]
- Lund-Johansen, M.; Bjerkvig, R.; Humphrey, P.A.; Bigner, S.H.; Bigner, D.D.; Laerum, O.D. Effect of epidermal growth factor on glioma cell growth, migration, and invasion in vitro. Cancer Res. 1990, 50, 6039–6044. [Google Scholar]
- Hu, B.; Guo, P.; Fang, Q.; Tao, H.Q.; Wang, D.; Nagane, M.; Huang, H.J.; Gunji, Y.; Nishikawa, R.; Alitalo, K.; et al. Angiopoietin-2 induces human glioma invasion through the activation of matrix metalloprotease-2. Proc. Natl. Acad. Sci. USA 2003, 100, 8904–8909. [Google Scholar]
- Ram, R.; Lorente, G.; Nikolich, K.; Urfer, R.; Foehr, E.; Nagavarapu, U. Discoidin domain receptor-1a (DDR1a) promotes glioma cell invasion and adhesion in association with matrix metalloproteinase-2. J. Neurooncol. 2006, 76, 239–248. [Google Scholar]
- Paulus, W.; Baur, I.; Beutler, A.S.; Reeves, S.A. Diffuse brain invasion of glioma cells requires beta 1 integrins. Lab. Invest. 1996, 75, 819–826. [Google Scholar]
- Belot, N.; Rorive, S.; Doyen, I.; Lefranc, F.; Bruyneel, E.; Dedecker, R.; Micik, S.; Brotchi, J.; Decaestecker, C.; Salmon, I.; et al. Molecular characterization of cell substratum attachments in human glial tumors relates to prognostic features. Glia 2001, 36, 375–390. [Google Scholar]
- Fukushima, Y.; Ohnishi, T.; Arita, N.; Hayakawa, T.; Sekiguchi, K. Integrin alpha3beta1-mediated interaction with laminin-5 stimulates adhesion, migration and invasion of malignant glioma cells. Int. J. Cancer 1998, 76, 63–72. [Google Scholar]
- Ranuncolo, S.M.; Ladeda, V.; Specterman, S.; Varela, M.; Lastiri, J.; Morandi, A.; Matos, E.; Bal de Kier Joffe, E.; Puricelli, L.; Pallotta, M.G. CD44 expression in human gliomas. J. Surg. Oncol. 2002, 79, 30–35, discussion 35-36. [Google Scholar]
- Okamoto, I.; Kawano, Y.; Tsuiki, H.; Sasaki, J.; Nakao, M.; Matsumoto, M.; Suga, M.; Ando, M.; Nakajima, M.; Saya, H. CD44 cleavage induced by a membrane-associated metalloprotease plays a critical role in tumor cell migration. Oncogene 1999, 18, 1435–1446. [Google Scholar]
- Okamoto, I.; Tsuiki, H.; Kenyon, L.C.; Godwin, A.K.; Emlet, D.R.; Holgado-Madruga, M.; Lanham, I.S.; Joynes, C.J.; Vo, K.T.; Guha, A.; et al. Proteolytic cleavage of the CD44 adhesion molecule in multiple human tumors. Am. J. Pathol. 2002, 160, 441–447. [Google Scholar]
- Nagano, O.; Saya, H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004, 95, 930–935. [Google Scholar]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar]
- Chuang, Y.Y.; Tran, N.L.; Rusk, N.; Nakada, M.; Berens, M.E.; Symons, M. Role of synaptojanin 2 in glioma cell migration and invasion. Cancer Res. 2004, 64, 8271–8275. [Google Scholar]
- Salhia, B.; Rutten, F.; Nakada, M.; Beaudry, C.; Berens, M.; Kwan, A.; Rutka, J.T. Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res. 2005, 65, 8792–8800. [Google Scholar]
- Huang, P.H.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Uncovering therapeutic targets for glioblastoma: A systems biology approach. Cell Cycle 2007, 6, 2750–2754. [Google Scholar]
- Ramnarain, D.B.; Park, S.; Lee, D.Y.; Hatanpaa, K.J.; Scoggin, S.O.; Otu, H.; Libermann, T.A.; Raisanen, J.M.; Ashfaq, R.; Wong, E.T.; et al. Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells. Cancer Res. 2006, 66, 867–874. [Google Scholar]
- Sunayama, J.; Matsuda, K.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Demuth, T.; Ross, K.R.; Berens, T.; Coons, S.W.; Watts, G.; Trent, J.M.; et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: A cDNA microarray analysis. J. Neurooncol. 2001, 53, 161–176. [Google Scholar]
- Kita, D.; Yonekawa, Y.; Weller, M.; Ohgaki, H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 2007, 113, 295–302. [Google Scholar]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J. Cell Sci. Suppl. 1985, 3, 161–172. [Google Scholar]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar]
- Yamazaki, H.; Fukui, Y.; Ueyama, Y.; Tamaoki, N.; Kawamoto, T.; Taniguchi, S.; Shibuya, M. Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-erbB) in human brain tumors. Mol. Cell. Biol. 1988, 8, 1816–1820. [Google Scholar]
- Biernat, W.; Huang, H.; Yokoo, H.; Kleihues, P.; Ohgaki, H. Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol. 2004, 14, 131–136. [Google Scholar]
- Feldkamp, M.M.; Lala, P.; Lau, N.; Roncari, L.; Guha, A. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 1999, 45, 1442–1453. [Google Scholar]
- Barker, F.G., 2nd; Simmons, M.L.; Chang, S.M.; Prados, M.D.; Larson, D.A.; Sneed, P.K.; Wara, W.M.; Berger, M.S.; Chen, P.; Israel, M.A.; et al. EGFR overexpression and radiation response in glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 410–418. [Google Scholar]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar]
- Rich, J.N.; Rasheed, B.K.; Yan, H. EGFR mutations and sensitivity to gefitinib. N. Engl. J. Med. 2004, 351, 1260–1261, author reply 1260-1261. [Google Scholar]
- Haas-Kogan, D.A.; Prados, M.D.; Tihan, T.; Eberhard, D.A.; Jelluma, N.; Arvold, N.D.; Baumber, R.; Lamborn, K.R.; Kapadia, A.; Malec, M.; et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J. Natl. Cancer Inst. 2005, 97, 880–887. [Google Scholar]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar]
- Guha, A.; Dashner, K.; Black, P.M.; Wagner, J.A.; Stiles, C.D. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int. J. Cancer 1995, 60, 168–173. [Google Scholar]
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735. [Google Scholar]
- Reardon, D.A.; Egorin, M.J.; Quinn, J.A.; Rich, J.N.; Gururangan, S.; Vredenburgh, J.J.; Desjardins, A.; Sathornsumetee, S.; Provenzale, J.M.; Herndon, J.E., 2nd.; et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J. Clin. Oncol. 2005, 23, 9359–9368. [Google Scholar]
- Reardon, D.A.; Dresemann, G.; Taillibert, S.; Campone, M.; van den Bent, M.; Clement, P.; Blomquist, E.; Gordower, L.; Schultz, H.; Raizer, J.; et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br. J. Cancer 2009, 101, 1995–2004. [Google Scholar]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar]
- Workman, P. Inhibiting the phosphoinositide 3-kinase pathway for cancer treatment. Biochem. Soc. Trans. 2004, 32, 393–396. [Google Scholar]
- Furnari, F.B.; Huang, H.J.; Cavenee, W.K. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998, 58, 5002–5008. [Google Scholar]
- Choe, G.; Horvath, S.; Cloughesy, T.F.; Crosby, K.; Seligson, D.; Palotie, A.; Inge, L.; Smith, B.L.; Sawyers, C.L.; Mischel, P.S. Analysis of the phosphatidylinositol 3'-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 2003, 63, 2742–2746. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar]
- Tohma, Y.; Gratas, C.; Biernat, W.; Peraud, A.; Fukuda, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J. Neuropathol. Exp. Neurol. 1998, 57, 684–689. [Google Scholar]
- Haas-Kogan, D.; Shalev, N.; Wong, M.; Mills, G.; Yount, G.; Stokoe, D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998, 8, 1195–1198. [Google Scholar]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar]
- Bleeker, F.E.; Lamba, S.; Zanon, C.; van Tilborg, A.A.; Leenstra, S.; Troost, D.; Hulsebos, T.; Vandertop, W.P.; Bardelli, A. Absence of AKT1 mutations in glioblastoma. PLoS One 2009, 4, e5638. [Google Scholar]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a novel Akt inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [Google Scholar]
- Chang, S.M.; Wen, P.; Cloughesy, T.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; De Angelis, L.; Raizer, J.; et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest. New Drugs 2005, 23, 357–361. [Google Scholar]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F.; et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar]
- Bogler, O.; Huang, H.J.; Kleihues, P.; Cavenee, W.K. The p53 gene and its role in human brain tumors. Glia 1995, 15, 308–327. [Google Scholar]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar]
- Barak, Y.; Gottlieb, E.; Juven-Gershon, T.; Oren, M. Regulation of mdm2 expression by p53: Alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 1994, 8, 1739–1749. [Google Scholar]
- Zauberman, A.; Flusberg, D.; Haupt, Y.; Barak, Y.; Oren, M. A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res. 1995, 23, 2584–2592. [Google Scholar]
- Mao, L.; Merlo, A.; Bedi, G.; Shapiro, G.I.; Edwards, C.D.; Rollins, B.J.; Sidransky, D. A novel p16INK4A transcript. Cancer Res. 1995, 55, 2995–2997. [Google Scholar]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar]
- Watanabe, T.; Vital, A.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. Selective acquisition of IDH1 R132C mutations in astrocytomas associated with Li-Fraumeni syndrome. Acta Neuropathol. 2009, 117, 653–656. [Google Scholar]
- Kloosterhof, N.K.; Bralten, L.B.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar]
- Watanabe, K.; Sato, K.; Biernat, W.; Tachibana, O.; von Ammon, K.; Ogata, N.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin. Cancer Res. 1997, 3, 523–530. [Google Scholar]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar]
- Ichimura, K.; Bolin, M.B.; Goike, H.M.; Schmidt, E.E.; Moshref, A.; Collins, V.P. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res. 2000, 60, 417–424. [Google Scholar]
- Nakamura, M.; Watanabe, T.; Klangby, U.; Asker, C.; Wiman, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol. 2001, 11, 159–168. [Google Scholar]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar]
- Durocher, D.; Jackson, S.P. DNA-PK, ATM and ATR as sensors of DNA damage: Variations on a theme? Curr. Opin. Cell Biol. 2001, 13, 225–231. [Google Scholar]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar]
- Stracker, T.H.; Usui, T.; Petrini, J.H. Taking the time to make important decisions: The checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 2009, 8, 1047–1054. [Google Scholar]
- Hirao, A.; Kong, Y.Y.; Matsuoka, S.; Wakeham, A.; Ruland, J.; Yoshida, H.; Liu, D.; Elledge, S.J.; Mak, T.W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar]
- Hirao, A.; Cheung, A.; Duncan, G.; Girard, P.M.; Elia, A.J.; Wakeham, A.; Okada, H.; Sarkissian, T.; Wong, J.A.; Sakai, T.; et al. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol. Cell. Biol. 2002, 22, 6521–6532. [Google Scholar]
- Ino, Y.; Wahrer, D.C.; Bell, D.W.; Haber, D.A.; Louis, D.N. Mutation analysis of the hCHK2 gene in primary human malignant gliomas. Neurogenetics 2000, 3, 45–46. [Google Scholar]
- Sallinen, S.L.; Ikonen, T.; Haapasalo, H.; Schleutker, J. CHEK2 mutations in primary glioblastomas. J. Neurooncol. 2005, 74, 93–95. [Google Scholar]
- Schmidt, E.E.; Ichimura, K.; Reifenberger, G.; Collins, V.P. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994, 54, 6321–6324. [Google Scholar]
- Reifenberger, G.; Reifenberger, J.; Ichimura, K.; Meltzer, P.S.; Collins, V.P. Amplification of multiple genes from chromosomal region 12q13-14 in human malignant gliomas: Preliminary mapping of the amplicons shows preferential involvement of CDK4, SAS, and MDM2. Cancer Res. 1994, 54, 4299–4303. [Google Scholar]
- Nishikawa, R.; Furnari, F.B.; Lin, H.; Arap, W.; Berger, M.S.; Cavenee, W.K.; Su Huang, H.J. Loss of P16INK4 expression is frequent in high grade gliomas. Cancer Res. 1995, 55, 1941–1945. [Google Scholar]
- Burns, K.L.; Ueki, K.; Jhung, S.L.; Koh, J.; Louis, D.N. Molecular genetic correlates of p16, cdk4, and pRb immunohistochemistry in glioblastomas. J. Neuropathol. Exp. Neurol. 1998, 57, 122–130. [Google Scholar]
- Hayashi, Y.; Ueki, K.; Waha, A.; Wiestler, O.D.; Louis, D.N.; von Deimling, A. Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol. 1997, 7, 871–875. [Google Scholar]
- Hegi, M.E.; zur Hausen, A.; Ruedi, D.; Malin, G.; Kleihues, P. Hemizygous or homozygous deletion of the chromosomal region containing the p16INK4a gene is associated with amplification of the EGF receptor gene in glioblastomas. Int. J. Cancer 1997, 73, 57–63. [Google Scholar]
- Lee, M.J.; Stephenson, D.A. Recent developments in neurofibromatosis type 1. Curr. Opin. Neurol. 2007, 20, 135–141. [Google Scholar]
- Gottfried, O.N.; Viskochil, D.H.; Couldwell, W.T. Neurofibromatosis Type 1 and tumorigenesis: Molecular mechanisms and therapeutic implications. Neurosurg. Focus 2010, 28, E8. [Google Scholar]
- Bos, J.L. ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar]
- Guha, A.; Feldkamp, M.M.; Lau, N.; Boss, G.; Pawson, A. Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene 1997, 15, 2755–2765. [Google Scholar]
- Sievert, A.J.; Jackson, E.M.; Gai, X.; Hakonarson, H.; Judkins, A.R.; Resnick, A.C.; Sutton, L.N.; Storm, P.B.; Shaikh, T.H.; Biegel, J.A. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol. 2009, 19, 449–458. [Google Scholar]
- Adjei, A.A.; Hidalgo, M. Intracellular signal transduction pathway proteins as targets for cancer therapy. J. Clin. Oncol. 2005, 23, 5386–5403. [Google Scholar]
- Kapoor, G.S.; O'Rourke, D.M. Mitogenic signaling cascades in glial tumors. Neurosurgery 2003, 52, 1425–1434, discussion 1434-1425. [Google Scholar]
- Kapoor, G.S.; O'Rourke, D.M. Receptor tyrosine kinase signaling in gliomagenesis: Pathobiology and therapeutic approaches. Cancer Biol. Ther. 2003, 2, 330–342. [Google Scholar]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar]
- Jackson, E.L.; Garcia-Verdugo, J.M.; Gil-Perotin, S.; Roy, M.; Quinones-Hinojosa, A.; VandenBerg, S.; Alvarez-Buylla, A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 2006, 51, 187–199. [Google Scholar]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.K.; Kittappa, R.; McKay, R.D. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008, 13, 69–80. [Google Scholar]
- Kishimoto, T. Interleukin-6: From basic science to medicine—40 years in immunology. Annu. Rev. Immunol. 2005, 23, 1–21. [Google Scholar]
- Weissenberger, J.; Loeffler, S.; Kappeler, A.; Kopf, M.; Lukes, A.; Afanasieva, T.A.; Aguzzi, A.; Weis, J. IL-6 is required for glioma development in a mouse model. Oncogene 2004, 23, 3308–3316. [Google Scholar]
- Su, Y.; Li, G.; Zhang, X.; Gu, J.; Zhang, C.; Tian, Z.; Zhang, J. JSI-124 inhibits glioblastoma multiforme cell proliferation through G(2)/M cell cycle arrest and apoptosis augment. Cancer Biol. Ther. 2008, 7, 1243–1249. [Google Scholar]
- Matsuda, T.; Nakamura, T.; Nakao, K.; Arai, T.; Katsuki, M.; Heike, T.; Yokota, T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999, 18, 4261–4269. [Google Scholar]
- Hjelmeland, A.B.; Wu, Q.; Wickman, S.; Eyler, C.; Heddleston, J.; Shi, Q.; Lathia, J.D.; Macswords, J.; Lee, J.; McLendon, R.E.; et al. Targeting A20 decreases glioma stem cell survival and tumor growth. PLoS Biol. 2010, 8, e1000319. [Google Scholar]
- Ligon, K.L.; Kesari, S.; Kitada, M.; Sun, T.; Arnett, H.A.; Alberta, J.A.; Anderson, D.J.; Stiles, C.D.; Rowitch, D.H. Development of NG2 neural progenitor cells requires Olig gene function. Proc. Natl. Acad. Sci. USA 2006, 103, 7853–7858. [Google Scholar]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar]
- Wurdak, H.; Zhu, S.; Romero, A.; Lorger, M.; Watson, J.; Chiang, C.Y.; Zhang, J.; Natu, V.S.; Lairson, L.L.; Walker, J.R.; et al. An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell 2010, 6, 37–47. [Google Scholar]
- Corsten, M.F.; Miranda, R.; Kasmieh, R.; Krichevsky, A.M.; Weissleder, R.; Shah, K. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007, 67, 8994–9000. [Google Scholar]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; diPierro, C.G.; Li, Y.; Chiocca, E.A.; et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 2009, 29, 15161–15168. [Google Scholar]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. microRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle 2010, 9, 1031–1036. [Google Scholar]
- Chi, A.S.; Norden, A.D.; Wen, P.Y. Antiangiogenic strategies for treatment of malignant gliomas. Neurotherapeutics 2009, 6, 513–526. [Google Scholar]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol. 2008, 7, 1152–1160. [Google Scholar]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E., 2nd; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A new alternative mechanism in glioblastoma vascularization: Tubular vasculogenic mimicry. Brain 2010, 133, 973–982. [Google Scholar]
- Weller, M. Angiogenesis in glioblastoma: Just another moving target? Brain 2010, 133, 955–956. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.Y.; et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C.; et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J.-I. Aberrant Signaling Pathways in Glioma. Cancers 2011, 3, 3242-3278. https://doi.org/10.3390/cancers3033242
Nakada M, Kita D, Watanabe T, Hayashi Y, Teng L, Pyko IV, Hamada J-I. Aberrant Signaling Pathways in Glioma. Cancers. 2011; 3(3):3242-3278. https://doi.org/10.3390/cancers3033242
Chicago/Turabian StyleNakada, Mitsutoshi, Daisuke Kita, Takuya Watanabe, Yutaka Hayashi, Lei Teng, Ilya V. Pyko, and Jun-Ichiro Hamada. 2011. "Aberrant Signaling Pathways in Glioma" Cancers 3, no. 3: 3242-3278. https://doi.org/10.3390/cancers3033242
APA StyleNakada, M., Kita, D., Watanabe, T., Hayashi, Y., Teng, L., Pyko, I. V., & Hamada, J.-I. (2011). Aberrant Signaling Pathways in Glioma. Cancers, 3(3), 3242-3278. https://doi.org/10.3390/cancers3033242