X Inactivation and Progenitor Cancer Cells

{kind=link}
Abstract
: In mammals, silencing of one of the two X chromosomes is necessary to achieve dosage compensation. The 17 kb non-coding RNA called Xist triggers X inactivation. Gene silencing by Xist can only be achieved in certain contexts such as in cells of the early embryo and in certain hematopoietic progenitors where silencing factors are present. Moreover, these epigenetic contexts are maintained in cancer progenitors in which SATB1 has been identified as a factor related to Xist-mediated chromosome silencing.1. Introduction
The genome contains all the information required to generate the different cell types that comprise an organism. In multicellular organisms, regulation of cell identity and cell proliferation is exquisitely coordinated to produce the different cell types that form different tissues and organs.
In particular, embryonic stem (ES) cells can give rise to a broad range of specialized cells. The flexibility to regulate gene expression patterns in parallel with more dynamic chromatin behavior provides these cells with the necessary developmental plasticity.
A variety of adult stem cells that occupy different cell niches are responsible for the maintenance of tissues and organs. Furthermore, cell identity has to be maintained in these cell niches.
However, exactly how stem cells maintain their own cell identity in these niches, while still possessing the ability to generate the mature cells that compose an organism, is not clear. This may be partly explained by the concept of epigenetic context, which can be defined as the sum of all epigenetic pathways and mechanisms for a given cell or cell type. Depending on its state of differentiation, the cell has a certain epigenetic context that can either prevent or allow it to change its identity. When the cell differentiates, the epigenetic programming of differential gene expression based on different epigenetic mechanisms (e.g., DNA methylation) constantly changes.
When determining cell identity, the “marks” that can be used to define these different epigenetic contexts need to be identified.
The presence of two X chromosomes (XX) in female mammals and one (XY) in males raises the question of how to equalize the X linked genes. This is adjusted by chromosome-wide transcriptional silencing of one of the two X chromosomes in females [1]. This chromosome wide-silencing is initiated by Xist, which is a 17 kb untranslated RNA that coats the X chromosome in cis [1].
Interestingly, in mouse ES cells, the transition from an active X chromosome to an inactive X chromosome (Xi) can be followed [2]. X chromosome inactivation is divided into an INITIATION and a MAINTENANCE phase. Using genetic manipulation of Xist expression, Xist was shown to have X chromosome silencing function only in a defined epigenetic window (48–72 h) after ES cells induced differentiation [2]. Moreover, within this window, gene silencing is reversible and genes will be reactivated once Xist expression is lost [2].
Upon differentiation, and after the initiation phase of X inactivation, Xist RNA coating is not essential for the maintenance of X inactivation. In the maintenance phase, Xi becomes subject to further modifications such as DNA methylation and histone hypoacetylation, thus keeping genes repressed without needing the constant expression of Xist [2-4].
Thus, the shift from Xist-dependent initiation to stabilization of the inactive state could be regarded as a change of epigenetic context. As a consequence, it can be used as a “mark” to analyze changes in the epigenetic contexts of different cells during development [5].
Previous work has demonstrated that the silencing function of Xist is re-established in immature hematopoietic precursors [6]. However, whether such a transition in the Xist silencing ability exists in neoplastic cells is not clear. The present review discusses recent findings showing that the pathway that enables the silencing function of Xist is re-established in lymphoma cells [7]. Moreover, the review discusses how the study of epigenetic context transitions in cancer cells allowed the identification of the special AT-rich binding protein SATB1 as a silencing factor [7].
2. A Particular Epigenetic Context of Progenitors Defined by Xist Silencing Ability Is Maintained in Neoplastic Cells
Although X inactivation is initiated in the cells of the early embryo, experiments using tetracycline-inducible Xist transgenes showed the presence of cells in the adult mouse in which Xist can initiate gene silencing [6]. These cells are lineage-restricted progenitors within the hematopoietic system, and this phenomenon has been clearly established for preB cells and CD4+ and CD8+ double positive T-cells [6].
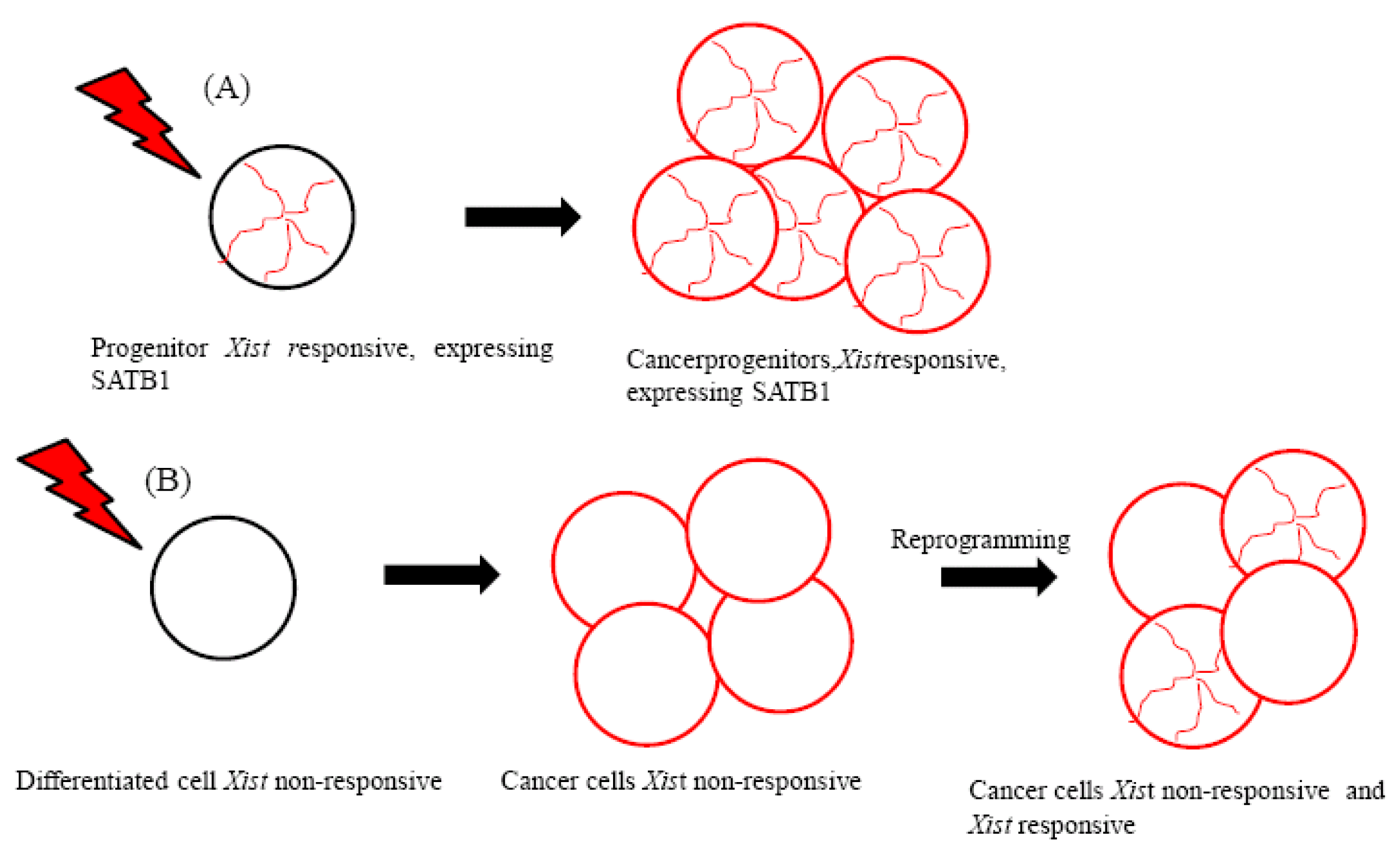
Interestingly, the hematopoietic stem cell (HSC) and mature blood cells do not have the cellular context for Xist-mediated silencing [6], which indicates that specific pathways used in the early embryo are reactivated transiently during the differentiation of the HSC. It could be argued that well-defined epigenetic pathways are shared among the early embryo and some kind of progenitor cells in which Xist can initiate chromosome-wide silencing [5]. A recent study demonstrated that Xist can trigger gene silencing in certain tumor cells [7]. Tumor formation in mice was induced by the expression of an oncogenic human lymphoma kinase fusion protein (NPM-ALK) that is expressed in the T cell compartment. This protein is generated from the fusion of the anaplastic lymphoma kinase (ALK) and the nucleophosmin gene (NPM/B23), and it is produced by a t(2;5) translocation in human anaplastic large cell lymphoma [8,9]. In this tumor model, ectopic induction of Xist expression caused X inactivation and suppressed tumor development [7].
Lymphoma cells derived from progenitors therefore maintain the epigenetic context that enables the initiation of Xist-mediated gene silencing (Figure 1A). These cells have been suggested to express silencing factors [7]. The special AT-rich binding protein (SATB1) was identified as an initiation factor for Xist-mediated silencing through a comparison of the expression profiles of Xist-responsive and Xist-resistant tumors [7]. Because SATB1 expression was found to be restricted in development, it has been proposed to characterize the cellular context for Xist-mediated silencing in ES cell differentiation and in T lymphocyte development [7].
SATB1 is a DNA binding protein that facilitates the organization of chromatin structure through its interaction with AT-rich DNA sequences.
SATB1 was first described in T cells, where SATB1 forms a network that overlaps the base of chromatin loops, circumscribing heterochromatin and regulating gene expression in a coordinate manner [10]. Moreover, it has the ability to recruit chromatin remodeling complexes to these anchorage sites, and is therefore capable of regulating histone modifications or nucleosomal positioning over large regions [11]. In addition, SATB1 activity is regulated by posttranslational modifications [12-14].
Recent and interesting work has shown that some of the genes SATB1 represses are upregulated by WNT signals [15]. The binding of WNT soluble proteins to their cell surface receptors initiates a signaling cascade that ultimately results in the accumulation of β-catenin in the nucleus, where it can bind and activate different transcription factors. In the absence of WNT signaling, β-catenin is phosphorylated and degraded. Direct interaction between SATB1 and β-catenin in Th2 cells has been detected. Moreover, β-catenin causes an increase in DNA binding by SATB1 by promoting its hypoacetilation (a modification that increases SATB1 affinity for DNA). Interestingly, β-catenin also binds to the same gene. Although SATB1 binds to DNA and subsequently recruits β-catenin to the DNA, once β-catenin is bound to SATB1 it can recruit additional proteins to help stimulate gene expression. β-catenin can modify the function of SATB1 from a repressor to an activator of gene function. In future studies, it would be interesting to investigate whether WNT signaling can influence Xist mediated gene silencing.
In summary, Xist-mediated gene silencing capacity is preserved in cancer progenitors in which SATB1 has been identified as a silencing factor.
SATB1 and Its Role in Xist-Mediated Silencing
The X chromosome comprises of a gene-rich outer rim and an internal core containing silenced non-genic sequences [5]. Upon silencing, genes are moved into the chromosome territory in a manner that is dependent on repeat A of Xist [16]. Immunolocalization studies in CD4+andCD8+ T cells in which SATB1 shows a ring- or cage-like staining pattern suggest that the sites where the SATB1 protein is localized do not overlap with Xist RNA domains. However, the induction of Xist RNA expression can change SATB1 localization patterns and SATB1 can influence Xist RNA localization. Based on these observations, it has been proposed that Xist can pull genes into the repressive compartment for gene silencing and that SATB1 may act as an anchor when this chromosome reorganization occurs. Interestingly, certain genes known as escapers remain external [7].
Moreover, it has been suggested that in the absence of SATB1, genes may be released to loop out from the Xist domain, and therefore oscillate between the Xist silencing domain and the transcription factories domain [17]. The association with transcription factories therefore results in gene transcription, and as a consequence, gene activity will continue despite the generation of a Xist RNA silencing domain [17]. The above model can explain why Xist is not able to silence outside narrow windows where SATB1 is expressed.
3. A Closer Look at This Particular Tumor Cell Context
Whether Xist-responsive cell types are exclusive to the embryo and the hematopoietic system, or if other adult stem cell niches such as skin are also characterized by re-establishing an appropriate epigenetic context, has not been elucidated. However, data suggest that the Xist-silencing pathway can be active in a wide variety of tumors. XIST expression is present in human testicular germ cell tumors, which is unusual considering that XIST is not normally expressed in male cells [18-20]. Moreover, the existence of multiple X chromosomes suggests that at a certain stage of tumor development, X inactivation must have been initiated [18-20]
On the other hand, an ectopic human XIST transgene has been found to induce chromosome inactivation in human HT-1080 fibrosarcoma cells [21] where SATB1 is expressed [22].
Importantly, SATB1 may be necessary for the epigenetic transition and reorganization of the genome in breast cancer cells that become highly metastatic [23-26] (Figure 1B). Supporting the role of SATB1 in promoting metastasis, SATB1 expression has been associated with the development and metastasis of bladder urothelial carcinoma and gastric carcinoma [27,28].
However, the findings of a recent study were in disagreement with previous results showing that SATB1 promotes metastasis in breast cancer [24]. Recently, it was suggested that this discrepancy may be due to differences in experimental conditions between these studies [25].
Taken together, these observations suggest that the context for Xist-mediated silencing might be found in a wider range of progenitors in different tissues, and in different cancer cells that may originate from them.
Following this line of thought, similarities between gene expression patterns of ES cells and poorly differentiated aggressive forms of cancer have been observed by bioinformatic analysis of expression data for a set of tumors [29]. Furthermore, chromatin organization and composition similarities between ES cells and tumor cells have also been reported [30].
As a result, it could be argued that tumor cells use chromatin regulation and epigenetic pathways that are normally active in ES cells.
4. Concluding Remarks
Analysis of the Xist gene silencing pathway could improve our understanding of the pathways that are active in narrow developmental windows in which cell identity is unstable and are conserved in neoplastic cells. One reason for restricting the activity of the silencing pathway could be to safeguard against illegitimate changes of cell identity that could lead to malignant transformation. The neoplastic cell could exploit these pathways, for example, to gain metastasizing potential. However, it still remains a challenge to analyze the Xist-silencing function in a wider spectrum of tumors to gain knowledge on how general the silencing context is in tumor formation. Furthermore, the possibility of classifying tumors according to Xist responsiveness is questionable. If possible, it would be important to characterize these tumors for metastatic potential and resistance to chemotherapy. Following this line of thought, recent observations have revealed that SATB1 expression is upregulated in multidrug-resistant breast cancer cells [31].
Further studies of these pathways could provide important information for the development of new therapies.
Acknowledgments
I would like to thank Anton Wutz for introducing me to the field of X inactivation and for generating such a great scientific atmosphere in his lab during all these years. I would also like to thank the reviewers of this manuscript for their interesting comments and useful suggestions.
This work was supported in part by a grant from the WWTF and the IMP through Boehringer Ingelheim and by Agencia Nacional de Investigacion e Inovacion (Program INNOVA URUGUAY-DCI-ALA/2007/19.040 URU-UE).
Reference
- Payer, B.; Lee, J.T. X chromosome dosage compensation: How mammals keep the balance. Ann. Rev. Genet. 2008, 42, 733–772. [Google Scholar]
- Wutz, A.; Jaenisch, R. A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol. Cell 2000, 5, 695–705. [Google Scholar]
- Brown, C.J.; Willard, H.F. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature 1994, 368, 154–156. [Google Scholar]
- Csankovszki, G.; Panning, B.; Bates, B.; Pehrson, J.R.; Jaenisch, R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 1999, 22, 323–324. [Google Scholar]
- Wutz, A. Xist function: Bridging chromatin and stem cells. Trends Genet. 2007, 23, 457–464. [Google Scholar]
- Savarese, F.; Flahndorfer, K.; Jaenisch, R.; Busslinger, M.; Wutz, A. Haematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Mol. Cell. Biol. 2006, 26, 7167–7177. [Google Scholar]
- Agrelo, R.; Souabni, A.; Novatchkova, M.; Haslinger, C.; Leeb, M.; Komnenovic, V.; Kishimoto, H.; Gresh, L.; Kohwi-Shigematsu, T.; Kenner, L.; et al. SATB1 defines the developmental context for gene silencing by Xist in lymphoma and embryonic cells. Dev. Cell 2009, 16, 507–516. [Google Scholar]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1994, 263, 1281–1284. [Google Scholar]
- Cai, S.; Lee, C.C.; Kohwi-Shigematsu, T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat. Genet. 2006, 38, 1278–1288. [Google Scholar]
- Yasui, D.; Miyano, M.; Cai, S.; Varga-Weisz, P.; Kohwi-Shigematsu, T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 2002, 419, 641–645. [Google Scholar]
- Pavan Kumar, P.; Purbey, P.K.; Sinha, C.K.; Notani, D.; Limaye, A.; Jayani, R.S.; Galande, S. Phosphorylation of SATB1, a global gene regulator, acts as a molecular switch regulating its transcriptional activity in vivo. Mol. Cell. 2006, 22, 231–243. [Google Scholar]
- Purbey, P.K.; Singh, S.; Notani, D.; Kumar, P.P.; Limaye, A.S.; Galande, S. Acetylation-dependent interaction of SATB1 and CtBP1 mediates transcriptional repression by SATB1. Mol. Cell. Biol. 2009, 29, 1321–1337. [Google Scholar]
- Tan, J.A.; Sun, Y.; Song, J.; Chen, Y.; Krontiris, T.G.; Durrin, L.K. SUMO conjugation to the matrix attachment region-binding protein, special AT-rich sequencebinding protein-1 (SATB1), targets SATB1 to promyelocytic nuclear bodies where it undergoes caspase cleavage. J. Biol. Chem. 2008, 283, 18124–18134. [Google Scholar]
- Notani, D.; Gottimukkala, K.P.; Jayani, R.S.; Limaye, A.S.; Damle, M.V.; Mehta, S.; Purbey, P.K.; Joseph, J.; Galande, S. Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol. 2010, 8, e1000296. [Google Scholar]
- Wutz, A.; Rasmussen, T.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar]
- Brockdorff, N. SAT in silence. Dev. Cell 2009, 16, 483–484. [Google Scholar]
- Kawakami, T.; Okamoto, K.; Ogawa, O.; Okada, Y. XIST unmethylated DNA fragments in male-derived plasma as a tumour marker for testicular cancer. Lancet 2004, 363, 40–42. [Google Scholar]
- Kawakami, T.; Okamoto, K.; Sugihara, H.; Hattori, T.; Reeve, A.E.; Ogawa, O.; Okada, Y. The roles of supernumerical X chromosomes and XIST expression in testicular germ cell tumors. J. Urol. 2003, 169, 1546–1552. [Google Scholar]
- Looijenga, L.H.; Gillis, A.J.; van Gurp, R.J.; Verkerk, A.J.; Oosterhuis, J.W. X inactivation in human testicular tumors. XIST expression and androgen receptor methylation status. Am. J. Pathol. 1997, 151, 581–590. [Google Scholar]
- Hall, L.L.; Byron, M.; Sakai, K.; Carrel, L.; Willard, H.F.; Lawrence, J.B. An ectopic human XIST gene can induce chromosome inactivation in postdifferentiation human HT-1080 cells. Proc. Natl. Acad. Sci. USA 2002, 99, 8677–8682. [Google Scholar]
- Agrelo, R. Institut Pasteur de Montevideo, Mataojo 2020, 1140 Montevideo, Uruguay, Unpublished results, 2011.
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar]
- Hnatyszyn, H.J.; Seo, P.; Clarke, J.; Ward, T.; Lippman, M. The role of SATB1 in breast cancer pathogenesis. J. Natl. Cancer Inst. 2010, 102, 1284–1296. [Google Scholar]
- Kohwi-Shigematsu, T.; Han, H.J.; Russo, J.; Kohwi, Y. Re: The role of SATB1 in breast cancer pathogenesis. J. Natl. Cancer Inst. 2010, 102, 1879–1880. [Google Scholar]
- Richon, V.M. A new path to the cancer epigenome. Nat. Biotechnol. 2008, 26, 655–656. [Google Scholar]
- Lu, X.; Cheng, C.; Zhu, S.; Yang, Y.; Zheng, L.; Wang, G.; Shu, X.; Wu, K.; Liu, K.; Tong, Q. SATB1 is an independent prognostic marker for gastric cancer in a Chinese population. Oncol. Rep. 2010, 4, 981–987. [Google Scholar]
- Liu, C.X.; Wen, Y.; Xu, K.; Zheng, S.B.; Xu, Y.W.; Chen, B.S. Expression of special AT-rich sequence-binding protein in bladder urothelial carcinoma and its clinical significance. Nan Fang Yi Ke Da Xue Xue Bao 2010, 30, 1389–1391. [Google Scholar]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 37, 239–342. [Google Scholar]
- Li, Q.Q.; Chen, Z.Q.; Xu, J.D.; Cao, X.X.; Chen, Q.; Liu, X.P.; Xu, Z.D. Overexpression and involvement of special AT-rich sequence binding protein 1 in multidrug resistance in human breast carcinoma cells. Cancer Sci. 2010, 101, 80–86. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Agrelo, R. X Inactivation and Progenitor Cancer Cells. Cancers 2011, 3, 2169-2175. https://doi.org/10.3390/cancers3022169
Agrelo R. X Inactivation and Progenitor Cancer Cells. Cancers. 2011; 3(2):2169-2175. https://doi.org/10.3390/cancers3022169
Chicago/Turabian StyleAgrelo, Ruben. 2011. "X Inactivation and Progenitor Cancer Cells" Cancers 3, no. 2: 2169-2175. https://doi.org/10.3390/cancers3022169
APA StyleAgrelo, R. (2011). X Inactivation and Progenitor Cancer Cells. Cancers, 3(2), 2169-2175. https://doi.org/10.3390/cancers3022169