Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy


Abstract
: Cellular FLICE (FADD-like IL-1beta-converting enzyme)-inhibitory protein (c-FLIP) is a major resistance factor and critical anti-apoptotic regulator that inhibits tumor necrosis factor-alpha (TNF-alpha), Fas-L, and TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis as well as chemotherapy-triggered apoptosis in malignant cells. c-FLIP is expressed as long (c-FLIPL), short (c-FLIPS), and c-FLIPR splice variants in human cells. c-FLIP binds to FADD and/or caspase-8 or -10 in a ligand-dependent and-independent fashion, which in turn prevents death-inducing signaling complex (DISC) formation and subsequent activation of the caspase cascade. Moreover, c-FLIPL and c-FLIPS are known to have multifunctional roles in various signaling pathways, as well as activating and/or upregulating several cytoprotective signaling molecules. Upregulation of c-FLIP has been found in various tumor types, and its downregulation has been shown to restore apoptosis triggered by cytokines and various chemotherapeutic agents. Hence, c-FLIP is an important target for cancer therapy. For example, small interfering RNAs (siRNAs) that specifically knockdown the expression of c-FLIPL in diverse human cancer cell lines augmented TRAIL-induced DISC recruitment and increased the efficacy of chemotherapeutic agents, thereby enhancing effector caspase stimulation and apoptosis. Moreover, small molecules causing degradation of c-FLIP as well as decreasing mRNA and protein levels of c-FLIPL and c-FLIPS splice variants have been found, and efforts are underway to develop other c-FLIP-targeted cancer therapies. This review focuses on (1) the functional role of c-FLIP splice variants in preventing apoptosis and inducing cytokine and drug resistance; (2) the molecular mechanisms that regulate c-FLIP expression; and (3) strategies to inhibit c-FLIP expression and function.1. Introduction
Cytotoxic anticancer agents continue to serve as the mainstay modality of systemic therapy in treating human malignancies that have disseminated from the primary tumor site and cannot be managed solely by surgical removal or radiation. The major limitation of cancer chemotherapy has proven to be drug resistance, whether acquired by the malignancy after transient disease remission (e.g., after breast cancer chemotherapy), or intrinsic to the malignancy (e.g., colon cancer, pancreatic cancer, glioblastoma, and prostate cancer are typically refractory to cancer chemotherapy). Several mechanisms have been found to cause chemotherapeutic resistance in cancer cells in vitro [1-4], but whether these mechanisms are also operative in the same manner in vivo requires further study. Understanding the mechanisms of resistance to chemotherapeutic agents will assist in the design of more effective strategies to overcome resistance in cancer cells.
Defects in apoptotic signaling and redundant survival mechanisms in malignant cells contribute to drug resistance in various cancer types [5,6]. Therefore, strategies to lower the threshold for triggering apoptosis in various cancers may lead to new and more effective therapeutic regimens. Acutely induced chemosensitization occurs when a pro-apoptotic signaling program induced in neoplastic cells by a chemotherapy drug includes disabling of a cytoprotective anti-apoptotic response. This is illustrated by our discovery that acute exposure of human leukemia cells to Taxol induced a pro-apoptotic program that entails coordinate caspase activation and downregulation of the anti-apoptotic protein cellular FLICE-like inhibitory protein (c-FLIP), a catalytically inactive caspase-8/-10 homologue [7]. c-FLIP variants are involved in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and chemotherapeutic drug resistance in a wide range of human malignancies [7-13]. The fact that Taxol has the added benefit of disabling a specific cytoprotective signal in neoplastic cells in conjunction with inducing apoptosis signaling is consistent with its often superior efficacy compared with other apoptosis-inducing chemotherapy drugs in managing diverse neoplastic diseases. Furthermore, a combination of Taxol/c-FLIP targeted therapy may improve the therapeutic response to Taxol by enhancing downregulation of c-FLIP variants in concert with drug-induced apoptosis signaling [7].
We have reported that upregulation of the pro-apoptotic TRAIL receptor DR5 may actually occur during the development of chemotherapy-induced drug resistance phenotype in cancer cells [14]. Moreover, upregulation of the pro-apoptotic signaling proteins or suppression of specific anti-survival signaling pathways by agents directed to increase pro-apoptotic proteins may acutely induce chemosensitization of resistant cancer cells. For instance, we previously demonstrated TRAIL treatment selectively triggered apoptosis in P-glycoprotein (P-gp ABCB1)-overexpressing multidrug resistant (MDR) cells [14-16]. Moreover, hypersensitivity to TRAIL was either due to (1) increased TRAIL binding to the TRAIL receptor DR5 in these cells compared to their drug sensitive counterparts [15]; or (2) up-regulation of DR5 and concomitant degradation of P-gp [14], the release of cytochrome c from mitochondria, activation of caspases-9 and -3 [14], as well as down-regulation of c-FLIP and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) by activation of caspase-3 [17]. These data also provided important determinants of TRAIL-induced sensitization of MDR cells to MDR-related agents [14,17]. Therefore, these results hold significant clinical implications for the use of TRAIL or TRAIL and chemotherapeutic drugs for treating cancers with the MDR phenotype. TRAIL holds enormous promise as a cancer therapeutic due to its highly selective apoptosis-inducing action on neoplastic versus normal cells [18,19]. Moreover, a recently published Phase I clinical trial revealed that recombinant TRAIL administration is safe and well tolerated, and that dose escalation achieved peak TRAIL serum concentrations equivalent to those associated with preclinical antitumor efficacy [20] However, to exploit the opportunity to successfully treat cancers with TRAIL, the problems of TRAIL resistance in a variety of tumor cells must first be overcome [21-23].
It is now recognized that the mechanism of action of chemotherapy drugs often involves the induction of cancer cell apoptosis, and that apoptosis resistance is a major contributing factor in chemotherapeutic drug resistance. Therefore, restoring apoptosis signaling in cancer cells with targeted therapeutics has enormous potential to improve the outcome of cancer chemotherapy by reversing a major mechanism of drug resistance. As we previously reported [24], c-FLIP is a critical target for therapeutic intervention aimed at inhibiting its transcription and posttranscription. In this review, we assess the outlook for improving the outcome of cancer therapy by targeting c-FLIP and exploring the possibility of increasing its degradation and/or decreasing its expression in order to provide a potentially safe approach to treat cancer. Novel modalities of cancer therapy that improve the efficacy of TRAIL as well as chemotherapeutic drugs and lessen the toxicity of these agents by targeting specific c-FLIP isoforms is discussed.
2. Apoptosis Signaling Pathways
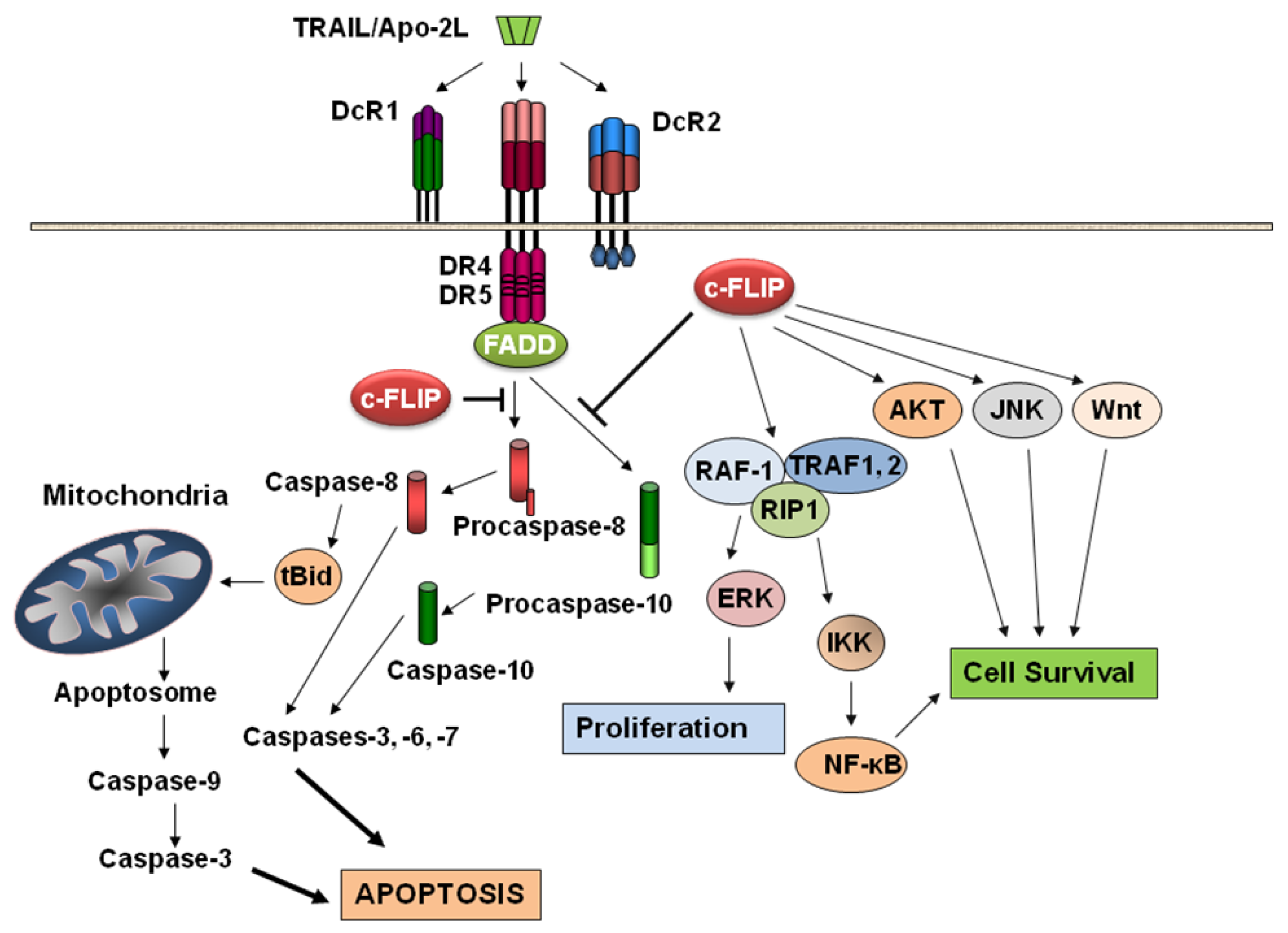
Apoptosis is a mechanism of programmed cell death involving signal transduction pathways that induce cells to self-destruct in response to organismal cues, e.g., digit formation in vertebrate limbs during embryonic development, environmental hazards (e.g., radiation-induced DNA damage), or anticancer therapeutics (e.g., chemotherapeutic agents and cytokines). Two well-studied pathways are involved in apoptosis: the mitochondrion-initiated pathway (Figure 1) and the cell surface death receptors pathway (Figure 1) [25-27]. In the mitochondrial pathway, cytochrome c, certain caspases, apoptosis-inducing factor, Smac/DIABLO, and other apoptosis-inducing factors are released from the mitochondrial intramembrane space to the cytosol [28]. Once released, cytochrome c and dATP bind to apoptotic proteinase-activating factor-1 (Apaf-1), and this complex along with adenine nucleotides promote procaspase-9 autoactivation [29], which in turn activates caspases-2, -3, -6, -7, -8, and -10. Apoptosis triggered by various stimuli requires direct activation of Bax and BAK at the mitochondria by a member of the Bcl-2 homology domain-3 (BH3)-only family of proteins including Bid, Bim, or PUMA [30]. The various anti- and pro-apoptotic members of the Bcl-2 family form a network of interactions that ultimately regulate the permeabilization of the mitochondrial outer membrane and release of apoptogenic factors such as cytochrome c to the cytoplasm [31]. Apoptosis initiated by the endoplasmic reticulum (ER) stress signaling pathway is also mainly dependent on the release of cytochrome c from the mitochondrial intermembrane space into the cytosol [32]. This release is associated with the opening of the permeability transition pore (PTP) and a collapse in the mitochondrial transmembrane potential (ΔΨm) due to the intake of Ca2+ following its release into the cytosol from the ER. Recent work has demonstrated that certain members of the Bcl-2 family are present on the ER where they appear to have a comprehensive function in the maintenance of ER homeostasis, participation in ER stress signal transduction pathways, and apoptosis [32].
In the death receptor-mediated apoptosis pathway (e.g., Fas/Fas ligand interaction or TRAIL/DR5 interaction and cell death), the initiator caspases-8 and -10 activate the downstream caspases including caspase-3 [18,19,24]. Active caspases-8 and -10 are known to cleave a pro-apoptotic Bcl-2 family member, Bid, and the truncated Bid induces mitochondrial cytochrome c release [26-29], thereby linking the two pathways. After activation, both caspases-8 and -9 activate caspase-3, which in turn cleaves other caspases and many cellular proteins including fodrin, various kinases, poly(ADP-ribose) polymerase, gelsolin, and DNA fragmentation factor-45 (DFF45) [26,33-37]. A third pathway has also been identified [38]. In this pathway, as Slee et al. [38] showed, BID is cleaved in response to several death-inducing stimuli (staurosporine, UV radiation, cycloheximide, etoposide) and that BID cleavage was blocked by Bcl-2, suggesting that degradation of BID occurred distal to cytochrome c release. Moreover, addition of cytochrome c to Jurkat post-nuclear extracts triggered cleavage of BID at Asp-59 which was catalysed by caspase-3 rather than caspase-8. These results provide evidence that caspase-3 mediated cleavage of BID represents a feedback loop for the amplification of mitochondrial cytochrome c release during cytotoxic drug and UV radiation-induced apoptosis [38].
3. Cellular FLICE-Like Inhibitory Protein (c-FLIP)
3.1. Structure of c-FLIP
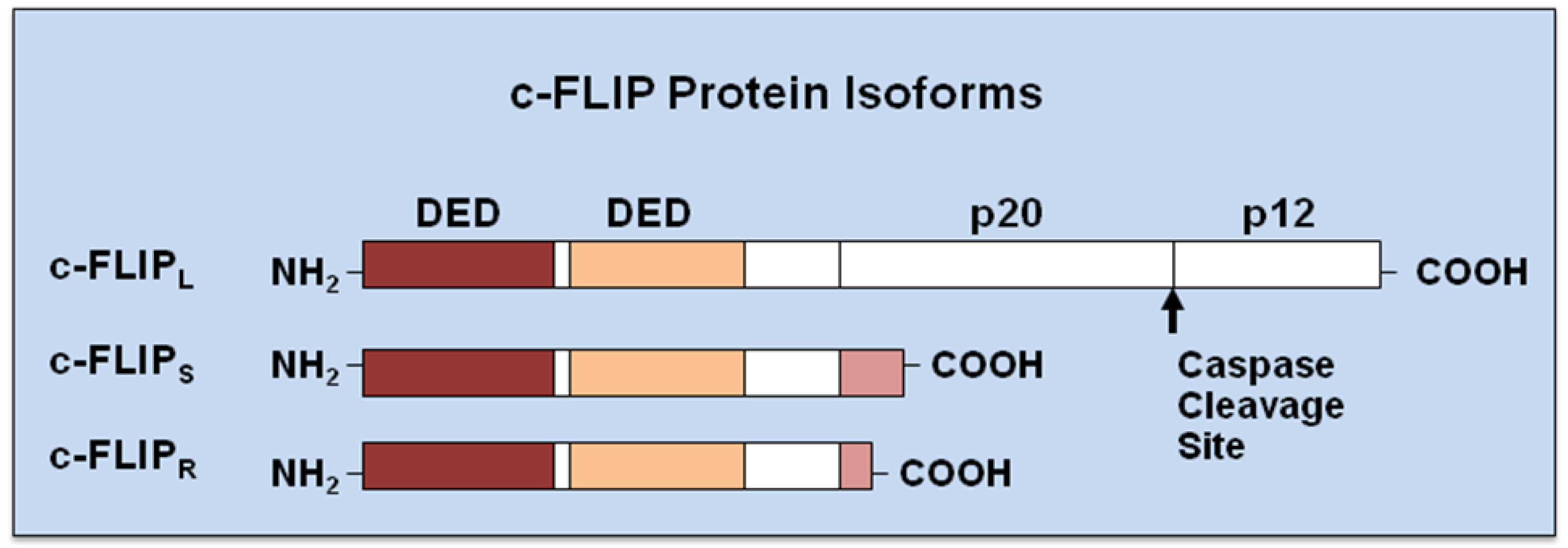
Originally, viral FLICE-inhibitory proteins (v-FLIPs) were identified by a bioinformatic search for novel virus-encoded apoptotic regulatory molecules containing a death effector domain (DED) [39-42]. Following the characterization of v-FLIPs, the mammalian cellular homologue was identified and called c-FLIP [43]. c-FLIP, also known as Casper, iFLICE, FLAME-1, CASH, CLARP, MRIT or usurpin [44,45], has 13 distinct splice variants [44], three of which are expressed as proteins: the 26 kDa short form (c-FLIPS), the 24 kDa form of c-FLIP (c-FLIPR), and the 55 kDa c-FLIPL [24,44,45] (Figure 2). The structures of c-FLIPS and the v-FLIP proteins are similar, except that the two DEDs of c-FLIPS are followed by 20 amino acids that appear to be crucial for its ubiquitaation and targeting for proteasomal degradation [46]. c-FLIPR also contains two DEDs but lacks the additional carboxy (C)-terminal amino acids that are present in c-FLIPS. The C-terminus of c-FLIPL is longer than that of c-FLIPS and closely resembles the structure of caspases-8 and -10 [47,48], but this region of c-FLIPL does not contain a functional caspase domain. This lack of caspase activity is the result of several amino acids substitutions, particularly the crucial cysteine residue in the catalytic domain which is necessary for the catalytic activity of caspases [47]. Additionally, c-FLIPL harbors a caspase-8 cleavage site at position Asp-376 (LEVD); c-FLIPL cleavage at this site produces the proteolytic fragment variant p43c-FLIP [48,49]. The C-terminal region of c-FLIPS and c-FLIPR play a crucial role in ubiquitnation and degradation as well as the anti-apoptotic function of these isoforms [50,51]. All three isoforms of c-FLIP can be recruited to the DISC through an interaction of their tandem DED domains with the adaptor protein FADD. Recently, Ueffing et al. [52] reported a functional single nucleotide polymorphism (SNP) in the human c-FLIP gene (rs10190751 A/G), located in the 3′ splice consensus of intron 6, which determines c-FLIPS production. Analysis of the rs101900751 G/A variation in follicular lymphoma patients indicates that rs10190751 A, which directs expression of the c-FLIPR isoform, is associated with increased risk for this disease [52].
3.2. Transcription and Translation of c-FLIP
Transcriptional activation of c-FLIP can be mediated by various signal transducers, including TNF ligands, growth factors, interleukins, chemokines, and chemotherapeutic agents [24]. Several transcription factors are known to transcriptionally regulate the c-FLIP gene [24,45]. These include NF-κB, p53 tumor suppressor protein, p63, E2F1, c-myc, IRF5, c-Fos, nuclear factor of activated T cells (NFAT), heterogeneous nuclear ribonucleoprotein K (hnRNP k), the forkhead transcription factor FOXO3a [42], Early growth response-1(EGR1), androgen receptor (AR), E2F, AP-1, and SP1 [24,45]. While NF-kB, p63, NFATc2, EGR1, hnRNP K, AR and SP1 are known to induce c-FLIP expression, c-myc, Foxo3a, c-Fos, IRF5, and SP3 suppress c-FLIP transcription [24,45]. p53 may transcriptionally upregulate the c-FLIP gene and also promote the degradation of c-FLIP protein [53,54]. Intriguingly, c-FLIPS was highly induced upon activation of T cells, primarily via the calcineurin-NFAT pathway [55]. Moreover, the human T-cell leukemia virus type 1 (HTLV-1)Tax protein up-regulating c-FLIP expression in HTLV-1-infected cells through activation of NF-κB [56].
Li et al. [57] reported that c-FLIPL is transcriptionally regulated by the activator protein-1 (AP-1) family member protein c-Fos, and that MG-132, an inhibitor of the proteasome, sensitizes TRAIL-resistant prostate cancer cells by inducing c-Fos and repressing c-FLIPL. Moreover, c-Fos, which is activated by MG-132, negatively regulates c-FLIPL by direct binding to the putative promoter region of the c-FLIPL gene. In addition to activating c-Fos, MG-132 also activates c-Jun, another AP-1 family member. c-Fos heterodimerizes with c-Jun to form an AP-1 complex that represses transcription of c-FLIPL.
E2F1, a transcription factor that plays a crucial role during S phase progression and apoptosis, triggers apoptosis in various lung adenocarcinoma cell lines by specifically downregulating of c-FLIPS leading to caspase-8 activation at the DISC [58]. Moreover, the SC35 splicing factor, which is a direct transcriptional target of E2F1, is involved in downregulation of c-FLIPS [59]. The specific overexpression of c-FLIPS is also seen in human lung adenocarcinomas with low levels of E2F1 [58]. Delineating the role of SC35 in regulating the expression of c-FLIPS will be very significant, not only for understanding how alternative splicing of the c-FLIP gene occurs, but also to possibly decrease the level of c-FLIPS by modulating SC35 expression.
c-FLIPS is also regulated at the translational level. Panner et al. [60,61] showed that TRAIL resistance in glioblastoma multiforme cells (GBM) is the result of c-FLIPS overexpression, and that activation of the Akt mammalian target of rapamycin (mTOR)-p70 S6 kinase 1 (S6K1) pathway leads to increased translation of the c-FLIPS protein. Conversely, inhibition of mTOR or its target S6K1 suppressed polyribosomal accumulation of c-FLIPS mRNA, c-FLIPS protein expression, and promoted TRAIL resistance in GBM cells. An mTOR-independent pathway can also act through a Ral effector protein, RalBP1 to suppress cdc42-mediated activation of S6 kinase and the translation of the c-FLIPS protein [61,62]. Moreover, it has been shown that Rocaglamide (Roc) sensitizes resistant adult T-cell leukemia/lymphoma (ATL) cells to DR4- and DR5-mediated apoptosis by translational suppression of c-FLIPS [63,64] through inactivation of the translation initiation factor 4E (eIF4E) [64].
3.3. c-FLIP Degradation
c-FLIP isoforms are short lived proteins whose stability is subject to isoform-specific regulation. c-FLIP is predominately degraded by the ubiquitin-proteasome degradation system [24,45,50]. Both c-FLIP isoforms can be degraded by the proteasome, but c-FLIPS appears to be particularly sensitive to ubiquitination and proteasomal degradation, partly due to two crucial lysine residues in the C-terminal 20 amino acids that are unique to c-FLIPS [50]. The sensitivity of c-FLIPS to ubiquitin-mediated degradation adds a novel concept to DISC regulation and its control of apoptosis [50].
Expression of c-FLIPL and c-FLIPS is also regulated by JNK activation via the E3 ubiquitin ligase Itch [24,45,65] under the control of JNK, polyubiquitinates c-FLIP to target it for degradation at the proteasome [65]. Phosphorylation events also play important roles in the regulation of c-FLIP protein levels. For instance, protein kinase C phosphorylation at the serine 193 (S193) residue of the c-FLIPS isoform inhibits its polyubiquitination, stabilizes c-FLIPS levels, and increases cell survival [66]. S193 phosphorylation was markedly increased by treatment with the PKC activator 12-O-tetradecanoylphorbol-13-acetate and decreased by inhibition of PKCα and PKCβ. Phosphorylation of S193 residue also decreased the ubiquitination of c-FLIPL but did not affect its stability, indicating that S193 phosphorylation has a different function in c-FLIPL than c-FLIPS. Moreover, Wang et al. [67] showed that pretreatment with the PKCδ-selective inhibitor rottlerin or transfection with PKCδ siRNA inhibited phorbol myristate acetate (PMA)-induced c-FLIP expression, which identifies a role for PKCδ in c-FLIP induction. These authors demonstrated a critical role for PKCδ/NF-κB in the induction of c-FLIP in human colon cancer cells. Downregulation of AMP-activated protein kinase also triggers ubiquitination and proteasome degradation of c-FLIP [68].
3.4. Upregulation of c-FLIP in Human Cancers
Increased expression of c-FLIP has been shown in cell lines from various types of cancers including colorectal [69,70], pancreatic [71,72], ovarian [73,74], gastric [75], breast [76,77], prostate [78], melanoma [79], glioblastoma [80], and it is implicated in TRAIL resistance and chemotherapy resistance. Gastric cancer SNU-216 cells [12], some pancreatic cancer cell lines [71], breast cancer cells [76,77], and leukemia cells [7,16] express high levels of c-FLIPL and c-FLIPS. FLIPS is also a key suppressor of TRAIL-induced apoptosis in human glioblastoma multiforme (GBM) cell lines and xenografts [60]. Furthermore, elevated levels of c-FLIP in tumor tissue from patients with colorectal cancer [80,81], bladder urothelial cancer [82], cervical cancer [83], Burkitt's lymphoma [84], non-Hodgkin's lymphoma [85], and head and neck squamous cell carcinoma (HNSCC) [86], and have been correlated with a poor clinical outcome and could be a reliable prognostic factor in these type of cancer. Overexpression of c-FLIP is also seen in gastric cancer and plays an important role in lymph node metastasis, which ultimately contributes to the tumor progression [87]. c-FLIP is expressed in pancreatic intraepithelial neoplasm lesions as well as in pancreatic ductal adenocarcinomas, whereas normal pancreatic ducts were consistently negative for c-FLIP expression [71].
3.5. c-FLIP Function
3.5.1. c-FLIP prevents apoptosis
Studies with animal models have revealed that c-FLIP plays an important role in T cell proliferation and heart development [88,89]. Moreover, abnormal c-FLIP expression has been found in various diseases such as cancer, multiple sclerosis, Alzheimer's disease, diabetes mellitus, and rheumatoid arthritis [24,44]. c-FLIP is also thought to be the main causal factor of “immune escape” [90]. c-FLIP is involved in TRAIL, Fas, TNF-α, and chemotherapeutic drug resistance in a wide range of human malignancies [24,44,72,91]. Moreover, studies using c-FLIP-deficient mice support a dual function for c-FLIPL by confirming a role for c-FLIP in Fas L, TNF-α-induced and apoptosis and revealing that c-FLIP has a similar function to caspase-8 in heart development [89]. Nonetheless, a now extensive literature encompassing diverse types of human cancer cells indicates that the action of c-FLIP is generally anti-apoptotic in cancer cells. Furthermore, interference with c-FLIP expression sensitizes tumor cells to death ligands and chemotherapy in experimental models [24,45,91-94]. In addition to its function as an apoptosis modulator, c-FLIP exerts other cellular functions including increased cell proliferation and tumorigenesis [24,45,48,94-96] (Figure 1).
While the precise mechanism of c-FLIP regulation of apoptosis remains elusive, the profound structural differences between human c-FLIP variants clearly indicate distinct regulatory roles for c-FLIPL and c-FLIPS in apoptosis. In fact, c-FLIPS inhibits TRAIL-induced DISC formation and apoptosis [24,45,48], while c-FLIPL is responsible for the above described dual functions whereby it inhibits Fas-induced caspase-8 activation when expressed at high levels, but enhances caspase-8 activation when its expression level is low [24,45]. These opposing c-FLIPL functions may reflect observations that c-FLIPL activates caspases-8 and -10 in vitro by forming heterodimeric enzyme molecules with a substrate specificity and catalytic activity indistinguishable from caspase-8 homodimers, despite the fact that c-FLIPL is protease dead [24,97,98]. Recent reports have clearly demonstrated that c-FLIPS also plays a central role in preventing cancer cell apoptosis. c-FLIPS has been shown to inhibit oxaliplatin-induced apoptosis through the sustained XIAP protein level and Akt activation [99]. c-FLIPS also suppresses apoptosis by inhibiting caspase-8 activation [100-102], although at different levels of procaspase-8 process [103,104]. c-FLIPL induces a conformation of procaspase-8 that triggers partial but incomplete proteolytic processing, while in contrast, c-FLIPS even prevents partial procaspase-8 activation at the DISC [8]. Using an in vitro induced proximity assay, Boatright et al. [97] provide evidence that c-FLIPL is an activator of caspase-8/-10 and demonstrate that the resulting heterodimer is enzymatically active with a substrate specificity identical to that of the caspase-8 homodimer.
We recently discovered that c-FLIPL interacts with DR5, FADD, and caspase-8 forming an apoptotic inhibitory complex (AIC) in MCF-7 breast cancer cells [76]. Moreover, silencing the c-FLIP gene by a specific siRNA leads to death ligand-independent but DR5-, FADD-, and caspase-8- and -9-dependent apoptosis in these cells. Furthermore, we showed that the knockdown of c-FLIP expression inhibits breast cancer cell proliferation and triggers spontaneous apoptosis by activating both the death receptor and mitochondrial pathways [76]. Our data support the previous report by Jin et al. [105] demonstrating that the peptide corresponding to the DR5 binding domain of c-FLIPL induces apoptosis in cancer cells. Therefore, inhibiting the interaction of DR5 and c-FLIPL by peptides or small molecule inhibitors should provide a mechanism by which tumor selective apoptosis can be achieved. Interestingly, a recent report also identified a checkpoint of the autophagy pathway where cellular and viral FLIPs could limit the Atg3-mediated step of LC3 ubiquitin-like protein conjugation to regulate autophagosome biogenesis. Furthermore, the c-FLIP-derived short peptides hold promise as new cancer therapeutic agents since they induced growth inhibition by binding to and effectively suppressing Atg3-c-FLIP interactions [106].
3.5.2. c-FLIP augments cytoprotective pathways
As shown in Figure 1, c-FLIP activates several cytoprotective signaling pathways involved in regulating cell survival, proliferation, and carcinogenesis. Overexpression of c-FLIPL activates NF-κB and ERK signaling by binding to adaptor proteins in each pathway, such as TNFR-associated factors 1 (TRAF1) and 2 (TRAF2), receptor-interacting protein 1 (RIP), and Raf-1 [48,107] (Figure 1). The caspase-8 processed N-terminal fragment of c-FLIPL (p43cFLIP) is more efficient than c-FLIPL at recruiting TRAF2 and RIP1, leading to more robust NF-κB activation [48,49,108,109]. Golks et al. [110] showed that in nonapoptotic cells, c-FLIP and the procaspase-8 heterodimer result in a novel NH2-terminal fragment of c-FLIP (p22-FLIP) which is the key mediator of NF-κB activation by binding directly to the IKK complex. These results provide a new mechanism of c-FLIP-mediated NF-κB activation. Recently, Chang et al. [111] demonstrated that TNF-α-mediated JNK activation increases turnover of the NF-κB-induced c-FLIP. This is not the result of direct c-FLIP phosphorylation, but rather depends on JNK-mediated phosphorylation and activation of the E3 ubiquitin ligase Itch which specifically ubiquitinates c-FLIP and induces its proteasomal degradation. Thus, JNK antagonizes NF-κB during TNF-α signaling by promoting the proteasomal elimination of c-FLIPL.
Akt is a serine-threonine kinase that plays a major role in transducing cellular survival signals and also regulates a number of proteins involved in the apoptotic signaling pathways. Recent results showed that Akt interacts with c-FLIPL protein and that c-FLIPL enhances anti-apoptotic Akt functions by modulating Gsk3β activity. Moreover, through its effects on Gsk3β, c-FLIPL overexpression in cancer cells induced resistance to TRAIL. This effect is mediated by regulation of p27(Kip1) and caspase-3 expression [112]. Downregulation of the DNA-PK/Akt pathway was also reported to correlate with high responsiveness to TRAIL-mediated growth inhibition and apoptosis [113]. siRNA-mediated suppression of DNA-PKcs or treatment with 4,5-dimethoxy-2-nitrobenzaldehyde (DMNB), a specific inhibitor of DNA-PK, led to decreased phosphorylation of Akt and Bad (a target molecule of Akt), increased expression of DR4/DR5, and down-regulation of c-FLIP [113]. Therefore, inhibition of the DNA-PK/Akt pathway may have clinical usefulness in treating TRAIL-resistant cancer cells. [97].
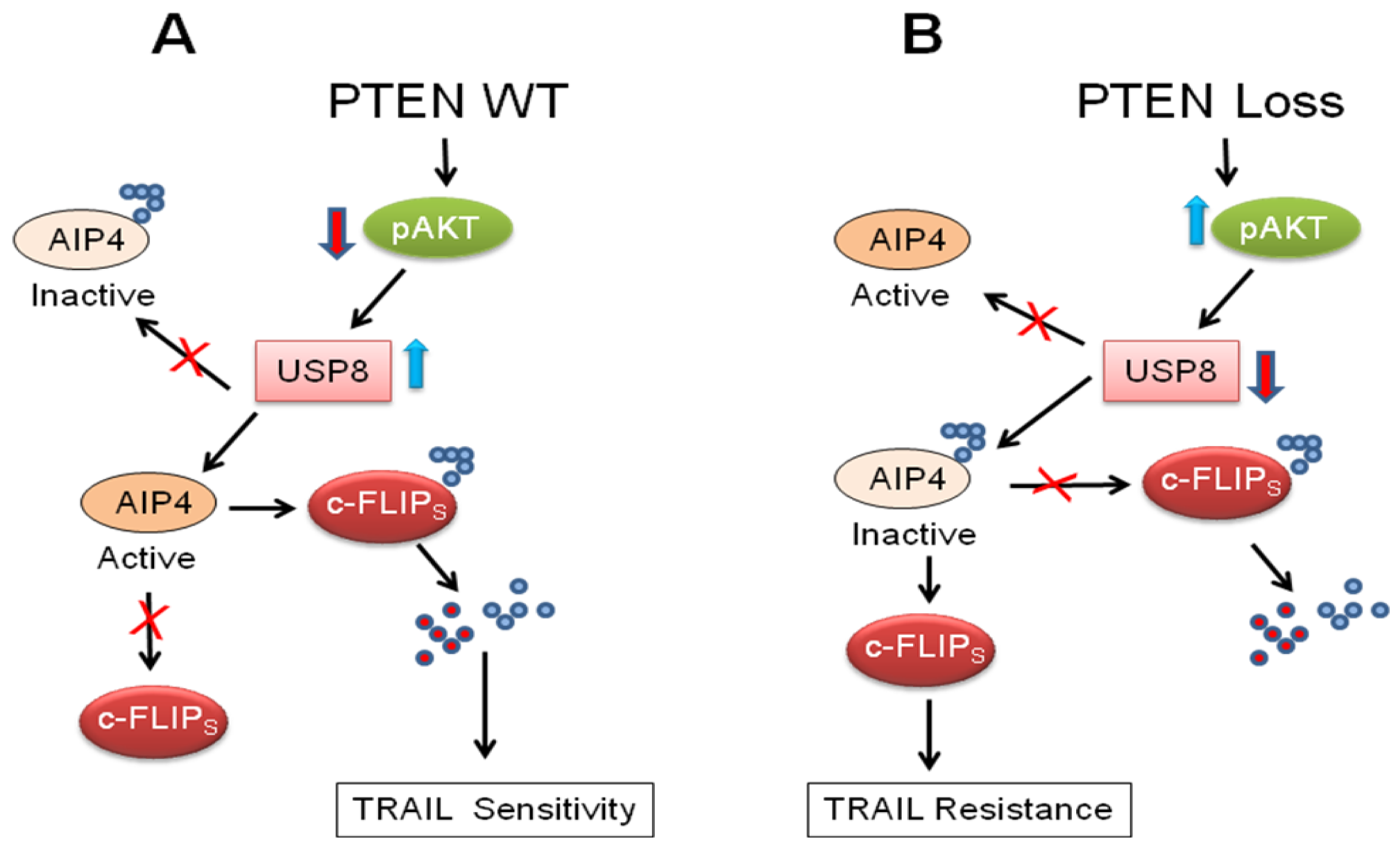
Panner et al. [114] initially reported that a novel phosphatase and tensin homologue (PTEN)-Akt-atrophin-interacting protein 4 (AIP4) pathway regulates c-FLIPS ubiquitination and stability in glioblastoma multiforme (GBM) cell lines and xenografts. However, how PTEN and Akt are linked to AIP4 activity was unclear. Recently, these authors described a second regulator of ubiquitin metabolism, the ubiquitin-specific protease 8 (USP8) which is a downstream target of Akt, and it links Akt to AIP4 and the regulation of c-FLIPS stability [115] (Figure 3). Overexpression of USP8 increased c-FLIPS ubiquitination, decreased FLIPS half-life, decreased FLIPS steady-state levels, and decreased TRAIL resistance (Figure 3). Therefore, PTEN appears to use control of ubiquitination to regulate TRAIL sensitivity in GBM cells.
c-FLIPL also interacts with Daxx (a death domain-associated protein that has been implicated in proapoptosis and transcriptional regulation) and prevents Fas-induced JNK activation [116]. Thus, c-FLIPL acting on both the FADD- and Daxx-mediated signaling pathways may be involved in completely inhibiting Fas-induced cell death. Furthermore, Nakajima et al. [117] demonstrated that c-FLIPL directly interacts with a JNK activator, MAP kinase kinase 7 (MKK7), in a TNF-α-dependent manner and inhibits the interactions of MKK7 with MAP/ERK kinase kinase 1 (MEKK1), apoptosis-signal-regulating kinase 1, (ASK1) and TGF-β-activated kinase 1. This interaction of c-FLIPL with MKK7 might selectively suppress JNK activation (Figure 1).
Another regulator of the c-FLIP expression is the calcium/calmodulin-dependent protein kinase II (CaMK II) which mediates the upregulation of c-FLIP, thereby protecting cancer cells from TRAIL-induced apoptosis. Treating resistant cells with the CaMK II inhibitor KN-93 inhibited CaMK II activity, reduced c-FLIP expression, inhibited c-FLIP phosphorylation, and rescued Fas agonistic antibody (CH-11) sensitivity [118,119]. Targeting this pathway may provide novel therapeutic strategies in treating cancers with upregulated CaMK II. Interestingly, phosphorylation of c-FLIP variants by CaMK II appears to promote c-FLIPL recruitment to the DISC and inhibit TRAIL-induced apoptosi [118,119], but phosphorylation of c-FLIPL by protein kinase C or the bile acid glycochenodeoxycholate results in decreased c-FLIPL recruitment to the DISC and increased the sensitivity of hepatocellular carcinoma cells to TRAIL-triggered apoptosis [120]. Thus, the particular site of phosphorylation on c-FLIPL appears to influence the functional outcome of this protein on apoptosis.
Increased expression of c-FLIP can alter cell cycle progression and enhance cell proliferation and carcinogenesis [121,122] (Figure 1). Overexpression of c-FLIPL inhibited the ubiquitination and proteasomal degradation of β-catenin, resulting in an increase in the target gene cyclin D1, colony formation, and invasive activity in prostate cancer cells. The c-FLIP/β-catenin/cyclin D1 signals contributing to colony formation and invasion were reversed by selective silencing of c-FLIP expression [123]. Similarly, c-FLIPL, in cooperation with FADD, enhances canonical Wnt signaling by inhibiting proteasomal degradation of β-catenin, thus suggesting a new mechanism involved with tumorigenesis [123]. Recent results also suggest a role for nuclear c-FLIPL in the modulation of Wnt signaling [124]. Interestingly, a deficiency in the adenomatous polyposis coli (APC) gene and subsequent activation of β- catenin can also lead to repression of c-FLIP expression through activation of c-Myc [125], c-FLIP upregulation may contribute to the carcinogenesis and aggressiveness of endometrial carcinomas and may serve as a useful prognostic factor for this tumor [24,126]. Wang et al. [127] demonstrated that c-FLIP overexpression is also significantly related to the presence of high-risk human papillomavirus (HR-HPV) infection during the progression of cervical squamous cell cancer and that c-FLIP is an early marker of cervical carcinogenesis. Moreover, HPV16 E2 protein interacts with and abrogates the apoptosis inhibitory function of c-FLIP and renders cervical cancer cell lines hypersensitive to Fas/FasL apoptosis. Overexpression of c-FLIP rescues cervical cancer cells from apoptosis induced by human HPV16 E2 protein expression [112]. This observation is greatly significant for developing therapeutic strategies to silence c-FLIP for intervention with cervical carcinogenesis [128]. Furthermore, overexpression of c-FLIPL also increases the hypoxia-inducible factor-1α (HIF1α) [129]. Overexpression of HIF1α can result in regulation of genes responsible for global changes in cell proliferation, metastasis, and invasion. Moreover, c-FLIP overexpression accelerated progression to androgen independence by inhibiting apoptosis in LNCaP prostate tumors implanted in nude mice [130].
Accumulating information clearly demonstrates that c-FLIPS plays a major role in causing resistance to death ligands and chemotherapeutic agents. Park et al. [131] reported that MEK1/2 inhibitors synergistically interacted with the heat shock protein 90 (HSP90) inhibitor, geldanamycins [17-allylamino-17-demethoxygeldanamycin (17AAG) and 17-dimethylaminoethylamino-17-demethoxy-geldanamycin], to kill hepatoma and pancreatic carcinoma cells. Treatment of cells with MEK1/2 inhibitors and 17AAG reduced expression of c-FLIPS that was connected to loss of MEK1/2 and AKT function. Moreover, overexpression of c-FLIPS or Inhibition of caspase-8 abolished cell killing by MEK1/2 inhibitors and 17AAG. Interestingly, Panner et al. [132] reported that HSP90α recruits c-FLIPS to the death-inducing signaling complex (DISC) and contributes to TRAIL resistance. Furthermore, combinations of low doses of sorafenib and vorinostat increased CD95 surface levels and CD95 association with caspase-8 and knockdown of CD95 or FADD expression reduced sorafenib/vorinostat cell death [133]. Signaling by CD95 caused protein kinase R (PKR)-like endoplasmatic reticulum kinase (PERK) activation that was responsible for both promoting caspase-8 association with CD95 and increased eIF2α phosphorylation. Suppression of eIF2α function abolished drug combination lethality. Cell killing was paralleled by PERK-and eIF2α-dependent lowering of c-FLIPs protein levels while overexpression of c-FLIPS maintained cell viability [133]. Similarly, Zhang et al. [134] showed that expression of phosphorylation-insensitive eIF2α-S51A blocked sorafenib- and vorinostat-induced suppression of c-FLIPS levels and overexpression of c-FLIPS abolished lethality. Overexpression of c-FLIPS function suppressed cell death by the multinuclear platinum chemotherapeutic BBR3610 [135].
3.5.3. c-FLIP increases cell motility
Another important role of c-FLIP is its involvement in increasing cancer cell motility. The role of c-FLIP in cell motility has been investigated using a c-FLIP-specific siRNA. Shim et al. [136] showed that siRNA-mediated down-regulation of c-FLIPL correlated with increased levels of reactive oxygen species (ROS), while over-expression of c-FLIPL triggered the opposite effect. ROS generated by silencing c-FLIP induced phosphorylation of Akt and impaired cell motility [136]. The role of c-FLIP in the motility of HeLa cells was also shown using siRNA directed against c-FLIP. Silencing c-FLIPL but not c-FLIPS inhibited the adhesion and motility of the cells by activating FAK and extracellular regulated kinase (ERK), and increasing MMP-9 expression [137]. Additional evidence demonstrating the role of c-FLIPL in triggering cell motility was recently provided in ovarian tumors [73]. In these tumors, c-FLIPL played a role in chaperoning tumor cells from immunosurveillance and increasing their invasive potential by augmenting cell motility [73].
3.5.4. c-FLIP triggers epithelial-mesenchymal transition (EMT)
EMT is a process that induces morphological and genetic changes of cancer cells from an epithelial to a mesenchymal phenotype, which forms the basis for the metastatic potential of tumor cells [138]. Various tumor microenvironmental factors, including cytokines, growth factors, and chemotherapeutic agents trigger EMT [138], and this process is believed to be partly responsible for the chemotherapy-resistant phenotype [138,139]. A cancer-associated antigen gene (CAGE) which is widely expressed in various cancer tissues and cancer cell lines regulates expression of EMT-related proteins through ERK, Akt and NF-kB [140,141]. Snail, an EMT-related protein, mediates the effect of CAGE by inducing matrix metalloproteinase-2 (MMP-2) and cancer cell motility. Interestingly, c-FLIP mediates the effect of CAGE on the induction of MMP-2 and cell motility by the induction of Snail [141].
3.6. c-FLIP as a Therapeutic Target for Cancer Treatment
Ectopic expression of c-FLIP variants decreased apoptosis caused by death ligands and anticancer agents [24], indicating that overexpression of these proteins may cause resistance to multiple anticancer drugs. Therapeutic modalities that lower the threshold of cancer cell apoptosis should lead to more effective cancer treatment. For example, strategies to inhibit the expression of c-FLIP variants not only trigger apoptosis in certain cancer types, but also sensitize cancer cells to chemotherapeutic agents, potentially allowing lower doses to be administered to patients and decreasing drug-induced systemic toxicities. Therefore, c-FLIP variants are critical apoptosis regulators that can serve as targets for small molecule inhibitors that downregulate their expression and serve as effective targeted therapeutics for cancer treatment. In order to support this hypothesis, our in vivo results showed that injecting liposomal complexes of c-FLIP-specific siRNA into MCF-7 xenografts eliminated the neoplastic cells without affecting the normal stromal and fibroblastic cells [142]. There does not appear to be a “handle” to inhibit c-FLIP function with small molecule ligands since, as discussed above, c-FLIP has significant structural similarity to caspase-8. This resemblance with caspase-8 makes c-FLIP protein a very difficult target for drugs to inhibit its function, since small molecules capable of blocking c-FLIP's recruitment to the DISC could also inhibit the recruitment of caspase-8, and as a result inhibit apoptosis. Therefore, to reduce or inhibit c-FLIP expression, small molecules which target c-FLIP without inhibiting caspases-8 and -10 are needed.
Small molecule therapeutics that selectively downregulate c-FLIPS or c-FLIPL and gene therapy strategies that knock down a specific c-FLIP variant have been used to downregulate these variants. Developing these innovative therapeutic strategies in conjunction with TRAIL and chemotherapeutic agents could potentially overcome the barrier of dose-limiting toxicity in cancer chemotherapy. TRAIL or chemotherapy resistance in diverse cancer cell types can be reversed by parallel treatment with agents known to downregulate c-FLIP variants. As discussed below and shown in Table 1 and Table 2, c-FLIP variants can be inhibited by compounds that inhibit their transcription or translation, trigger their degradation, or by c-FLIP-specific small interfering RNA (siRNA) which sensitize a wide range of cancer cell types to TRAIL and chemotherapy-induced apoptosis.
3.6.1. c-FLIP transcriptional regulators for cancer therapy
As shown in Table 1, DNA damaging agents are promising drugs with regard to downregulating levels c-FLIP variants. Pretreatment with chemotherapeutic drugs including cisplatin, doxorubicin, or topoisomerase I inhibitors (camptothecin, 9-NC, irinotecan) downregulated c-FLIP variants expression in various tumor cells by inhibiting its transcription and rendering cells sensitive to death receptor-triggered apoptosis (Table 1) [69,143-148]. Successful inhibition of malignant cell growth and apoptosis induction using histone deacetylase inhibitor (HDACi) compounds has highlighted the potential use of these compounds as anticancer agents. Several HDACi have been shown to downregulate c-FLIP expression in various cancer cells at the transcriptional and translational levels [149-152]. Among these, suberoylanilide hydroxamic acid (SAHA, vorinostat) is the most promising HDACi that causes robust inhibition of c-FLIP variants [149]. Recent results demonstrated that TRAIL-triggered apoptosis in breast cancer cells is blocked at the level of apical activation of caspase-8, and that SAHA enhances the TRAIL-induced processing and activation of procaspase-8. Interestingly, degradation of c-FLIPL and c-FLIPS by an ubiquitin/proteasome-dependent Itch/AIP4-independent mechanism is observed upon exposure to SAHA [149]. We recently showed that a new HDACi 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) or droxinostat [151,152], identified using a highthroughput chemical library screen [153,154], triggered apoptosis in the breast cancer cell line MCF-7 through c-FLIPL and c-FLIPS mRNA as well as protein downregulation [151]. Interestingly, this agent induced more robust apoptosis in a doxorubicin-resistant variant of MCF-7 cells [151]. As shown in Table 1, a number of agents with modulating effects on Akt, PI3K, NF-κB, and Ras pathways, as well as an inhibitor of STAT3 have also been shown to transcriptionally silence c-FLIP expression.
3.6.2. Oligonucleotide and RNAi-targeting of c-FLIP for cancer therapy
We have shown that CCRF-HSB-2 human lymphoblastic leukemia cells transfected with an antisense c-FLIP plasmid abrogated c-FLIPS and c-FLIPL expression and triggered a significant increase in Taxol-induced apoptosis [7]. Logan et al. [143] investigated whether using an antisense oligonucleotide to target c-FLIP was a clinically feasible approach (Table 2). These authors developed a novel c-FLIP-targeted antisense phosphorothioate oligonucleotide (AS PTO) and recently used it in vitro in transient transfection experiments and in vivo using xenograft models in Balb/c nude mice [143]. The AS PTO downregulated c-FLIP and resulted in caspase-8 activation and apoptosis induction in non-small cell lung cancer (NSCLC) cells, but not in normal lung cells. Similar results were observed in colorectal and prostate cancer cells. The AS PTO also sensitized cancer cells but not normal lung cells to apoptosis induced by TRAIL and increased chemotherapy-triggered apoptosis in NSCLC cells. Importantly, compared to a control non-targeted PTO, intraperitoneal delivery of c-FLIP AS PTO inhibited the growth of NSCLC xenografts and enhanced the in vivo antitumor effects of cisplatin. Therefore, this c-FLIP-targeted AS PTO may have potential for further pre-clinical development.
The development of RNA interference (RNAi)-based therapeutics to target the c-FLIP gene in vivo may change the way cancers are treated by inducing apoptosis [79] or by sensitizing cancers to chemotherapeutic agents. However, difficulties in siRNA design, delivery, and stability must be solved before RNAi-based therapeutics will be feasible for clinical use. We have used lipocomplexes of c-FLIP siRNA to successfully knock down the c-FLIP gene and induce spontaneous apoptosis in MCF-7 breast cancer cells in vitro [142] (Table 2), and in vivo by directly injecting the c-FLIP siRNA lipocomplexes into MCF-7 mouse xenografts [142]. Lipocomplexes of c-FLIP siRNA have also been used to successfully silence the c-FLIP gene and trigger spontaneous apoptosis in A549 lung cancer cells [165], HCT116 colorectal cancer cells [166], and LNCaP and PC3 prostate cancer cells [13]. Furthermore, c-FLIP siRNA lipocomplexes injected into HCT116 colorectal tumor xenografts decreased tumor growth [166]. These studies show that c-FLIP siRNA lipocomplex formulations can be used to successfully knock down the c-FLIP gene in various cancer cell types [76,95].
3.6.3. c-FLIP degradation as a target for cancer therapy
As discussed above, c-FLIP is predominately degraded by the ubiquitin-proteasome system. Downregulation of c-FLIPL and c-FLIPS due to degradation is observed in cells treated with various apoptosis-inducing agents (Table 2). Cycloheximide [167] and anisomycin [153], two protein synthesis inhibitors, as well as the RNA synthesis inhibitor actinomycin D [168] have been shown to downregulate c-FLIPL and c-FLIPS. Treating cancer cells with fluorouracil (5-FU) was also demonstrated to downregulate both isoforms in colon cancer cell lines [106,169] (Table 2).
Peroxisome proliferator-activated receptor γ (PPARγ) agonists sensitize cancer cells to TRAIL by ubiquitination and proteasome-dependent c-FLIP degradation [170-174]. Tiwary et al. [77] recently reported that α-tocopherol ether-linked acetic acid analogue (α-TEA) downregulation of c-FLIP is mediated by ER stress-dependent JNK/CHOP/DR5 signaling via JNK activation of Itch E3 ligase ubiquitination and involved in activation of the ER-stress-dependent events via reducing the inhibitory effect of c-FLIP on caspase-8.
Proteasome inhibitors are a new class of drugs that decrease proliferation and induce apoptosis in a variety of hematologic and solid malignancies [175-186]. Interestingly, several proteasome inhibitors lead to the downregulation of c-FLIPL and c-FLIPS [24,45,172]. The induction of apoptosis by the proteasome inhibitors MG-132 and PS-341 (bortezomib, Velcade®) in primary chronic lymphocytic leukemia (CLL) cells and the Burkitt lymphoma cell line BJAB was associated with upregulation of TRAIL and its death receptors, DR4 and DR5, and decreased c-FLIP protein expression [179]. Similarly, bortezomib decreased c-FLIP expression in multiple myeloma and human esophageal squamous cell carcinoma cell lines [180,181]. However, the effect of PS-341 on the regulation of c-FLIP expression may be cancer cell-type specific. In contrast to what was observed human esophagil cancer cell lines, Liu et al. reported that PS-341 upregulates DR5 as well as c-FLIP and survivin in human non-small cell lung carcinomas (NSCLC) cells [183]. As discussed earlier, c-FLIP is degraded via a ubiquitin-proteosome system. Therefore, PS-341 should increase c-FLIP and prevent apoptosis. Interestingly, Zhao et al. have shown that PS-341 decreases c-FLIP at the gene level [184].
The Bcr-Abl kinase inhibitor imatinib mesylate (formerly known as CGP 57148B, STI571, or Gleevec) is currently the standard therapy for chronic myeloid leukemia (CML). Hamaï et al. [187] reported that Imatinib mesylate increases human melanoma cell sensitivity to TRAIL-induced cell death by directly downregulating protein levels of c-FLIP variants. Interestingly, Park et al. [188] showed that silencing the Bcr-Abl in K562 leukemia cells led to the downregulation of c-FLIPL and subsequent increase to TRAIL sensitivity.
As shown in Table 2, a number of agents known to affect various targets and signaling pathways in cancer cells also cause degradation of c-FLIP variants (189-205). Moreover, several compounds have been shown to inhibit expression of c-FLIP variants, but whether these agents cause degradation of these proteins or silence their transcription remain to be found. Nutlin-3, a small molecule antagonist of MDM2 which inhibit the p53-MDM2 interaction and activates p53 signaling was recently shown to decrease expression of c-FLIPS and c-FLIPL and was synergistic with TRAIL in triggering cell death [206]. Moreover, Ozarelix, a gonadotropin-releasing hormone antagonist [207], celecoxib, a cyclooxygenase-2 inhibitor [208], the chemopreventive agent, all-trans-retinyl acetate (RAc) [125], smac mimetic compounds (SMC) [180], and sunitinib, an orally administered tyrosine kinase inhibitor (TKI) [210] decreased expression of c-FLIP. Furthermore, downregulation of c-FLIP by a specific microRNAs (i.e., miR-512-3p) increased taxol-induced apoptosis [211], supporting our previous report that silencing c-FLIP variants increases Taxol-triggered apoptosis [7]. Gemcitabine was also recently shown to inhibit expression of c-FLIP variant in pancreatic cancer cells [71], but whether it inhibits the transcription, enhances degradation, or prevents translation of c-FLIP remains to be found.
Recent data [122,212,213] clearly demonstrate that ataxia telangiectasia mutated (ATM) kinase activity modulates c-FLIPL and c-FLIPS protein levels in response to DNA damage (Figure 4). Moreover, the radiomimetic drug Neocarzinostatin (NCS) may trigger the down-regulation of c-FLIP isoforms by inducing the activation of the ATM kinase in response to DNA damage [122]. ATM kinase activity negatively modulates the stability of c-FLIPL and c-FLIPS at the protein level, thereby promoting the sensitivity to apoptosis induction by Fas (CD95/APO-1), a TRAIL-R1/R2-related death receptor [213]. NCS-triggered decrease in c-FLIP resulted in increased sensitivity to TRAIL which was inhibited by ATM kinase activity inhibition [122]. Upon NCS treatment, ATM promotes c-FLIPL protein degradation through the ubiquitin-proteasome system but the mechanism of degradation of c-FLIPS that ATM is linked to remains to be determined [122].
4. Conclusions
It is now evident that c-FLIP variants induce resistance to death receptor ligands and chemotherapeutic agents in various cancer cells and that c-FLIP may be a relevant clinical target for counteracting therapy resistant human malignancies. The current state of the art reviewed in this article suggests that targeting c-FLIP in combination with TRAIL or standard chemotherapies has therapeutic potential for treating human cancers. As discussed, various classes of agents can downregulate c-FLIP expression. However, c-FLIP has significant structural similarity to caspase-8; this makes c-FLIP a very difficult target for developing drugs that inhibit this protein directly, since small molecules capable of blocking c-FLIP's recruitment to the DISC could simultaneously inhibit the recruitment of caspase-8 and thereby inhibit apoptosis. Therefore, to reduce or inhibit c-FLIP expression, small molecules which target c-FLIP without inhibiting caspases-8 and -10 are needed. Compounds that inhibit or downregulate c-FLIP mRNA expression will particularly be of interest. As discussed above, employing a high-throughput chemical screening strategy, a small molecule inhibitor of c-FLIP, 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) or droxinostat has been identified that downregulates c-FLIPL and c-FLIPS mRNA and protein levels, reduces cell survival, and induces apoptosis. The foregoing discussion justifies optimism that future cancer therapy will be improved by innovations that combine chemotherapy with drug resistance-reversing multi-targeted therapy, e.g., combination regimens of chemotherapy and small molecule drugs that downregulate c-FLIP.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of action | Agent | References |
|---|---|---|
| Generation of DNA adduct formation, intra- and interstrand crosslinks | Cisplatin, oxaliplatin | [69,70,143-146] |
| DNA intercalator | Doxorubicin | [147] |
| Topoisomerase I inhibitor | Camptothecin, 9-nitrocamptothecin (9-NC), irinotecan | [69,148] |
| Histone deacetylase inhibitor | Vorinostat, trichostatin, droxinostat (CMH), valproic acid, NCH-51, MS-275, romidepsin (FK228/depsipeptide), and AR-42 | [149-154] |
| Anti-microtubule targeting agent | Lupeol (triterpene) | [155-157] |
| Inhibitor of IκB kinase (IKK) and NF-κB pathways | Celastrol, zerumbone, withaferin A, quinacrine | [158-160] |
| Inhibitor of TNFα-mediated NF-κB activation | Chrysin (flavonoid) | [161] |
| Inhibitor of mitogen-induced proliferative response | S-adenosylmethionine (SAMe) 5′-methylthioadenosine (MTA) | [162] |
| Inhibitor of signal transducer and activator of transcription 3 (STAT3) | CDDO-imidazolide (synthetic triterpenoid) | [163] |
| Ras/mTOR inhibitor | Salirasib | [164] |
| Mechanism of action | Agent | References |
|---|---|---|
| Blockade of mRNA translation and RNaseH-mediated cleavage, with subsequent degradation of the mRNA:antisense DNA heteroduplex | Antisense oligonucleotide | [60] |
| c-FLIP RNA interference | SiRNAs | [24,45,116, 165,166] |
| Inhibitor of mammalian target of rapamycin (mTOR) | Rapamycin | [83] |
| Inhibitor of phosphorylation of (Roc) translation initiation factor 4E (eIF4E) Disrupts the eIF4E/eIF4G association | Rocaglamide | [87] |
| Antimicrotubule agent | Taxol (paclitaxel) | [7] |
| Protein synthesis inhibitors | Cyclohexamide and anisomycin | [167] |
| RNA synthesis inhibitor | Actinomycin D | [168,169] |
| Thymidylate synthase (TS) inhibitor | 5-fluorouracil (5-FU) | [8,122] |
| PPARγ modulation | CDDO, 15-deoxy-δ (12,14)-prostaglandin J2 (15d-PGJ2), Rosiglitazone, Troglitazone | [170-174] |
| Proteasome inhibitors | PS-34 (bortezomib), MG-132 | [80,175-186] |
| Bcr-Abl kinase inhibitor | Imatinib mesylate | [187,188] |
| Blocks the activation of NF-κB and TGF-β1/Smad signaling pathways | Silibinin (flavonoid) | [189] |
| Induces cyclin D1 phosphorylation, Increases ubiquitination of c-FLIP | SHetA2 | [190] |
| Akt and NF-κB downregulation | Genistin (isoflavone) | [191] |
| Multikinase inhibitor | Sorafenib | [192] |
| Inhibitor of AKT phosphorylation | Eupatolide | [193] |
| Elevation of c-Jun N-terminal kinase (JNK) and its substrate c-Jun | α-TEA | [194,195] |
| Inhibitor of transglutaminase 2 (TG2) | Cystamine | [196] |
| Proteasome-mediated degradation of c-FLIPS | Paxilline | [197] |
| Inhibitor of phosphorylation of AKT (pAKT) and cellular FLICE-like inhibitory protein (c-FLIP) | Tamoxifen | [198] |
| Increases reactive oxygen species (ROS) and induces proteasome-dependent degradation of c-FLIP | Isoquinoline alkaloid, Berberine (BBR) | [199] |
| Activator of signal transduction cascades | INF-γ | [200] |
| Induce ROS, activator of both death receptor- and mitochondrial-mediated apoptotic pathways | β-elemene piperazine derivatives | [201] |
| EGFR tyrosine kinase inhibitor | AG1478 | [202] |
| Down-regulates STAT3 | Kahweol, a coffee-specific diterpene | [203] |
| Phosphodiesterase inhibitor | Pentoxifylline (PTX) | [204] |
| Phosphatidylinositol 3′-kinase inhibitor | LY294002 | [205] |
Acknowledgments
We would like to thank Mary D. Kraeszig for her editorial assistance. The work in Safa's laboratory was supported by research grants from the National Cancer Institute (CA 080734, CA 90878, and CA 101743), Department of Defense (DOD) (OC 06095), the Lung Cancer Working Group (LCWG), and the Indiana University Cancer Center Translational Research Acceleration Collaboration (ITRAC) initiative.
References
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar]
- Clarke, R.; Leonessa, F.; Trock, B. Multidrug resistance/P-glycoprotein and breast cancer: Review and meta-analysis. Semin. Oncol. 2005, 32, S9–S15. [Google Scholar]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar]
- Roberti, A.; La Sala, D.; Cinti, C. Multiple genetic and epigenetic interacting mechanisms contribute to clonally selection of drug-resistant tumors: Current views and new therapeutic prospective. J. Cell. Physiol. 2006, 207, 571–581. [Google Scholar]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar]
- Mashima, T.; Tsuruo., T. Defects of the apoptotic pathway as therapeutic target against cancer. Drug Resist. Updat. 2005, 8, 339–343. [Google Scholar]
- Day, T.W.; Najafi, F.; Wu, C.H.; Safa, A.R. Cellular FLICE-like inhibitory protein (c-FLIP): A novel target for taxol-induced apoptosis. Biochem. Pharmacol. 2006, 71, 1551–1561. [Google Scholar]
- Galligan, L.; Longley, D.B.; McEwan, M.; Wilson, T.R.; McLaughlin, K.; Johnston, P.G. Chemotherapy and TRAIL-mediated colon cancer cell death: The roles of P53, TRAIL receptors, and c-FLIP. Mol. Cancer Ther. 2005, 4, 2026–2036. [Google Scholar]
- Longley, D.B.; Wilson, T.R.; McEwan, M.; Allen, W.L.; McDermott, U.; Galligan, L.; Johnston, P.G. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 2006, 25, 838–848. [Google Scholar]
- Mathas, S.; Lietz, A.; Anagnostopoulos, I.; Hummel, F.; Wiesner, B.; Janz, M.; Jundt, F.; Hirsch, B.; Johrens-Leder, K.; Vornlocher, H.P.; Bommert, K.; Stein, H.; Dorken, B. c-FLIP mediates resistance of hodgkin/reed-sternberg cells to death receptor-induced apoptosis. J. Exp. Med. 2004, 199, 1041–1052. [Google Scholar]
- Van Geelen, C.M.; de Vries, E.G.; de Jong, S. Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: Paving the road to patient-tailored therapy. Drug Resist. Updat. 2004, 7, 345–358. [Google Scholar]
- Xiao, C.; Yang, B.F.; Song, J.H.; Schulman, H.; Li, L.; Hao, C. Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp. Cell Res. 2005, 304, 244–255. [Google Scholar]
- Zhang, X.; Jin, T.G.; Yang, H.; DeWolf, W.C.; Khosravi-Far, R.; Olumi, A.F. Persistent c-FLIP(L) expression is necessary and sufficient to maintain resistance to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in prostate cancer. Cancer Res. 2004, 64, 7086–7091. [Google Scholar]
- Park, S.J.; Bijangi-Vishehsaraei, K.; Safa, A.R. Selective TRAIL-triggered apoptosis due to overexpression of TRAIL death receptor 5 (DR5) in P-glycoprotein-bearing multidrug resistant CEM/VBL1000 human leukemia cells. Int. J. Biochem. Mol. Biol. 2010, 1, 90–100. [Google Scholar]
- Park, S.J.; Wu, C.H.; Choi, M.R.; Najafi, F.; Emami, A.; Safa, A.R. P-glycoprotein enhances trail-triggered apoptosis in multidrug resistant cancer cells by interacting with the death receptor DR5. Biochem. Pharmacol. 2006, 72, 293–307. [Google Scholar]
- Wu, C.H.; Kao, C.H.; Safa, A.R. TRAIL recombinant adenovirus triggers robust apoptosis in multidrug-resistant Hl-60/Vinc cells preferentially through death receptor DR5. Hum. Gene Ther. 2008, 19, 731–143. [Google Scholar]
- Seo, S.B.; Hur, J.G.; Kim, M.J.; Lee, J.W.; Kim, H.B.; Bae, J.H.; Kim, D.W.; Kang, C.D.; Kim, S.H. TRAIL sensitizes MDR cells to MDR-related drugs by down-regulation of P-glycoprotein through inhibition of DNA-PKcs/Akt/GSK-3β pathway and activation of caspases. Mol. Cancer 2010, 9, 199. [Google Scholar]
- Daniel, D.; Wilson, N.S. Tumor necrosis factor: Renaissance as a cancer therapeutic? Curr. Cancer Drug Targets 2008, 8, 124–131. [Google Scholar]
- Yang, A.; Wilson, N.S.; Ashkenazi, A. Proapoptotic DR4 and DR5 signaling in cancer cells: Toward clinical translation. Curr. Opin. Cell Biol. 2010, 22, 837–844. [Google Scholar]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O'Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; Ashkenazi, A.; Jubb, A.M.; Mendelson, D.S. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar]
- Rippo, M.R.; Moretti, S.; Vescovi, S.; Tomasetti, M.; Orecchia, S.; Amici, G.; Catalano, A.; Procopio, A. FLIP Overexpression inhibits death receptor-induced apoptosis in malignant mesothelial cells. Oncogene 2004, 23, 7753–7760. [Google Scholar]
- Morales, J.C.; Ruiz-Magana, M.J.; Ruiz-Ruiz, C. Regulation of the resistance to TRAIL-induced apoptosis in human primary T lymphocytes: Role of NF-κB inhibition. Mol. Immunol. 2007, 44, 2587–2597. [Google Scholar]
- Shankar, S.; Srivastava, R.K. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: Mechanisms and clinical implications. Drug Resist. Updat. 2004, 7, 139–156. [Google Scholar]
- Safa, A.R.; Day, T.W.; Wu, C.H. Cellular FLICE-like inhibitory protein (c-FLIP): A novel target for cancer therapy. Curr. Cancer Drug Targets 2008, 8, 37–46. [Google Scholar]
- Cereghetti, G.M.; Scorrano, L. The many shapes of mitochondrial death. Oncogene 2006, 25, 4717–4724. [Google Scholar]
- Gogvadze, V.; Orrenius, S. Mitochondrial regulation of apoptotic cell death. Chem. Biol. Interact. 2006, 163, 4–14. [Google Scholar]
- Meng, X.W.; Lee, S.H.; Kaufmann, S.H. Apoptosis in the treatment of cancer: A promise kept? Curr. Opin. Cell Biol. 2006, 18, 668–676. [Google Scholar]
- Ferri, K.F.; Jacotot, E.; Blanco, J.; Este, J.A.; Zamzami, N.; Susin, S.A.; Xie, Z.; Brothers, G.; Reed, J.C.; Penninger, J.M.; Kroemer, G. Apoptosis control in syncytia induced by the HIV type 1-envelope glycoprotein complex: Role of mitochondria and caspases. J. Exp. Med. 2000, 192, 1081–1092. [Google Scholar]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar]
- Ren, D.; Tu, H.C.; Kim, H.; Wang, G.X.; Bean, G.R.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 2010, 330, 1390–1393. [Google Scholar]
- Danial, N.N.; Gimenez-Cassina, A.; Tondera, D. Homeostatic functions of BCL-2 proteins beyond apoptosis. Adv. Exp. Med. Biol. 2010, 687, 1–32. [Google Scholar]
- Szegezdi, E.; Macdonald, D.C.; Ni Chonghaile, T.; Gupta, S.; Samali, A. Bcl-2 family on guard at the ER. Am. J. Physiol. Cell Physiol. 2009, 296, C941–C953. [Google Scholar]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar]
- Strasser, A.; O'Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar]
- Wolf, B.B.; Green, D.R. Suicidal tendencies: Apoptotic cell death by caspase family proteinases. J. Biol. Chem. 1999, 274, 20049–20052. [Google Scholar]
- He, B.; Lu, N.; Zhou, Z. Cellular and nuclear degradation during apoptosis. Curr. Opin. Cell Biol. 2009, 21, 900–912. [Google Scholar]
- Kurokawa, M.; Kornbluth, S. Caspases and kinases in a death grip. Cell 2009, 138, 838–854. [Google Scholar]
- Slee, E.A.; Keogh, S.A.; Martin, S.J. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: A potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 2000, 7, 556–565. [Google Scholar]
- Bertin, J.; Armstrong, R.C.; Ottilie, S.; Martin, D.A.; Wang, Y.; Banks, S.; Wang, G.H.; Senkevich, T.G.; Alnemri, E.S.; Moss, B.; Lenardo, M.J.; Tomaselli, K.J.; Cohen, J.I. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc. Natl. Acad. Sci. USA 1979, 94, 1172–1176. [Google Scholar]
- Bélanger, C.; Gravel, A.; Tomoiu, A.; Janelle, M.E.; Gosselin, J.; Tremblay, M.J.; Flamand, L. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J. Hum. Virol. 2001, 4, 62–73. [Google Scholar]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schröter, M.; Scaffidi, C.; Krammer, P.H.; Peter, M.E.; Tschopp, J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; Rimoldi, D.; French, L.E.; Tschopp, J. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar]
- Hu, S.; Vincenz, C.; Buller, M.; Dixit, V.M. A novel family of viral death effector domain-containing molecules that inhibit both CD-95 and tumor necrosis factor receptor-1-induced apoptosis. J. Biol. Chem. 1997, 272, 9621–9624. [Google Scholar]
- Micheau, O. Cellular FLICE-inhibitory protein: An attractive therapeutic target? Expert Opin. Ther. Targets 2003, 7, 559–573. [Google Scholar]
- Shirley, S.; Micheau, O. Targeting c-FLIP in cancer. Cancer Lett. 2010. [Google Scholar] [CrossRef]
- Palacios, C.; Yerbes, R.; Lopez-Rivas, A. Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res. 2006, 66, 8858–8869. [Google Scholar]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar]
- Kataoka, T.; Budd, R.C.; Holler, N.; Thome, M.; Martinon, F.; Irmler, M.; Burns, K.; Hahne, M.; Kennedy, N.; Kovacsovics, M.; Tschopp, J. The caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr. Biol. 2000, 10, 640–648. [Google Scholar]
- Kataoka, T.; Tschopp, J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-κB signaling pathway. Mol. Cell. Biol. 2004, 24, 2627–2636. [Google Scholar]
- Poukkula, M.; Kaunisto, A.; Hietakangas, V.; Denessiouk, K.; Katajamaki, T.; Johnson, M.S.; Sistonen, L.; Eriksson, J.E. Rapid turnover of c-FLIPshort is determined by its unique C-terminal tail. J. Biol. Chem. 2005, 280, 27345–27355. [Google Scholar]
- Ueffing, N.; Keil, E.; Freund, C.; Kuhne, R.; Schulze-Osthoff, K.; Schmitz, I. Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ. 2008, 15, 773–782. [Google Scholar]
- Ueffing, N.; Singh, K.K.; Christians, A.; Thorns, C.; Feller, A.C.; Nagl, F.; Fend, F.; Heikaus, S.; Marx, A.; Zotz, R.B.; Brade, J.; Schulz, W.A.; Schulze-Osthoff, K.; Schmitz, I.; Schwerk, C.A. A single nucleotide polymorphism determines protein isoform production of the human c-FLIP protein. Blood 2009, 114, 572–579. [Google Scholar]
- Bartke, T.; Siegmund, D.; Peters, N.; Reichwein, M.; Henkler, F.; Scheurich, P.; Wajant, H. p53 upregulates cFLIP, inhibits transcription of NF-κB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene 2001, 20, 571–580. [Google Scholar]
- Chandrasekaran, Y.; McKee, C.M.; Ye, Y.; Richburg, J.H. Influence of TRP53 status on FAS membrane localization, CFLAR (c-FLIP) ubiquitinylation, and sensitivity of GC-2spd (ts) cells to undergo FAS-mediated apoptosis. Biol. Reprod. 2006, 74, 560–568. [Google Scholar]
- Ueffing, N.; Schuster, M.; Keil, E.; Schulze-Osthoff, K.; Schmitz, I. Up-regulation of c-FLIP short by NFAT contributes to apoptosis resistance of short-term activated T cells. Blood 2008, 112, 690–698. [Google Scholar]
- Okamoto, K.; Fujisawa, J.; Reth, M.; Yonehara, S. Human T-cell leukemia virus type-I oncoprotein Tax inhibits fas-mediated apoptosis by inducing cellular FLIP through activation of NF-κB. Genes Cells 2006, 11, 177–191. [Google Scholar]
- Li, W.; Zhang, X.; Olumi, A.F. MG-132 Sensitizes TRAIL-resistant prostate cancer cells by activating c-Fos/c-Jun heterodimers and repressing c-FLIPL. Cancer Res. 2007, 67, 2247–2255. [Google Scholar]
- Salon, C.; Eymin, B.; Micheau, O.; Chaperot, L.; Plumas, J.; Brambilla, C.; Brambilla, E.; Gazzeri, S. E2F1 induces apoptosis and sensitizes human lung adenocarcinoma cells to death-receptor-mediated apoptosis through specific downregulation of c-FLIPshort. Cell Death Differ. 2006, 13, 260–272. [Google Scholar]
- Merdzhanova, G.; Edmond, V.; De Seranno, S.; Van den Broeck, A.; Corcos, L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008, 15, 1815–1823. [Google Scholar]
- Panner, A.; James, C.D.; Berger, M.S.; Pieper, R.O. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multiforme cells. Mol. Cell. Biol. 2005, 25, 8809–8823. [Google Scholar]
- Panner, A.; Nakamura, J.L.; Parsa, A.T.; Rodriguez-Viciana, P.; Berger, M.S.; Stokoe, D.; Pieper, R.O. mTOR-independent translational control of the extrinsic cell death pathway by RalA. Mol. Cell. Biol. 2006, 26, 7345–7357. [Google Scholar]
- Panner, A.; Parsa, A.T.; Pieper, R.O. Translational regulation of TRAIL sensitivity. Cell Cycle 2006, 5, 147–150. [Google Scholar]
- Krueger, A.; Fas, S.C.; Giaisi, M.; Bleumink, M.; Merling, A.; Stumpf, C.; Baumann, S.; Holtkotte, D.; Bosch, V.; Krammer, P.H.; Li-Weber, M. HTLV-1 tax protects against CD95-mediated apoptosis by induction of the cellular FLICE-inhibitory protein (c-FLIP). Blood 2006, 107, 3933–3939. [Google Scholar]
- Bleumink, M.; Köhler, R.; Giaisi, M.; Proksch, P.; Krammer, P.H.; Li-Weber, M. Rocaglamide breaks TRAIL resistance in HTLV-1-associated adult T-Cell leukemia/lymphoma by translational suppression of c-FLIP expression. Cell Death Differ. 2011, 18, 362–370. [Google Scholar]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIPL turnover. Cell 2006, 124, 601–613. [Google Scholar]
- Kaunisto, A.; Kochin, V.; Asaoka, T.; Mikhailov, A.; Poukkula, M.; Meinander, A.; Eriksson, J.E. PKC-mediated phosphorylation regulates c-FLIP ubiquitylation and stability. Cell Death Differ. 2009, 16, 1215–1226. [Google Scholar]
- Wang, Q.; Wang, X.; Zhou, Y.; Evers, B.M. PKCδ-mediated regulation of FLIP expression in human colon cancer cells. Int. J. Cancer 2006, 118, 326–334. [Google Scholar]
- Jung, S.N.; Park, I.J.; Kim, M.J.; Kang, I.; Choe, W.; Kim, S.S.; Ha, J. Down-regulation of AMP-activated protein kinase sensitizes DU145 carcinoma to Fas-induced apoptosis via c-FLIP degradation. Exp. Cell Res. 2009, 315, 2433–2441. [Google Scholar]
- Longley, D.B.; Wilson, T.R.; McEwan, M.; Allen, W.L.; McDermott, U.; Galligan, L.; Johnston, P. G c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 2006, 25, 838–848. [Google Scholar]
- Wilson, T.R.; McLaughlin, K.M.; McEwan, M.; Sakai, H.; Rogers, K.M.; Redmond, K.M.; Johnston, P.G.; Longley, D.B. c-FLIP: A key regulator of colorectal cancer cell death. Cancer Res. 2007, 6, 5754–5762. [Google Scholar]
- Haag, C.; Stadel, D.; Zhou, S.; Bachem, M.G.; Möller, P.; Debatin, K.M.; Fulda, S. Identification of c-FLIPL and c-FLIPS as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut 2011, 60, 225–237. [Google Scholar]
- Kauh, J.; Fan, S.; Xia, M.; Yue, P.; Yang, L.; Khuri, F.R.; Sun, S.Y. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS One 2010, 5, e10376. [Google Scholar]
- El-Gazzar, A.; Wittinger, M.; Perco, P.; Anees, M.; Horvat, R.; Mikulits, W.; Grunt, T.W.; Mayer, B.; Krainer, M. The role of c-FLIP in ovarian cancer: Chaperoning tumor cells from immunosurveillance and increasing their invasive potential. Gynecol. Oncol. 2010, 117, 451–459. [Google Scholar]
- Park, S.J.; Kim, M.J.; Kim, H.B.; Sohn, H.Y.; Bae, J.H.; Kang, C.D.; Kim, S.H. Trichostatin A sensitizes human ovarian cancer cells to TRAIL-induced apoptosis by downregulation of c-FLIPL via inhibition of EGFR pathway. Biochem. Pharmacol. 2009, 77, 1328–1336. [Google Scholar]
- Nam, S.Y.; Jung, G.A.; Hur, G.C.; Chung, H.Y.; Kim, W.H.; Seol, D.W.; Lee, B.L. Upregulation of FLIPS by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Sci. 2003, 94, 1066–1073. [Google Scholar]
- Day, T.W.; Huang, S.; Safa, A.R. c-FLIP knockdown induces ligand-independent DR5-, FADD-, caspase-8-, and caspase-9-dependent apoptosis in breast cancer cells. Biochem. Pharmacol. 2008, 76, 1694–704. [Google Scholar]
- Tiwary, R.; Yu, W.; Li, J.; Park, S.K.; Sanders, B.G.; Kline, K. Role of endoplasmic reticulum stress in alpha-TEA mediated TRAIL/DR5 death receptor dependent apoptosis. PLoS One 2010, 5, e11865. [Google Scholar]
- Zhang, X.; Zhang, L.; Yang, H.; Huang, X.; Otu, H.; Libermann, T.A.; DeWolf, W.C.; Khosravi-Far, R.; Olumi, A.F. c-Fos as a proapoptotic agent in TRAIL-induced apoptosis in prostate cancer cells. Cancer Res. 2007, 67, 9425–9434. [Google Scholar]
- Yang, B.F.; Xiao, C.; Li, H.; Yang, S.J. Resistance to Fas-mediated apoptosis in malignant tumours is rescued by KN-93 and cisplatin via downregulation of c-FLIP expression and phosphorylation. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1245–1251. [Google Scholar]
- McLornan, D.P.; Barrett, H.L.; Cummins, R.; McDermott, U.; McDowell, C.; Conlon, S.J.; Coyle, V.M.; Van Schaeybroeck, S.; Wilson, R.; Kay, E.W.; Longley, D.B.; Johnston, P.G. Prognostic significance of TRAIL signaling molecules in stage II and III colorectal cancer. Clin. Cancer Res. 2010, 16, 3442–3451. [Google Scholar]
- Ullenhag, G.J.; Mukherjee, A.; Watson, N.F.; Al-Attar, A.H.; Scholefield, J.H.; Durrant, L.G. Overexpression of FLIPL is an independent marker of poor prognosis in colorectal cancer patients. Clin. Cancer Res. 2007, 13, 5070–5075. [Google Scholar]
- Korkolopoulou, P.; Goudopoulou, A.; Voutsinas, G.; Thomas-Tsagli, E.; Kapralos, P.; Patsouris, E.; Saetta, A.A. c-FLIP expression in bladder urothelial carcinomas: Its role in resistance to Fas-mediated apoptosis and clinicopathologic correlations. Urology 2004, 63, 1198–1204. [Google Scholar]
- Wang, W.; Wang, S.; Song, X.; Sima, N.; Xu, X.; Luo, A.; Chen, G.; Deng, D.; Xu, Q.; Meng, L.; Lu, Y.; Ma, D. The relationship between c-FLIP expression and human papillomavirus E2 gene disruption in cervical carcinogenesis. Gynecol. Oncol. 2007, 105, 571–577. [Google Scholar]
- Valnet-Rabier, M.B.; Challier, B.; Thiebault, S.; Angonin, R.; Margueritte, G.; Mougin, C.; Kantelip, B.; Deconinck, E.; Cahn, J.Y.; Fest, T. c-FLIP protein expression in Burkitt's lymphomas is associated with a poor clinical outcome. Br. J. Haematol. 2005, 128, 767–773. [Google Scholar]
- Valente, G.; Manfroi, F.; Peracchio, C.; Nicotra, G.; Castino, R.; Nicosia, G.; Kerim, S.; Isidoro, C. cFLIP expression correlates with tumour progression and patient outcome in non-hodgkin lymphomas of low grade of malignancy. Br. J. Haematol. 2006, 132, 560–570. [Google Scholar]
- Li, X.; Pan, X.; Zhang, H.; Lei, D.; Liu., D.; Xu., F.; Luan, X. Overexpression of cFLIP in head and neck squamous cell carcinoma and its clinicopathologic correlations. J. Cancer Res. Clin. Oncol. 2008, 134, 609–615. [Google Scholar]
- Zhou, X.D.; Yu, J.P.; Liu, J.; Luo, H.S.; Chen, H.X.; Yu, HG. HG Overexpression of cellular FLICE-inhibitory protein (FLIP) in gastric adenocarcinoma. Clin. Sci. (Lond.) 2004, 106, 397–405. [Google Scholar]
- Zhang, N.; Hopkins, K.; He, Y.W. The long isoform of cellular FLIP is essential for T lymphocyte Proliferation through an NF-κB-independent pathway. J. Immunol. 2008, 180, 5506–5511. [Google Scholar]
- Yeh, W.C.; Itie, A.; Elia, A.J.; Ng, M.; Shu, H.B.; Wakeham, A.; Mirtsos, C.; Suzuki, N.; Bonnard, M.; Goeddel, D.V.; Mak, T.W. Requirement for casper (c-FLIP) in regulation of death receptor-Induced apoptosis and embryonic development. Immunity 2000, 12, 633–642. [Google Scholar]
- Melki, M.T.; Saïdi, H.; Dufour, A.; Olivo-Marin, J.C.; Gougeon, M.L. Escape of HIV-1-infected dendritic cells from TRAIL-mediated NK cell cytotoxicity during NK-DC cross-talk—A pivotal role of HMGB1. PLoS Pathog. 2010, 6, e1000862. [Google Scholar]
- Bagnol., M.; Canevari, S.; Mezzanzanica, D. Cellular FLICE-inhibitory protein (c-FLIP) signaling: A key regulator of receptor-mediated apoptosis in physiologic context and cancer. Int. J. Biochem. Cell Biol. 2010, 42, 210–213. [Google Scholar]
- Testa, U. TRAIL/TRAIL-R in hematologic malignancies. J. Cell. Biochem. 2010, 110, 21–34. [Google Scholar]
- Yang, J.K. FLIP as an anti-cancer therapeutic target. Yonsei Med. J. 2008, 49, 19–27. [Google Scholar]
- Yu, J.W.; Shi, Y. FLIP and the death effector domain family. Oncogene 2008, 27, 6216–6227. [Google Scholar]
- Day, T.W.; Safa, A.R. RNA interference in cancer: Targeting the anti-apoptotic protein c-FLIP for drug discovery. Mini Rev. Med. Chem. 2009, 9, 741–748. [Google Scholar]
- Yu, J.W.; Jeffrey, P.D.; Shi, Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc. Natl. Acad. Sci. USA 2009, 106, 8169–8174. [Google Scholar]
- Boatright, K.M.; Deis, C.; Denault, J.-B.; Sutherlin, D.P.; Salvesen, G.S. Activation of caspases-8 and -10 by FLIPL. Biochem. J. 2004, 382, 651–657. [Google Scholar]
- Walczak, H.; Haas, T.L. Biochemical analysis of the native TRAIL death-inducing signaling complex. Methods Mol. Biol. 2008, 414, 221–239. [Google Scholar]
- Kim, S.; Lee, T.J.; Park, J.W.; Kwon, T.K. Overexpression of cFLIPs inhibits oxaliplatin-mediated apoptosis through enhanced XIAP stability and Akt activation in human renal cancer cells. J. Cell. Biochem. 2008, 105, 971–979. [Google Scholar]
- Li, F.Y.; Jeffrey, P.D.; Yu, J.W.; Shi, Y. Crystal structure of a viral FLIP: Insights into FLIP-mediated inhibition of death receptor signaling. J. Biol. Chem. 2006, 281, 2960–2968. [Google Scholar]
- Budd, R.C.; Yeh, W.C.; Tschopp, J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol. 2006, 6, 196–204. [Google Scholar]
- Misra, R.S.; Russell, J.Q.; Koenig, A.; Hinshaw-Makepeace, J.A.; Wen, R.; Wang, D.; Huo, H.; Littman, D.R.; Ferch, U.; Ruland, J.; Thome, M.; Budd, R.C. Caspase-8 and c-FLIPL associate in lipid rafts with NF-κB adaptors during T cell activation. J. Biol. Chem. 2007, 282, 19365–19374. [Google Scholar]
- Leverkus, M.; Diessenbacher, P.; Geserick, P. FLIPing the coin? Death receptor-mediated signals during skin tumorigenesis. Exp. Dermatol. 2008, 17, 614–622. [Google Scholar]
- Zhang, N.; Hopkins, K.; He, Y.W. The long isoform of cellular FLIP is essential for T lymphocyte proliferation through an NF-κB-independent pathway. J. Immunol. 2008, 180, 5506–5511. [Google Scholar]
- Jin, T.G.; Kurakin, A.; Benhaga, N.; Abe, K.; Mohseni, M.; Sandra, F.; Song, K.; Kay, B.K.; Khosravi-Far, R. Fas-associated protein with death domain (FADD)-independent recruitment of c-FLIPL to death receptor 5. J. Biol. Chem. 2004, 279, 55594–55601. [Google Scholar]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; Jung, J.U. FLIP-mediated autophagy regulation in cell death control. Nat. Cell. Biol. 2009, 11, 1355–1362. [Google Scholar]
- Chaudhary, P.M.; Eby, M.T.; Jasmin, A.; Kumar, A.; Liu, L.; Hood, L. Activation of the NF-κB pathway by caspase 8 and its homologs. Oncogene 2000, 19, 4451–4460. [Google Scholar]
- Fang, L.W.; Tai, T.S.; Yu, W.N.; Liao, F.; Lai, M.Z. Phosphatidylinositide 3-kinase priming couples c-FLIP to T cell activation. J. Biol. Chem. 2004, 279, 13–18. [Google Scholar]
- Dohrman, A.; Kataoka, T.; Cuenin, S.; Russell, J.Q.; Tschopp, J.; Budd, R.C. Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-kappa B activation. J. Immunol. 2005, 174, 5270–5278. [Google Scholar]
- Golks, A.; Brenner, D.; Krammer, P.H.; Lavrik, I.N. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-κB activation. J. Exp. Med. 2006, 203, 1295–1305. [Google Scholar]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIPL turnover. Cell 2006, 124, 601–613. [Google Scholar]
- Quintavalle, C.; Incoronato, M.; Puca, L.; Acunzo, M.; Zanca, C.; Romano, G.; Garofalo, M; Iaboni, M.; Croce, C.M.; Condorelli, G. c-FLIPL enhances anti-apoptotic Akt functions by modulation of Gsk3β activity. Cell Death Differ. 2010, 17, 1908–1916. [Google Scholar]
- Kim, M.J.; Kim, H.B.; Bae, J.H.; Lee, J.W.; Park, S.J.; Kim, D.W.; Park, S.I.; Kang, C.D.; Kim, S.H. Sensitization of human K562 leukemic cells to TRAIL-induced apoptosis by inhibiting the DNA-PKcs/Akt-mediated cell survival pathway. Biochem. Pharmacol. 2009, 78, 573–582. [Google Scholar]
- Panner, A.; Crane, C.A.; Weng, C.; Feletti, A.; Fang, S.; Parsa, A.T.; Pieper, R.O. Ubiquitin-specific protease 8 links the PTEN-Akt-AIP4 pathway to the control of FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res. 2010, 70, 5046–5053. [Google Scholar]
- Panner, A.; Crane, C.A.; Weng, C.; Feletti, A.; Parsa, A.T.; Pieper, R.O. A novel PTEN-dependent link to ubiquitination controls FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res. 2009, 69, 7911–7916. [Google Scholar]
- Kim, Y.Y.; Park, B.J.; Seo, G.J.; Lim, J.Y.; Lee, S.M.; Kimm, K.C.; Park, C.; Kim, J.; Park, S.I. Long form of cellular FLICE-inhibitory protein interacts with daxx and prevents Fas-induced JNK activation. Biochem. Biophys. Res. Commun. 2003, 312, 426–433. [Google Scholar]
- Nakajima, A.; Komazawa-Sakon, S.; Takekawa, M.; Sasazuki, T.; Yeh, W.C.; Yagita, H.; Okumura, K.; Nakano, H. An antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits the JNK pathway. EMBO J. 2006, 25, 5549–5559. [Google Scholar]
- Yang, B.F.; Xiao, C.; Roa, W.H.; Krammer, P.H.; Hao, C. Calcium/calmodulin-dependent protein kinase II regulation of c-FLIP expression and phosphorylation in modulation of Fas-mediated signaling in malignant glioma cells. J. Biol. Chem. 2003, 278, 7043–7050. [Google Scholar]
- Xiao, C.; Yang, B.F.; Song, J.H.; Schulman, H.; Li, L.; Hao, C. Inhibition of CaMKII-mediate c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp. Cell. Res. 2005, 304, 244–255. [Google Scholar]
- Higuchi, H.; Yoon, J.H.; Grambihler, A.; Werneburg, N.; Bronk, S.F.; Gores, G.J. Bile acids stimulate cFLIP phosphorylation enhancing TRAIL-mediated apoptosis. J. Biol. Chem. 2003, 278, 454–461. [Google Scholar]
- Gilot, D.; Serandour, A.L.; Ilyin, G.P.; Lagadic-Gossmann, D.; Loyer, P.; Corlu, A.; Coutant, A.; Baffet, G.; Peter, M.E.; Fardel, O.; Guguen-Guillouzo, C. A role for caspase-8 and c-FLIPL in proliferation and cell-cycle progression of primary hepatocytes. Carcinogenesis 2005, 26, 2086–2094. [Google Scholar]
- Stagni, V.; Mingardi, M.; Santini, S.; Giaccari, D.; Barilà, D. ATM kinase activity modulates cFLIP protein levels: Potential interplay between DNA damage signalling and TRAIL-induced apoptosis. Carcinogenesis 2010, 31, 1956–1963. [Google Scholar]
- Naito, M.; Katayama, R.; Ishioka, T.; Suga, A.; Takubo, K.; Nanjo, M.; Hashimoto, C.; Taira, M.; Takada, S.; Takada, R.; Kitagawa, M.; Matsuzawa, S.; Reed, J.C.; Tsuruo, T. Cellular FLIP inhibits β-catenin ubiquitylation and enhances Wnt signaling. Mol. Cell. Biol. 2004, 24, 8418–8427. [Google Scholar]
- Katayama, R.; Ishioka, T.; Takada, S.; Takada, R.; Fujita, N.; Tsuruo, T.; Naito, M. Modulation of Wnt signaling by the nuclear localization of cellular FLIP-L. J. Cell Sci. 2010, 123, 23–28. [Google Scholar]
- Zhang, L.; Ren, X.; Alt, E.; Bai, X.; Huang, S.; Xu, Z.; Lynch, P.M.; Moyer, M.P.; Wen, X.F.; Wu, X. Chemoprevention of colorectal cancer by targeting APC-deficient cells for apoptosis. Nature 2010, 464, 1058–1061. [Google Scholar]
- Chen, H.X.; Liu, Y.J.; Zhou, X.D.; Luo, R.Y. Expression of cellular FLICE/caspase-8 inhibitory protein is associated with malignant potential in endometrial carcinoma. Int. J. Gynecol. Cancer 2005, 15, 663–670. [Google Scholar]
- Wang, W.; Wang, S.; Song, X.; Sima, N.; Xu, X.; Luo, A.; Chen, G.; Deng, D.; Xu, Q.; Meng, L.; Lu, Y.; Ma, D. The relationship between c-FLIP expression and human papillomavirus E2 gene disruption in cervical carcinogenesis. Gynecol. Oncol. 2007, 105, 571–577. [Google Scholar]
- Wang, W.; Fang, Y.; Sima, N.; Li, Y.; Li, W.; Li, L.; Han, L.; Liao, S.; Han, Z.; Gao, Q.; Li, K.; Deng, D.; Meng, L.; Zhou, J.; Wang, S.; Ma, D. Triggering of death receptor apoptotic signaling by human papillomavirus 16 E2 protein in cervical cancer cell linesis mediated by interaction with c-FLIP. Apoptosis 2011, 16, 55–66. [Google Scholar]
- Ishioka, T.; Katayama, R.; Kikuchi, R.; Nishimoto, M.; Takada, S.; Takada, R.; Matsuzawa, S.; Reed, J.C.; Tsuruo, T.; Naito, M. Impairment of the ubiquitin-proteasome system by cellular FLIP. Genes Cells 2007, 12, 735–744. [Google Scholar]
- Gao, S.; Wang, H.; Lee, P.; Melamed, J.; Li, C.X.; Zhang, F.; Wu, H.; Zhou, L.; Wang, Z. Androgen receptor and prostate apoptosis response factor-4 target the c-FLIP gene to determine survival and apoptosis in the prostate gland. J. Mol. Endocrinol. 2006, 36, 463–483. [Google Scholar]
- Park, M.A.; Zhang, G.; Mitchell, C.; Rahmani, M.; Hamed, H.; Hagan, M.P.; Yacoub., A.; Curiel, D.T.; Fisher, P.B.; Grant, S.; Dent, P. Mitogen-activated protein kinase kinase 1/2 inhibitors and 17-allylamino-17-demethoxygeldanamycin synergize to kill human gastrointestinal tumor cells in vitro via suppression of c-FLIP-s levels and activation of CD95. Mol. Cancer Ther. 2008, 7, 2633–2648. [Google Scholar]
- Panner, A.; Murray, J.C.; Berger, M.S.; Pieper, R.O. Heat shock protein 90α recruits FLIPS to the death-inducing signaling complex and contributes to TRAIL resistance in human glioma. Cancer Res. 2007, 67, 9482–9489. [Google Scholar]
- Walker, T.; Mitchell, C.; Park, M.A.; Yacoub, A.; Graf, M.; Rahmani, M.; Houghton, P.J.; Voelkel-Johnson, C.; Grant, S.; Dent, P. Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Mol. Pharmacol. 2009, 76, 342–355. [Google Scholar]
- Zhang, G.; Park, M.A.; Mitchell, C.; Hamed, H.; Rahmani, M.; Martin, A.P.; Curiel, D.T.; Yacoub, A.; Graf, M.; Lee, R.; Roberts, J.D.; Fisher, P.B.; Grant, S.; Dent, P. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin. Cancer Res. 2008, 14, 5385–5399. [Google Scholar]
- Mitchell, C.; Kabolizadeh, P.; Ryan, J.; Roberts, J.D.; Yacoub, A.; Curiel, D.T.; Fisher, P.B.; Hagan, M.P.; Farrell, N.P.; Grant, S.; Dent, P. Low-dose BBR3610 toxicity in colon cancer cells is p53-independent and enhanced by inhibition of epidermal growth factor receptor (ERBB1)-phosphatidyl inositol 3 kinase signaling. Mol. Pharmacol. 2007, 72, 704–714. [Google Scholar]
- Shim, E.; Lee, Y.S.; Kim, H.Y.; Jeoung, D. Down-regulation of c-FLIP increases reactive oxygen species, induces phosphorylation of serine/threonine kinase Akt, and impairs motility of cancer cells. Biotechnol. Lett. 2007, 29, 141–147. [Google Scholar]
- Park, D.; Shim, E.; Kim, Y.; Kim, Y.M.; Lee, H.; Choe, J.; Kang, D.; Lee, Y.S.; Jeoung, D. c-FLIP promotes the motility of cancer cells by activating FAK and ERK, and increasing MMP-9 expression. Mol. Cells 2008, 25, 184–195. [Google Scholar]
- Cano, C.E.; Motoo, Y.; Iovanna, J.L. Epithelial-to-mesenchymal transition in pancreatic adenocarcinoma. Sci. World J. 2010, 10, 1947–1957. [Google Scholar]
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 27–33. [Google Scholar]
- Kim, Y.; Jeoung, D. Role of CAGE, a novel cancer/testis antigen, in various cellular processes, including tumorigenesis, cytolytic T lymphocyte induction, and cell motility. J. Microbiol. Biotechnol. 2008, 18, 600–610. [Google Scholar]
- Kim, Y.; Park, H.; Jeoung, D. CAGE, a cancer/testis antigen, induces c-FLIPL and snail to enhance cell motility and increase resistance to an anti-cancer drug. Biotechnol. Lett. 2009, 31, 945–952. [Google Scholar]
- Day, T.W.; Sinn, A.L.; Huang, S.; Pollok, K.E.; Sandusky, G.E.; Safa, A.R. c-FLIP gene silencing eliminates tumor cells in breast cancer xenografts without affecting stromal cells. Anticancer Res. 2009, 29, 3883–3886. [Google Scholar]
- Logan, A.E.; Wilson, T.R.; Fenning, C.; Cummins, R.; Kay, E.; Johnston, P.G.; Longley, D.B. In vitro and in vivo characterisation of a novel c-FLIP-targeted antisense phosphorothioate oligonucleotide. Apoptosis 2010, 15, 1435–1443. [Google Scholar]
- Kinoshita, H.; Yoshikawa, H.; Shiiki, K.; Hamada, Y.; Nakajima, Y.; Tasaka, K. Cisplatin (CDDP) sensitizes human osteosarcoma cell to Fas/CD95-mediated apoptosis by down-regulating FLIP-L expression. Int. J. Cancer 2000, 88, 986–991. [Google Scholar]
- Abedini, M.R.; Muller, E.J.; Brun, J.; Bergeron, R.; Gray, D.A.; Tsang, B.K. Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res. 2008, 68, 4511–4517. [Google Scholar]
- Song, J.H.; Song, D.K.; Herlyn, M.; Petruk, K.C.; Hao, C. Cisplatin down-regulation of cellular Fas-associated death domain-like interleukin-1β-converting enzyme-like inhibitory proteins to restore tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human melanoma cells. Clin. Cancer Res. 2003, 9, 4255–4266. [Google Scholar]
- El-Zawahry, A.; McKillop, J.; Voelkel-Johnson, C. Doxorubicin increases the effectiveness of Apo2L/TRAIL for tumor growth inhibition of prostate cancer xenografts. BMC Cancer 2005, 5, 2. [Google Scholar]
- Chatterjee, D.; Schmitz, I.; Krueger, A.; Yeung, K.; Kirchhoff, S.; Krammer, P.H.; Peter, M.E.; Wyche, J.H.; Pantazis, P. Induction of apoptosis in 9-nitrocamptothecin-treated DU145 human prostate carcinoma cells correlates with de novo synthesis of CD95 and CD95 ligand and down-regulation of c-FLIPshort. Cancer Res. 2001, 61, 7148–7154. [Google Scholar]
- Yerbes, R.; López-Rivas, A. Itch/AIP4-independent proteasomal degradation of cFLIP induced by the histone deacetylase inhibitor SAHA sensitizes breast tumour cells to TRAIL. Invest. New Drugs 2010. [Google Scholar] [CrossRef]
- Lucas, D.M.; Alinari, L.; West, D.A.; Davis, M.E.; Edwards, R.B.; Johnson, A.J.; Blum, K.A.; Hofmeister, C.C.; Freitas, M.A.; Parthun, M.R.; Wang, D.; Lehman, A.; Zhang, X.; Jarjoura, D.; Kulp, S.K.; Croce, C.M.; Grever, M.R.; Chen, C.S.; Baiocchi, R.A.; Byrd, J.C. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS One 2010, 5, e10941. [Google Scholar]
- Bijangi-Vishehsaraei, K.; Saadatzadeh, M.R.; Huang, S.; Murphy, M.P.; Safa, AR. 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) Targets mRNA of the c-FLIP variants and induces apoptosis in MCF-7 human breast cancer cells. Mol. Cell. Biochem. 2010, 342, 133–142. [Google Scholar]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Sukhai, M.A.; Hurren, R.; Anyiwe, K.; Mao, X.; Suarez Saiz, F.; Gronda, M.; Eberhard, Y; MacLean, N.; Ketela, T.; Reed, J.C.; Moffat, J; Minden, M.D; Batey, R.A.; Schimmer, A.D. Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol. Cancer Ther. 2010, 9, 246–256. [Google Scholar]
- Schimmer, A.D.; Thomas, M.P.; Hurren, R.; Gronda, M.; Pellecchia, M.; Pond, G.R.; Konopleva, M.; Gurfinkel, D.; Mawji, I.A.; Brown, E.; Reed, J.C. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res. 2006, 66, 2367–2375. [Google Scholar]
- Mawji, I.A.; Simpson, C.D.; Gronda, M.; Williams, M.A.; Hurren, R.; Henderson, C.J.; Datti, A.; Wrana, J.L.; Schimmer, A.D. A chemical screen identifies anisomycin as an anoikis sensitizer that functions by decreasing FLIP protein synthesis. Cancer Res. 2007, 67, 8307–8315. [Google Scholar]
- Saleem, M.; Murtaza, I.; Witkowsky, O.; Kohl, A.M.; Maddodi, N. Lupeol triterpene, a novel diet-based microtubule targeting agent: Disrupts survivin/cFLIP activation in prostate cancer cells. Biochem. Biophys. Res. Commun. 2009, 388, 576–582. [Google Scholar]
- Murtaza, I.; Saleem, M.; Adhami, V.M.; Hafeez, B.B.; Mukhtar, H. Suppression of cFLIP by lupeol, a dietary triterpene, is sufficient to overcome resistance to TRAIL-mediated apoptosis in chemoresistant human pancreatic cancer cells. Cancer Res. 2009, 69, 1156–1165. [Google Scholar]
- Siddique, H.R.; Saleem, M. Beneficial health effects of lupeol triterpene: A review of preclinical studies. Life Sci. 2011, 88, 285–293. [Google Scholar]
- Yodkeeree, S.; Sung, B.; Limtrakul, P.; Aggarwal, B.B. Zerumbone enhances TRAIL-induced apoptosis through the induction of death receptors in human colon cancer cells: Evidence for an essential role of reactive oxygen species. Cancer Res. 2009, 69, 6581–6589. [Google Scholar]
- Lee, T.J.; Um, H.J.; Min, D.S.; Park, J.W.; Choi, K.S.; Kwon, T.K. Withaferin a sensitizes TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Radic. Biol. Med. 2009, 46, 1639–1649. [Google Scholar]
- Jani, T.S.; Devecchio, J.; Mazumdar, T.; Agyeman, A.; Houghton, J.A. Inhibition of NF-κB signaling by quinacrine is cytotoxic to human colon carcinoma cell lines and is synergistic in combination with TRAIL or oxaliplatin. J. Biol. Chem. 2010, 285, 19162–19172. [Google Scholar]
- Li, X.; Huang, Q.; Ong, C.N.; Yang, XF.; Shen, H.M. Chrysin sensitizes tumor necrosis factor-alpha-induced apoptosis in human tumor cells via suppression of NF-κB. Cancer Lett. 2010, 293, 109–116. [Google Scholar]
- Li, T.W.; Zhang, Q.; Oh, P; Xia, M; Chen, H.; Bemanian, S.; Lastra, N.; Circ, M.; Moyer, M.P.; Mato, J.M.; Aw, T.Y.; Lu, S.C. S-adenosylmethionine and methylthioadenosine inhibit cellular FLICE inhibitory protein expression and induce apoptosis in colon cancer cells. Mol. Pharmacol. 2009, 76, 192–200. [Google Scholar]
- Petronelli, A.; Saulle, E.; Pasquini, L.; Petrucci, E.; Mariani, G.; Biffoni, M.; Ferretti, G.; Scambia, G.; Benedetti-Panici, P.; Greggi, S.; Cognetti, F.; Russo, M.A.; Sporn, M.; Testa, U. High sensitivity of ovarian cancer cells to the synthetic triterpenoid CDDO-imidazolide. Cancer Lett. 2009, 282, 214–228. [Google Scholar]
- Charette, N.; De Saeger, C.; Lannoy, V.; Horsmans, Y.; Leclercq, I.; Stärkel, P. Salirasib inhibits the growth of hepatocarcinoma cell lines in vitro and tumor growth in vivo through ras and mTOR inhibition. Mol. Cancer 2010, 9, 256. [Google Scholar]
- Sharp, D.A.; Lawrence, D.A.; Ashkenazi, A. Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2005, 280, 19401–19409. [Google Scholar]
- Wilson, T.R.; McLaughlin, K.M.; McEwan, M.; Sakai, H.; Rogers, K.M.; Redmond, K.M.; Johnston, P.G.; Longley, D.B. c-FLIP: A key regulator of colorectal cancer cell death. Cancer Res. 2007, 67, 5754–5762. [Google Scholar]
- Jeon, Y.K.; Kim, H.; Park, S.O.; Choi, H.Y.; Kim, Y.A.; Park, S.S.; Kim, J.E.; Kim, Y.N.; Kim, C.W. Resistance to Fas-mediated apoptosis is restored by cycloheximide through the downregulation of cellular FLIPL in NK/T-cell lymphoma. Lab. Invest. 2005, 85, 874–884. [Google Scholar]
- Olsson, A.; Diaz, T.; Aguilar-Santelises, M.; Osterborg, A.; Celsing, F.; Jondal, M.; Osorio, L.M. Sensitization to TRAIL-induced apoptosis and modulation of FLICE-inhibitory protein in B chronic lymphocytic leukemia by actinomycin D. Leukemia 2001, 15, 1868–1877. [Google Scholar]
- Ganten, T.M.; Haas, T.L.; Sykora, J.; Stahl, H.; Sprick, M.R.; Fas, S.C.; Krueger, A.; Weigand, M.A.; Grosse-Wilde, A.; Stremmel, W.; Krammer, P.H.; Walczak, H. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ. 2004, 11 (Suppl. 1), S86–S96. [Google Scholar]
- Plissonnier, M.L.; Fauconnet, S.; Bittard, H.; Lascombe, I. Insights on distinct pathways of thiazolidinediones (PPARgamma ligand)-promoted apoptosis in TRAIL-sensitive or -resistant malignant urothelial cells. Int. J. Cancer 2010, 127, 1769–1784. [Google Scholar]
- Kim, Y.H.; Jung, E.M.; Lee, T.J.; Kim, S.H.; Choi, Y.H.; Park, J.W.; Park, J.W.; Choi, K.S.; Kwon, T.K. Rosiglitazone promotes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Radic. Biol. Med. 2008, 44, 1055–1068. [Google Scholar]
- Han, H.; Shin, S.W.; Seo, C.Y.; Kwon, H.C.; Han, JY.; Kim, I.H.; Kwak, JY.; Park, J.I. 15-deoxy-delta 12,14-prostaglandin J2 (15d-PGJ 2) sensitizes human leukemic HL-60 cells to tumor necrosis factor-related bpoptosis-inducing ligand (TRAIL)-induced apoptosis through Akt downregulation. Apoptosis 2007, 12, 2101–2114. [Google Scholar]
- Zou, W.; Liu, X.; Yue, P.; Khuri, F.R.; Sun, S.Y. PPARgamma ligands enhance TRAIL induced apoptosis through DR5 upregulation and c-FLIP downregulation in human lung cancer cells. Cancer Biol. Ther. 2007, 6, 99–106. [Google Scholar]
- Plissonnier, M.L.; Fauconnet, S.; Bittard, H.; Lascombe, I. Insights on distinct pathways of thiazolidinediones (PPARγ ligand)-promoted apoptosis in TRAIL-sensitive or -resistant malignant urothelial cells. Int. J. Cancer 2010, 127, 1769–1784. [Google Scholar]
- Tobinai, K. Proteasome inhibitor, bortezomib, for myeloma and lymphoma. Int. J. Clin. Oncol. 2007, 12, 318–326. [Google Scholar]
- Wu, W.K.; Cho, C.H.; Lee, C.W.; Wu, K.; Fan, D.; Yu, J.; Sung, J.J. Proteasome inhibition: A new therapeutic strategy to cancer treatment. Cancer Lett. 2010, 293, 15–22. [Google Scholar]
- Choi, M.R.; Najafi, F.; Safa, A.R.; Drexler, H.C. Analysis of changes in the proteome of HL-60 promyeloid leukemia cells induced by the proteasome inhibitor PSI. Biochem. Pharmacol. 2008, 75, 2276–2288. [Google Scholar]
- Van Geelen, C.M.; de Vries, E.G.; de Jong, S. Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: Paving the road to patient-tailored therapy. Drug Resist. Updat. 2004, 7, 345–358. [Google Scholar]
- Kabore, A.F.; Sun, J.; Hu, X.; McCrea, K.; Johnston, J.B.; Gibson, S.B. The TRAIL apoptotic pathway mediates proteasome inhibitor induced apoptosis in primary chronic lymphocytic leukemia cells. Apoptosis 2006, 11, 1175–1193. [Google Scholar]
- Perez, L.E.; Parquet, N.; Meads, M.; Anasetti, C.; Dalton, W. Bortezomib restores stroma-mediated APO2L/TRAIL apoptosis resistance in multiple myeloma. Eur. J. Haematol. 2010, 84, 212–222. [Google Scholar]
- Seki, N.; Toh, U.; Sayers, T.J.; Fujii, T.; Miyagi, M.; Akagi, Y.; Kusukawa, J.; Kage, M.; Shirouzu, K.; Yamana, H. Bortezomib sensitizes human esophageal squamous cell carcinoma cells to TRAIL-mediated apoptosis via activation of both extrinsic and intrinsic apoptosis pathways. Mol. Cancer Ther. 2010, 9, 1842–1851. [Google Scholar]
- Sayers, T.J.; Brooks, A.D.; Koh, C.Y.; Ma, W.; Seki, N.; Raziuddin, A.; Blazar, B.R.; Zhang, X.; Elliott, P.J.; Murphy, W.J. The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood 2003, 102, 303–310. [Google Scholar]
- Liu, X.; Yue, P.; Chen, S.; Hu, L.; Lonial, S.; Khuri, F.R.; Sun, S.Y. The proteasome inhibitor PS-341 (Bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007, 67, 4981–4988. [Google Scholar]
- Zhao, X.; Qiu, W.; Kung, J.; Zhao, X.; Peng, X.; Yegappan, M.; Yen-Lieberman, B.; His, E.D. Bortezomib induces caspase-dependent apoptosis in hodgkin lymphoma cell lines and is associated with reduced c-FLIP expression: A gene expression profiling study with implications for potential combination therapies. Leuk. Res. 2008, 32, 275–285. [Google Scholar]
- Ge, R.; Wang, Z.; Zeng, Q.; Xu, X.; Olumi, A.F. F-box protein 10, an NF-κB-dependent anti-apoptotic protein, regulates TRAIL-induced apoptosis through modulating c-Fos/c-FLIP pathway. Cell Death Differ. 2011. [Google Scholar] [CrossRef]
- Ri, M.; Iida, S.; Ishida, T.; Ito, A.; Yano, H.; Inagaki, A.; Ding, J.; Kusumoto, S.; Komatsu, H.; Utsunomiya, A.; Ueda, R. Bortezomib-induced apoptosis in mature T-cell lymphoma cells partially depends on upregulation of noxa and functional repression of Mcl-1. Cancer Sci. 2009, 100, 341–348. [Google Scholar]
- Hamaï, A.; Richon, C.; Meslin, F.; Faure, F.; Kauffmann, A.; Lecluse, Y.; Jalil, A.; Larue, L.; Avril, M.F.; Chouaib, S.; Mehrpour, M. Imatinib enhances human melanoma cell susceptibility to TRAIL-induced cell death: Relationship to Bcl-2 family and caspase activation. Oncogene 2006, 25, 7618–7634. [Google Scholar]
- Park, S.J.; Kim, M.J.; Kim, H.B.; Kang, C.D.; Kim, S.H. Sensitization of imatinib-resistant CML cells to TRAIL-induced apoptosis is mediated through down-regulation of Bcr-Abl as well as c-FLIP. Biochem. J. 2009, 420, 73–81. [Google Scholar]
- Son, Y.G.; Kim, E.H.; Kim, J.Y.; Kim, S.U.; Kwon, T.K.; Yoon, A.R.; Yun, C.O.; Choi, K.S. Silibinin sensitizes human glioma cells to TRAIL-mediated apoptosis via DR5 up-regulation and down-regulation of c-FLIP and survivin. Cancer Res. 2007, 67, 8274–8284. [Google Scholar]
- Lin, Y.; Liu, X.; Yue, P.; Benbrook, D.M.; Berlin, K.D.; Khuri, F.R.; Sun, S.Y. Involvement of c-FLIP and survivin down-regulation in flexible heteroarotinoid-induced apoptosis and enhancement of TRAIL-initiated apoptosis in lung cancer cells. Mol. Cancer Ther. 2008, 7, 3556–3565. [Google Scholar]
- Siegelin, M.D.; Siegelin, Y.; Habel, A.; Gaiser, T. Genistein enhances proteasomal degradation of the short isoform of FLIP in malignant glioma cells and thereby augments TRAIL-mediated apoptosis. Neurosci. Lett. 2009, 453, 92–97. [Google Scholar]
- Katz, S.I.; Zhou, L.; Chao, G.; Smith, C.D.; Ferrara, T.; Wang, W.; Dicker, D.T.; El-Deiry, W.S. Sorafenib inhibits ERK1/2 and MCL-1 (L) phosphrylation levels resulting in caspase-independent cell death on malignant pleural mesothelioma. Cancer Biol. Ther. 2009, 8, 2406–2416. [Google Scholar]
- Lee, J.; Hwangbo, C; Lee, J.J.; Seo, J.; Lee, J.H. The sesquiterpene lactone eupatolide sensitizes breast cancer cells to TRAIL through down-regulation of c-FLIP expression. Oncol. Rep. 2010, 23, 229–237. [Google Scholar]
- Jia, L.; Yu, W.; Wang, P.; Sanders, BG.; Kline, K. In vivo and in vitro studies of anticancer actions of α-TEA for human prostate cancer cells. Prostate 2008, 68, 849–860. [Google Scholar]
- Yu, W.; Tiwary, R.; Li, J.; Park, S.K.; Jia, L.; Xiong, A.; Simmons-Menchaca, M.; Sanders, B.G.; Kline, K. α-TEA induces apoptosis of human breast cancer cells via activation of TRAIL/DR5 death receptor pathway. Mol. Carcinog. 2010, 49, 964–973. [Google Scholar]
- Jang, J.H.; Park, J.S.; Lee, T.J.; Kwon, T.K. Transglutaminase 2 expression levels regulate sensitivity to cystamine plus TRAIL-mediated apoptosis. Cancer Lett. 2010, 287, 224–230. [Google Scholar]
- Kang, Y.J.; Kim, I.Y.; Kim, E.H.; Yoon, M.J.; Kim, S.U.; Kwon, T.K.; Choi, K.S. Paxilline enhances TRAIL-mediated apoptosis of glioma cells via modulation of c-FLIP, survivin and DR5. Exp. Mol. Med. 2010, 43, 24–34. [Google Scholar]
- Pawar, P.; Ma, L.; Byon, C.H.; Liu, H.; Ahn, E.Y.; Jhala, N.; Arnoletti, J.P.; McDonald, J.M.; Chen, Y. Molecular mechanisms of tamoxifen therapy for cholangiocarcinoma: Role of calmodulin. Clin. Cancer Res. 2009, 15, 1288–1296. [Google Scholar]
- Lee, S.J.; Noh, H.J.; Sung, E.G.; Song, I.H.; Kim, J.Y.; Kwon, T.K.; Lee, T.J. Berberine sensitizes TRAIL-induced apoptosis through proteasome-mediated downregulation of c-FLIP and Mcl-1 proteins. Int. J. Oncol. 2011, 38, 485–492. [Google Scholar]
- Rogers, K.M.; Thomas, M.; Galligan, L.; Wilson, T.R.; Allen, W.L.; Sakai, H.; Johnston, P.G.; Longley, D.B. Cellular FLICE-inhibitory protein regulates chemotherapy-induced apoptosis in breast cancer cells. Mol. Cancer Ther. 2007, 6, 1544–1551. [Google Scholar]
- Iwase, M.; Takaoka, S.; Uchida, M.; Yoshiba, S.; Kondo, G.; Watanabe, H.; Ohashi, M.; Nagumo, M. Epidermal growth factor receptor inhibitors enhance susceptibility to Fas-mediated apoptosis in oral squamous cell carcinoma cells. Oral Oncol. 2008, 44, 361–368. [Google Scholar]
- Yu, Z.; Wang, R.; Xu, L.; Xie, S.; Dong, J.; Jing, Y. β-Elemene piperazine derivatives induce apoptosis in human leukemia cells through downregulation of c-FLIP and generation of ROS. PLoS One 2011, 6, e15843. [Google Scholar]
- Jang, J.H.; Lee, T.J.; Yang, E.S; Min. do, S.; Kim, Y.H.; Kim, S.H.; Choi, Y.H.; Park, J.W.; Choi, K.S.; Kwon, T.K. Compound C sensitizes caki renal cancer cells to TRAIL-induced apoptosis through reactive oxygen species-mediated down-regulation of c-FLIPL and Mcl-1. Exp. Cell Res. 2010, 316, 2194–2203. [Google Scholar]
- Rishi, L.; Gahlot, S.; Kathania, M.; Majumdar, S. Pentoxifylline induces apoptosis in vitro in cutaneous T cell lymphoma (HuT-78) and enhances FasL mediated killing by upregulating Fas expression. Biochem. Pharmacol. 2009, 77, 30–45. [Google Scholar]
- Liu, J.; Fu, X.Q.; Zhou, W.; Yu, HG.; Yu, J.P.; Luo, H.S. LY294002 potentiates the anti-cancer effect of oxaliplatin for gastric cancer via death receptor pathway. World J. Gastroenterol. 2011, 17, 181–190. [Google Scholar]
- Drakos, E.; Atsaves, V.; Schlette, E.; Li, J.; Papanastasi, I.; Rassidakis, G.Z.; Medeiros, L.J. The therapeutic potential of p53 reactivation by nutlin-3a in ALK+ anaplastic large cell lymphoma with wild-type or mutated p53. Leukemia 2009, 23, 2290–2299. [Google Scholar]
- Festuccia, C.; Dondi, D.; Piccolella, M.; Locatelli, A.; Gravina, G.L.; Tombolini, V.; Motta, M. Ozarelix, a fourth generation GnRH antagonist, induces apoptosis in hormone refractory androgen receptor negative prostate cancer cells modulating expression and activity of death receptors. Prostate 2010, 70, 1340–1349. [Google Scholar]
- Lu, G.; Liu, Y.; Ji, B.; Wei, F.; Hao, C; Wang, G. Synergistic effect of celecoxib on TRAIL-induced apoptosis in hepatocellular carcinoma cells. Cancer Invest. 2010, 28, 629–634. [Google Scholar]
- Cheung, H.H.; Mahoney, D.J.; Lacasse, E.C; Korneluk, R.G. Down-regulation of c-FLIP enhances death of cancer cells by SMAC mimetic compound. Cancer Res. 2009, 69, 7729–7738. [Google Scholar]
- Ding, W.; Cai, T.; Zhu, H.; Wu, R.; Tu, C.; Yang, L.; Lu, W.; He, Q.; Yang, B. Synergistic antitumor effect of TRAIL in combination with sunitinib in vitro and in vivo. Cancer Lett. 2010, 293, 158–166. [Google Scholar]
- Chen, F.; Zhu, H.H.; Zhou, L.F.; Wu, S.S.; Wang, J.; Chen, Z. Inhibition of c-FLIP expression by miR-512-3p contributes to taxol-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2010, 23, 1457–1462. [Google Scholar]
- Stagni, V.; di Bari, M.G.; Cursi, S.; Condò, I.; Cencioni, M.T.; Testi, R.; Lerenthal, Y.; Cundari, E.; Barilà, D. ATM kinase activity modulates Fas sensitivity through the regulation of FLIP in lymphoid cells. Blood 2008, 111, 829–837. [Google Scholar]
- Ivanov, V.N.; Zhou, H.; Partridge, M.A.; Hei, T.K. Inhibition of ataxia telangiectasia mutated kinase activity enhances TRAIL-mediated apoptosis in human melanoma cells. Cancer Res. 2009, 69, 3510–3519. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Safa, A.R.; Pollok, K.E. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers 2011, 3, 1639-1671. https://doi.org/10.3390/cancers3021639
Safa AR, Pollok KE. Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers. 2011; 3(2):1639-1671. https://doi.org/10.3390/cancers3021639
Chicago/Turabian StyleSafa, Ahmad R., and Karen E. Pollok. 2011. "Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy" Cancers 3, no. 2: 1639-1671. https://doi.org/10.3390/cancers3021639
APA StyleSafa, A. R., & Pollok, K. E. (2011). Targeting the Anti-Apoptotic Protein c-FLIP for Cancer Therapy. Cancers, 3(2), 1639-1671. https://doi.org/10.3390/cancers3021639